Redox-Mediated Polymer Catalyst for Lithium-Air Batteries with High Round-Trip Efficiency


Abstract

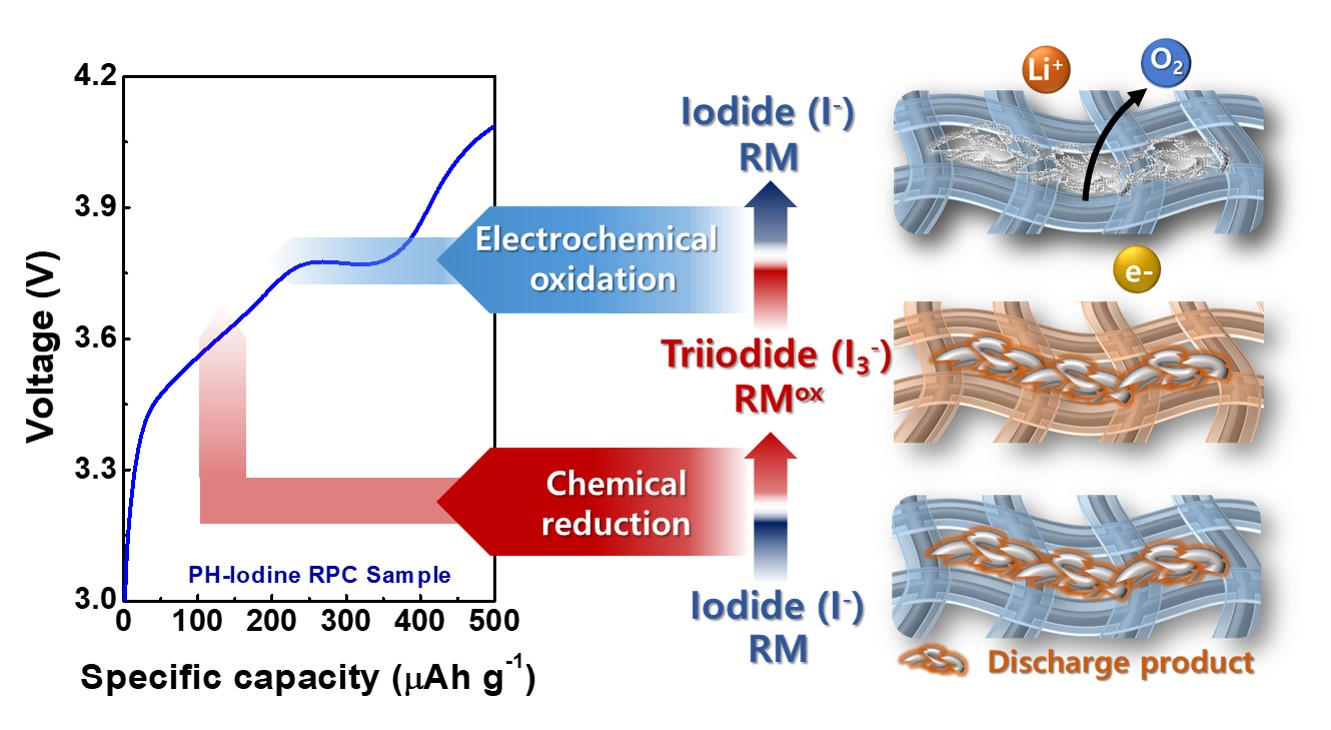{kind=link}
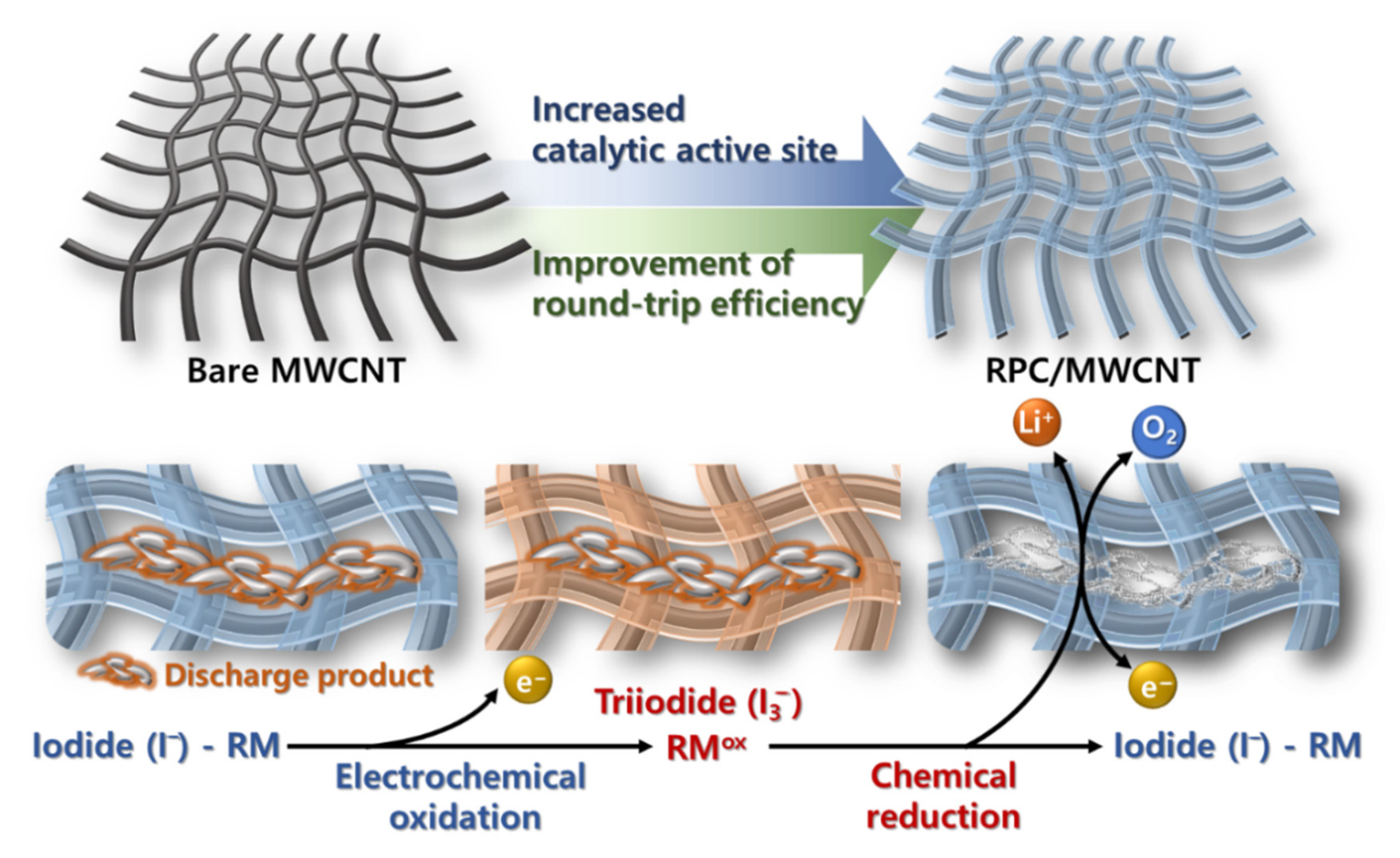{kind=link}
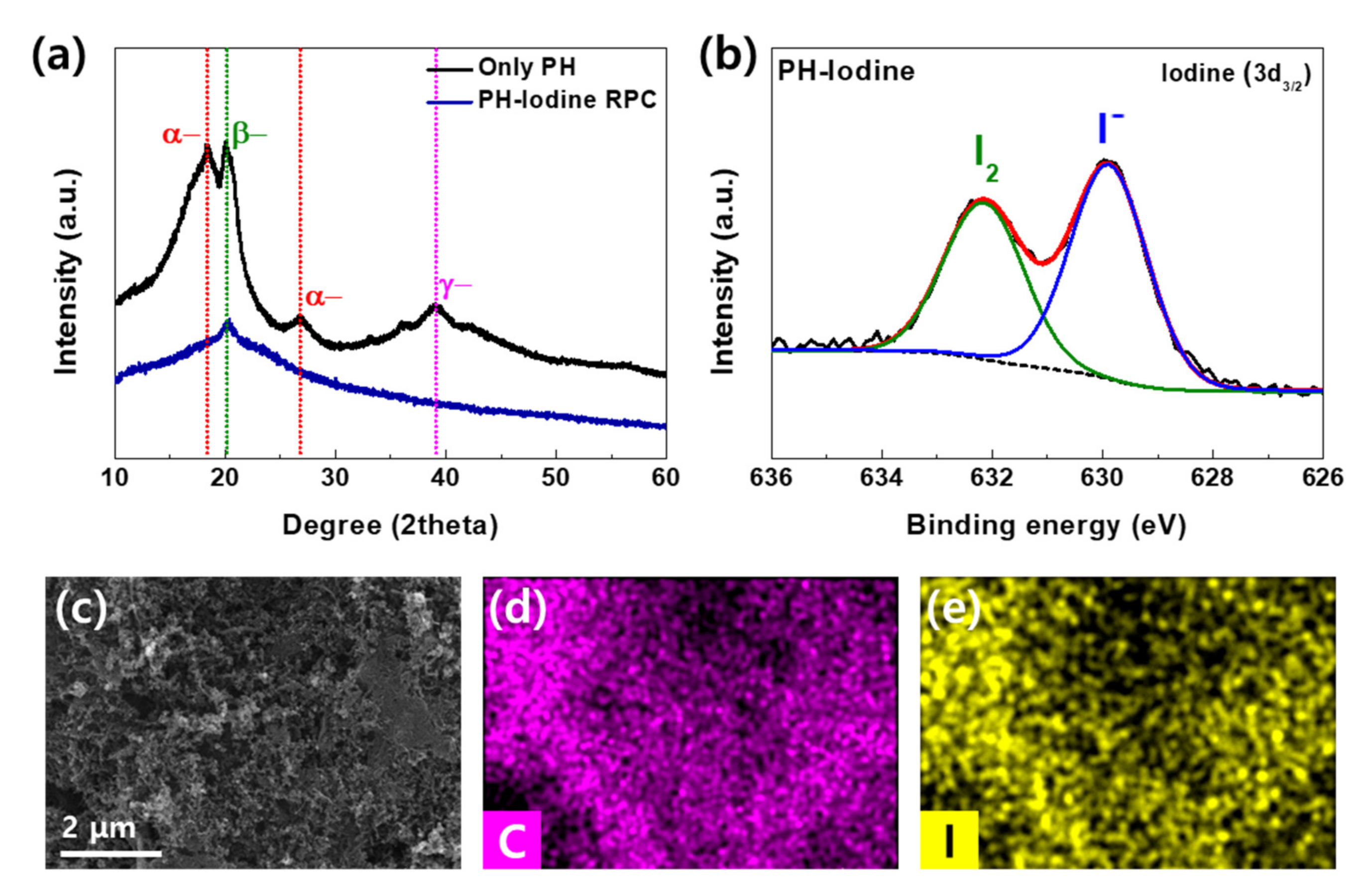{kind=link}
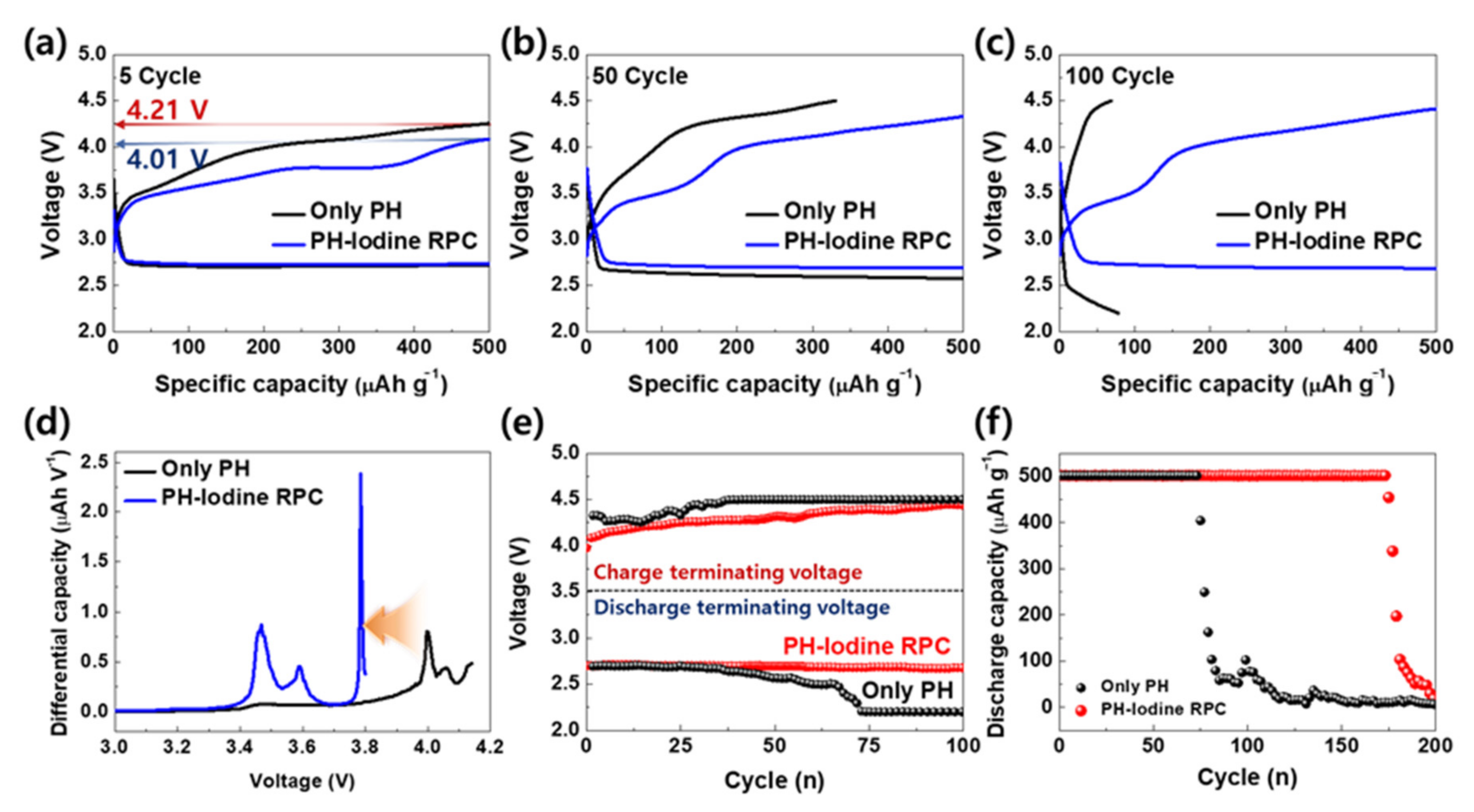{kind=link}
{kind=link}
{kind=link}
1. Introduction
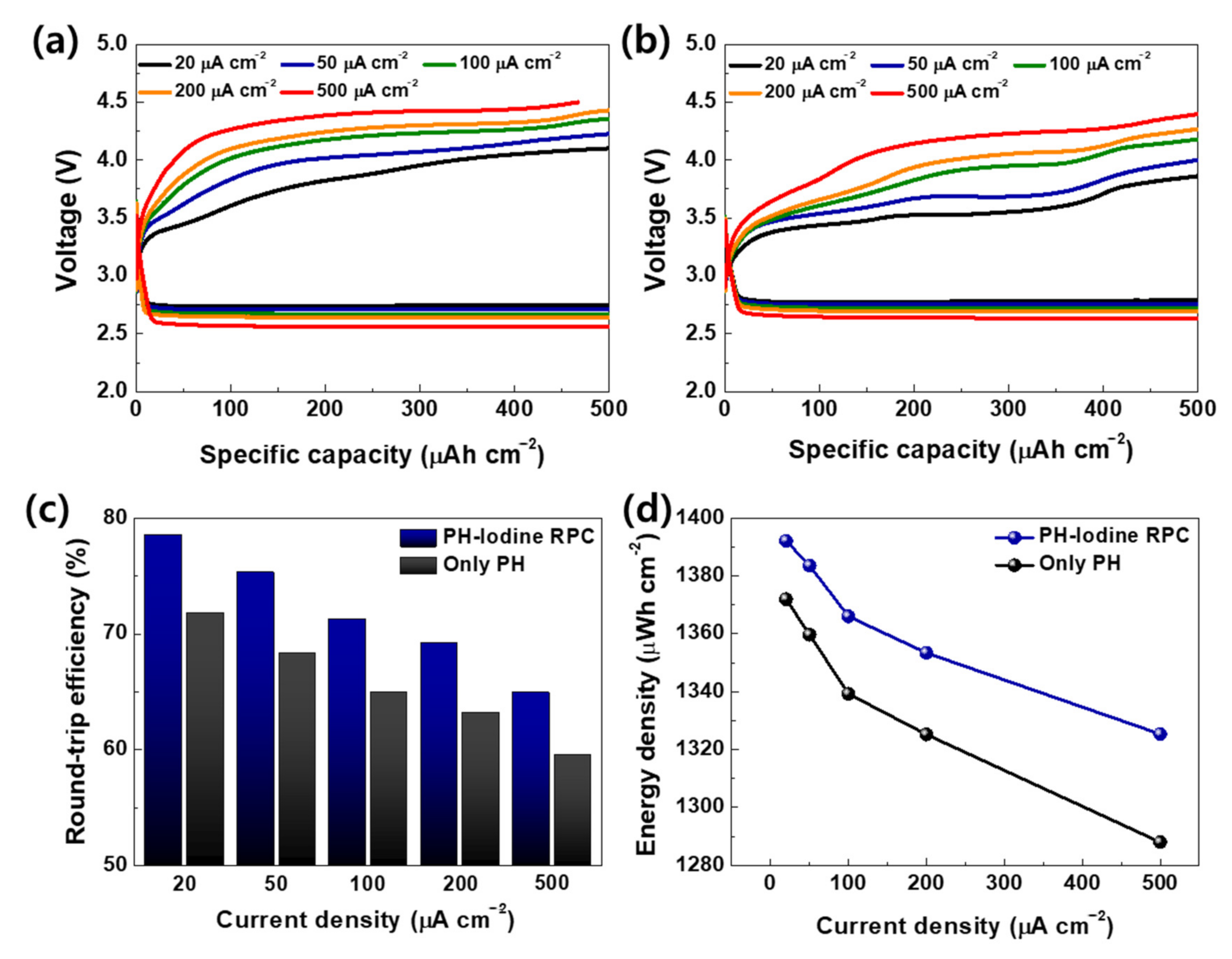
2. Results and Discussion
3. Materials and Methods
3.1. Preparations of Redox-Mediated Polymer Catalyst (RPC)
3.2. Fabrication of Electrodes
3.3. Materials Characterization
3.4. Cell Fabrication and Measurement
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bruce, P.G.; Freunberger, S.A.; Hardwick, J.-M. Tarascon, Li-O2 and Li-S Batteries with High Energy Storage. Nat. Mater. 2012, 11, 19–29. [Google Scholar] [CrossRef]
- Grande, L.; Paillard, E.; Hassoun, J.; Park, J.-B.; Lee, Y.-J.; Sun, Y.K.; Passerini, S.; Scrosati, B. The Lithium/Air Battery: Still an Emerging System or a Practical Reality? Adv. Mater. 2015, 27, 784–800. [Google Scholar] [CrossRef]
- Xu, S.; Yao, Y.; Guo, Y.; Zeng, X.; Lacey, S.D.; Song, H.; Chen, C.; Li, Y.; Dai, J.; Wang, Y.; et al. Textile Inspired Lithium-Oxygen Battery Cathode with Decoupled Oxygen and Electrolyte Pathways. Adv. Mater. 2018, 30, 1704907. [Google Scholar] [CrossRef]
- Choi, W.; Kim, M.; Park, J.O.; Kim, J.-H.; Choi, K.; Kim, Y.S.; Kim, T.Y.; Ogata, K.; Im, D.; Doo, S.-G.; et al. Ion-channel Aligned Gas-blocking Membrane for Lithium-air Batteries. Sci. Rep. 2017, 7, 12037. [Google Scholar] [CrossRef]
- Freunberger, S.A.; Chen, Y.; Peng, Z.; Griffin, J.M.; Hardwick, L.J.; Bardé, F.; Novák, P.; Burce, P.G. Reactions in the Rechargeable Lithium-O2 Battery with Alkyl Carbonate Electrolytes. J. Am. Chem. Soc. 2011, 133, 8040–8047. [Google Scholar] [CrossRef]
- McCloskey, B.D.; Bethune, D.S.; Shelby, R.M.; Girishkumar, G.; Luntz, A.C. Solvents’ Critical Role in Nonaqueous Lithium-Oxygen Battery Electrochemistry. J. Phys. Chem. Lett. 2011, 2, 1161–1166. [Google Scholar] [CrossRef]
- Feng, N.; He, P.; Zhou, H. Critical Challenges in Rechargeable Aprotic Li-O2 Batteries. Adv. Energy Mater. 2016, 6, 1502303. [Google Scholar] [CrossRef]
- Han, J.; Guo, X.; Ito, Y.; Liu, P.; Hojo, D.; Aida, T.; Hirata, A.; Fujita, T.; Adschiri, T.; Zhou, H.; et al. Effect of Chemical Doping on Cathodic Performance of Bicontinuous Nanoporous Graphene for Li-O2 Batteries. Adv. Energy Mater. 2016, 6, 1501870. [Google Scholar] [CrossRef]
- Shu, C.; Lin, Y.; Su, D. N-doped onion-like carbon as an efficient oxygen electrode for long-life Li-O2 battery. J. Mater. Chem. A 2016, 4, 2128–2136. [Google Scholar] [CrossRef]
- Shao, Y.; Ding, J.; Xiao, J.; Zhang, J.; Xu, W.; Park, S.; Zhang, J.-G.; Wang, Y.; Liu, J. Making Li-Air Batteries Rechargeable: Material Challenges. Adv. Funct. Mater. 2013, 23, 987–1004. [Google Scholar] [CrossRef]
- Bergner, B.J.; Schűrmann, A.; Peppler, K.; Garsuch, A.; Janek, J. A Mobile Catalyst for Rechargeable Li-O2 Batteries. J. Am. Chem. Soc. 2014, 136, 15054–15064. [Google Scholar] [CrossRef]
- Ko, Y.; Park, H.; Kim, J.; Lim, H.-D.; Lee, B.; Kwon, G.; Lee, S.; Bae, Y.; Park, S.K.; Kang, K. Biological Redox Mediation in Electron Transport Chain of Bacteria for Oxygen Reduction Reaction Catalysts in Lithium-Oxygen Batteries. Adv. Funct. Mater. 2019, 29, 1805623. [Google Scholar] [CrossRef]
- Truong, T.T.; Liu, Y.; Ren, Y.; Trahey, L.; Sun, Y. Morphological and Crystalline Evolution of Nanostructured MnO2 and Its Application in Lithium-Air Batteries. ACS Nano 2012, 6, 8067–8077. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Nasybulin, E.N.; Engelhard, M.H.; Kovarik, L.; Bowden, M.E.; Li, X.S.; Gaspar, D.J.; Xu, W.; Zhang, J.-G. Dendrimer-Encapsulated Ruthenium Oxide Nanoparticles as Catalysts in Lithium-Oxygen Batteries. Adv. Funct. Mater. 2014, 24, 7510–7519. [Google Scholar] [CrossRef]
- Jung, H.-G.; Jeong, Y.S.; Park, J.-B.; Sun, Y.-K.; Scrosati, B.; Lee, Y.J. Ruthenium-Based Electrocatalysts Supported on Reduced Graphene Oxide for Lithium-Air Batteries. ACS Nano 2013, 7, 3532–3539. [Google Scholar] [CrossRef]
- McCloskey, B.D.; Scheffler, R.; Speidel, A.; Bethune, D.S.; Shelby, R.M.; Luntz, A.C. On the Efficacy of Electrocatalysis in Nonaqueous Li-O2 Batteries. J. Am. Chem. Soc. 2011, 133, 18038–18041. [Google Scholar] [CrossRef]
- Chen, Y.; Freunberger, S.A.; Peng, Z.; Fontaine, O.; Bruce, P.G. Charging a Li–O2 battery using a redox mediator. Nat. Chem. 2013, 5, 489–494. [Google Scholar] [CrossRef]
- Kwak, W.-J.; Hirshberg, D.; Sharon, D.; Shin, H.-J.; Afri, H.; Park, J.-B.; Garsuch, A.; Chesneau, F.F.; Frimer, A.A.; Aurbach, D.; et al. Understanding the Behavior of Li-Oxygen Cells Containing LiI. J. Mater. Chem. A 2015, 3, 8855–8864. [Google Scholar] [CrossRef]
- Kim, B.G.; Jo, C.; Shin, J.; Mun, Y.; Lee, J.; Choi, J.W. Ordered Mesoporous Titanium Nitride as a Promising Carbon-Free Cathode for Aprotic Lithium-Oxygen Batteries. ACS Nano 2017, 11, 1736–1746. [Google Scholar] [CrossRef]
- Adams, B.D.; Black, R.; Radtke, C.; Williams, Z.; Mehdi, B.L.; Browning, N.D.; Nazar, L.F. The Importance of Nanometric Passivating Films on Cathodes for Li-Air Batteries. ACS Nano 2014, 8, 12483–12493. [Google Scholar] [CrossRef]
- Kim, M.-C.; Choi, S.; Kim, H.; Han, S.-B.; Moon, S.-H.; Kim, E.-S.; Kim, Y.-S.; Park, K.-Y. Polymeric redox mediator as a stable cathode catalyst for lithium-O2 batteries. J. Power Source 2020, 453, 227850. [Google Scholar] [CrossRef]
- Wang, X.; Xiao, C.; Liu, H.; Huang, Q.; Fu, H. Fabrication and properties of PVDF and PVDF-HFP microfiltration membranes. J. Appl. Polym. Sci. 2018, 135, 46711. [Google Scholar] [CrossRef]
- Sun, D.; Shen, Y.; Zhang, W.; Yu, L.; Yi, Z.; Yin, W.; Wang, D.; Huang, Y.; Wang, J.; Wang, D.; et al. A Solution-Phase Bifunctional Catalyst for Lithium-Oxygen Batteries. J. Am. Chem. Soc. 2014, 136, 8941–8946. [Google Scholar] [CrossRef]
- Oh, W.J.; Lim, H.S.; Won, J.S.; Lee, S.G. Preparation of PVDF/PAR Composites with Piezoelectric Properties by Post-Treatment. Polymers 2018, 10, 1333. [Google Scholar] [CrossRef]
- Guo, Q.; Han, Y.; Wang, H.; Xiong, S.; Sun, W.; Zheng, C.; Xie, K. Flame Retardant and Stable Li1.5Al0.5Ge1.5(PO4)3-Supported Ionic Liquid Gel Polymer Electrolytes for High Safety Rechargeable Solid-State Lithium Metal Batteries. J. Phys. Chem. C 2018, 122, 10334–10342. [Google Scholar] [CrossRef]
- Lim, H.-D.; Lee, B.; Zheng, Y.; Hong, J.; Kim, J.; Gwon, H.; Ko, Y.; Lee, M.; Cho, K.; Kang, K. Rational Design of Redox Mediators for Advanced Li-O2 batteries. Nat. Energy 2016, 1, 16066. [Google Scholar] [CrossRef]
- Bruke, C.M.; Black, R.; Kochetkov, I.R.; Giordani, V.; Addison, D.; Nazar, L.F.; McCloskey, B.D. Implications of 4e− Oxygen Reduction via Iodide Redox Mediation in Li-O2 Batteries. ACS Energy Lett. 2016, 1, 747–756. [Google Scholar] [CrossRef]
- Lim, H.-D.; Song, H.; Kim, J.; Gwon, H.; Bae, Y.; Park, K.-Y.; Hong, J.; Kim, H.; Kim, T.; Kim, Y.H.; et al. Superior Rechargeability and Efficiency of Lithium-Oxygen Batteries: Hierarchical Air Electrode Architecture Combined with a Soluble Catalyst. Angew. Chem. Int. Ed. 2014, 53, 3926–3931. [Google Scholar] [CrossRef]
- Li, F.; Chen, J. Mechanistic Evolution of Aprotic Lithium-Oxygen Batteries. Adv. Energy Mater. 2017, 7, 1602934. [Google Scholar] [CrossRef]
- Liu, T.; Leskes, M.; Yu, W.; Moore, A.J.; Zhou, L.; Bayley, P.M.; Kim, G.; Grey, C.P. Cycling Li-O2 batteries via LiOH formation and decomposition. Science 2015, 350, 530–533. [Google Scholar] [CrossRef]
- Dutta, A.; Ito, K.; Nomura, A.; Kubo, Y. Quantitative Delineation of the Low Energy Decomposition Pathway for Lithium Peroxide in Lithium–Oxygen Battery. Adv. Sci. 2020, 7, 2001660. [Google Scholar] [CrossRef]
- Aurbach, D.; McCloskey, B.D.; Nazar, L.F.; Bruce, P.G. Advances in understanding mechanisms underpinning lithium–air batteries. Nat. Energy 2016, 1, 16128. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.-C.; Song, J.H.; Lee, Y.-W.; Sohn, J.I. Redox-Mediated Polymer Catalyst for Lithium-Air Batteries with High Round-Trip Efficiency. Catalysts 2020, 10, 1479. https://doi.org/10.3390/catal10121479
Kim M-C, Song JH, Lee Y-W, Sohn JI. Redox-Mediated Polymer Catalyst for Lithium-Air Batteries with High Round-Trip Efficiency. Catalysts. 2020; 10(12):1479. https://doi.org/10.3390/catal10121479
Chicago/Turabian StyleKim, Min-Cheol, Jung Hyun Song, Young-Woo Lee, and Jung Inn Sohn. 2020. "Redox-Mediated Polymer Catalyst for Lithium-Air Batteries with High Round-Trip Efficiency" Catalysts 10, no. 12: 1479. https://doi.org/10.3390/catal10121479
APA StyleKim, M.-C., Song, J. H., Lee, Y.-W., & Sohn, J. I. (2020). Redox-Mediated Polymer Catalyst for Lithium-Air Batteries with High Round-Trip Efficiency. Catalysts, 10(12), 1479. https://doi.org/10.3390/catal10121479