Abstract
Carbon dioxide is an intrinsically stable molecule; however, it can readily react with various nucleophilic reagents. In the presence of a cyanide source, CO2 was proven to be useful to promote addition reactions. Here we report the use of CO2 to facilitate 1,4-conjugate cyanide addition reaction to chalcones to generate organonitriles. Nitriles are key component in organic synthesis due to their utility in numerous functional group transformation, however, conjugation addition of cyanide has been a challenge in this substrate class due to side reactions. To mitigate this, we employed simple ammonium and metal cyanide sources as nucleophiles under carbon dioxide atmosphere where high selectivity toward the desired product was obtained. The presented reaction is not feasible under inert atmosphere, which highlights the important role of CO2, as a Lewis and Brøndsted acidic catalyst. Further derivatization of organonitriles compounds were performed to showcase the utility of the reaction, while an unprecedented dimerization reaction was identified and characterized, affording a cyclopentanone scaffold.
1. Introduction
Carbon dioxide (CO2) is a thermodynamically stable and kinetic inert molecule however utilization and functionalization of CO2 found many unique modes of actions [1,2]. For example, CO2 can be used as temporary protecting group, thus preventing polymerization of acrylonitriles initiated by cyanide anion [3,4]. In principle, the nucleophilicity of cyanide is sufficiently high however it requires catalytic species to enhance reaction rates under controlled manner to improve selectivity [5]. This is particularly the case with insoluble metal cyanides, such as NaCN and KCN, when reactions are performed in organic solvents. Nevertheless, the obtained organonitriles are ubiquitous functional group in organic synthesis, enabling facile synthesis of various molecular scaffolds via meta-selective C-H activation [6,7], reduction (amines) [8], hydrolysis (carbonyls) [9], radical reactions (cyanide abstraction) [10,11] and umpolung chemistry (cyanohydrins) [12]. Among many reaction pathways, hydrocyanation with gaseous HCN showed the most atom economic reaction with olefin substrates [13]. The use of solid metal cyanide sources is desirable to avoid volatile HCN, however, it often requires high reaction temperature due to the low solubility and reactivity of alkali metal cyanides [14]. Recently our group demonstrated the employment of catalytic amounts of CO2 for 1,4-conjugate cyanide addition reaction of coumarin substrates [15]. The use of ammonium cyanide as a nucleophile was sufficient to quantitatively convert the starting materials to the desired products under 1 atm of CO2 (Scheme 1A). In addition, we found that cyanohydrin synthesis can be facilitated under CO2 atmosphere, implying potential catalysis mediated by CO2. Here we expand our system to a general Michael acceptor namely chalcones, which exhibit broad application potentials after cyanation reaction under CO2 atmosphere. Previous studies on chalcone cyanation reactions were limited to metal catalysts, including a Ni-catalyzed cyano-borrowing process [16] a Sm-mediated reaction [17] and a Sc(OTf)3 mediated reaction with an ammonium cyanide nucleophile [18]. Organic and inorganic bases, tetraarylphosphonium inner salts and Cs2CO3 were reported to catalyze addition of TMSCN (trimethylsilyl cyanide) to chalcones [19,20]. To the best of our knowledge, the CO2-promoted cyanation reaction of chalcones exhibit the first metal-free 1,4-cyanation reaction without generating HCN under practical reaction conditions by using cyanide salts [21,22,23].
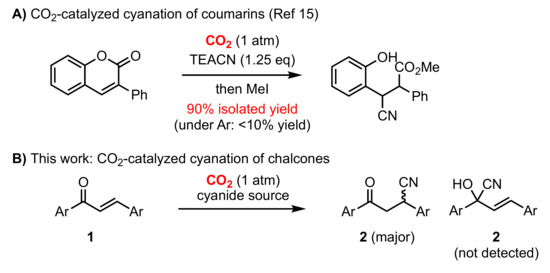
Scheme 1.
(A) Previous work: CO2-catalyzed cyanation of coumarins, TEACN: tetraethylammonium cyanide (B) cyanation of chalcone (1) under CO2 atmosphere (1 atm).
2. Results
We commenced our investigation on chalcone cyanation reactions by optimizing reaction conditions under CO2 atmosphere. It was clear that the reaction showed no product formation under inert atmosphere (N2) although the starting material was fully consumed. This can be ascribed to the reversibility of the reaction and the decomposition of the cyanated product (2) to form the corresponding cyanohydrin (1,2-adduct), dehydration products and oligomers. We hypothesized that the presence of CO2 diminished the polymerization of chalcone substrates by forming carboxylate intermediates on the α-position of the product [5]. It is noteworthy that the formation of cyanohydrin of 1,4-conjugate addition products 2 can occur, without the formation of 1,2-adducts (2′). The desired products can be obtained after standard work-up.
2.1. Optimization
To understand the cyanation process, we attempted to optimize reaction conditions. At the outset, we observed that the presence of CO2 is critical to obtain the desired product (entry 1, Table 1). The structure of the cyanation product 2a was unambiguously confirmed by isolating crystalline product which was recrystallized and analyzed by X-ray crystallography. On the other hand, the crude reaction mixture, under nitrogen atmosphere, often showed complicated mixtures of byproducts and insoluble particles, indicating formation of polymeric species. Under carbon dioxide atmosphere (1 atm), it was found that 2–3 equivalents of the cyanide nucleophile were necessary to afford satisfactory yields of the desired product (up to 85% isolated yield, entries 2 and 3). Interestingly, the employment of KCN instead of tetraethylammonium cyanide (TEACN) showed a lower yield (19%) confirming the importance of solubility of cyanide nucleophile. This can be controlled by adding an additional phase-transfer reagent (NMe4Cl) in the presence of excess amounts of KCN (3.3 equiv) as a nucleophile, affording the product in a good yield (72%). Solvent screening and temperature screening showed no further improvement of the reaction conditions (entries 6–9). However, we concluded that the positive CO2 effect was quite general in some of the tested organic solvents, for example, under nitrogen atmosphere, un-optimal solvents afforded lower yields of the product compared to the reactions under CO2.

Table 1.
Optimization of reaction conditions a.
2.2. Substrate Scope
With optimized reaction conditions in our hands, we turned our attention to evaluate our methodology in electronically differentiated substrates 1b-m to show generality of the process (Table 2). The corresponding chalcone substrates were readily prepared via condensation reactions (See Supplementary Materials). In general our reaction conditions was proven to be applicable for most of chalcones affording conjugated addition product in good yields (59–73% isolated yield) within short reaction time. When the reaction time was prolonged, the reaction proved to form various byproducts, presumably via dimerization and oligomerization. It is noteworthy here that some of cyanation product were obtained in low yields (2d: 13%, 2g: 20%), however, these are unique cases with highly electron-withdrawing group (-NO2) and a free amine (-NH2), which are difficult to control for other types of side reactions. Further investigations with cyclic enone substrates (1h-1l) showed promising reactivity for cyanation under CO2, delivering the desired products in moderate to good yields. Indanone-derived product 2h was spontaneous crystallized (15% from the reaction mixture), which confirmed the product unambiguously by X-ray crystallography as a single diastereomer. Interestingly, chroman-4-one derived chalcones (1k and 1l, entries 10 and 11) showed different reaction pathways: chalcone 1k was converted to ring-opened form (2k), presumably after a cyanation-induced ring opening reaction and then double bond isomerization to afford thermodynamically stable products. For the reaction with chalcone 1l, we tentatively assigned the main product as chromone derivative (2l) without the incorporation of cyanide, indicating many potential reaction pathways, which can lead to the formation of unexpected byproducts.
Table 2.
Substrate scope for the cyanation of different chalcone derivates under CO2 a.
Among many possible side reaction pathways, we found out that a dimerization reaction occurred at a higher concentration (7.5 g-scale, 36.0 mmol, 0.36 M) to generate cyclopentenone scaffold 8 in 50% isolated yield. A proposed reaction mechanism includes Robinson annulation (1,4-conjugate cyanation reaction and subsequent aldol condensation reaction). We confirmed that the obtained cyclopentenone 8 showed reasonable stability in the reaction mixture. Based on our analysis of the reaction mixture, the cyclic enone (8) showed no reactivity in the presence of additional cyanide nucleophiles under the same reaction conditions. In 1959, R. B. Davis reported the isolation of a similar compound from a reaction of benzaldehyde, sodium cyanide and acetophenone in methanol without detailed characterization of the compound [24]. To verify this, we investigated the reaction mechanism by analyzing the reaction mixture at earlier stages and isolated dimer 8-int (Scheme 2), which was formed after the cyanation reaction. This dimerization process can be controlled by increasing the concentration of CO2 (dry ice, 25 g for 2.4 mmol chalcone) or in the presence of stoichiometric amounts of a proton donor (2,6-diisopropylphenol): the desired product was obtained in good yield 80% and 81% respectively. The role the additional proton source needs further investigation, however, we presumed that the formation of ammonium enolate (i.e., 1a-CN) prohibits the selective conversion of the starting material to the product 2. In the presence of a sterically demanding phenol, a facile protonation process of the enolate is expected affording the desired products. Higher concentration of CO2 and water is also beneficial to render acidic reaction conditions, therefore minimizing oligomerization and polymerization reactions (Scheme 2C). We presume that the carboxylated product (2a-CO2) is responsible for the successful cyanation reactions under our optimized reaction conditions despite the substrate dependency.
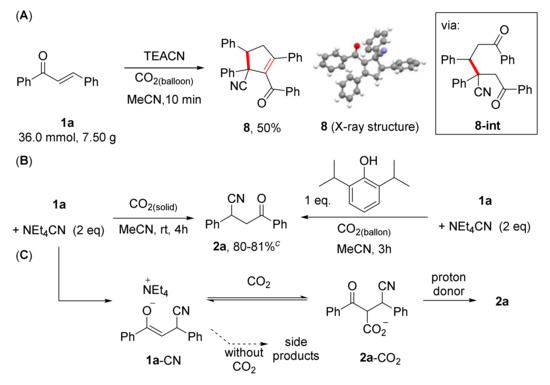
Scheme 2.
(A) Dimerization of chalcone (1a) to cyclopentanone 8 and the single crystal X-ray structure of dimer 8 (B) large scale cyanation reactions with dry ice and a proton donor. (C) a plausible reaction mechanism with CO2 and without CO2 (oligomerization and other side products formation reactions).
2.3. Functionalization of Cyanated Chalcones
We further demonstrated the utility of the CO2-mediated conjugate cyanation reaction by functionalizing the β-cyanoketone product 2a. The racemic cyanated product was prepared in a larger scale reaction based on the above-mentioned studies (Scheme 2B,C) and then subjected to various types of organic transformation as illustrated in Figure 1. Reduction of the ketone functional group was performed with NaBH4 in methanol to give selectively β-nitrile alcohol albeit low isolate yield of the product presumably due to the high acidity of the α-proton. Nevertheless, this reduction reaction shows an interesting synthetic route toward functionalized alcohols with a nitrile group untouched. We also performed transformation of nitrile group to tetrazole in the presence of ZnCl2 promoter, smoothly affording the desired product in 39% yield [25]. For hydrolysis, we found out that with the choice of a strong Brønsted acid directed to different hydrolysis products, amide product 4a was obtained in good yield (71%) in concentrated sulfuric acid at room temperature. On the other hand, carboxylic acid product 5a was obtained under reflux conditions with concentrated HCl under CO2 atmosphere to prevent a potential decarboxylation reaction.
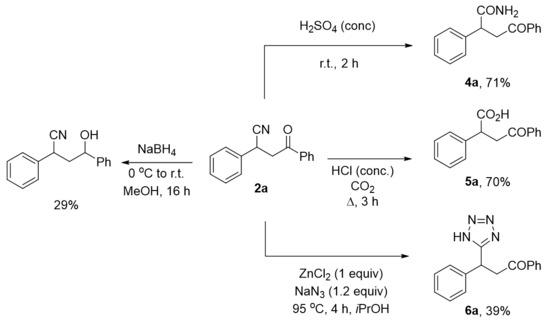
Figure 1.
Functionalization of cyanation product 2a via hydrolysis, tetrazole formation, reduction and amide bond formation reactions.
3. Discussion
The use of CO2 for organic synthesis is appealing for many reasons [26] (1) as a cheap and non-innocent inert gas, (2) often accelerate and control reactions, (3) enabling chemistry with insoluble metal cyanide reagents in organic solvents. However, it is important to note that the employment of CO2 can complicate the outcome of reactions due to undesired binding of nucleophiles therefore reducing the reactivity. Based on our investigation and previous studies [15,27,28], we presumed that the positive effects of CO2 in catalyzing chemical reactions can be general, particularly in reactions involving reversible steps. In the current investigation with chalcones, the reaction is highly controlled by thermodynamics that allowed us to access various products in high yield. The importance of CO2, however, cannot be neglected due to the severe side products’ formation in the absence of CO2. This phenomenon—a cleaner reaction mixture under CO2—has been sporadically revealed in synthetic chemistry [29,30]. Further investigation will pave the way to understand the true capacity of CO2 in organic synthesis, potentially providing new ways of mitigating anthropogenic CO2 via CO2 capture and sequestration in a catalytic manner.
4. Materials and Methods
All the chemicals, unless stated otherwise were purchased from commercial suppliers in the highest purity and used without further purification. Solvents used were HPLC (high performance liquid chromatography) grade either as it is or dried on molecular sieves (4 Å) prior to use. The water concentrations of all the solvents used in the present study were measured on a Karl Fischer titrator (831 KF Coulometer). Analytical thin layer chromatography was done on Merck DC-Alufolien SIO2 60 F254 0.2 mm thick pre-coated TLC plates. Column chromatography was performed using SiO2(SI 1721, 60 Å, 40–63 µm). 1H NMR and 13C NMR spectra were recorded with 500 MHz Ultrashield Plus 500 spectrometer and 126 MHz on a Bruker instrument. All chemical shifts (d) are given in ppm using the solvent residual peak as reference. X-ray crystallography was performed by the crystallography service of the Department of Chemistry, University of Copenhagen, Denmark on a Bruker/Nonius Kappa CCD 4-circle diffractometer.
5. Conclusions
In conclusion, we have demonstrated a facile cyanation reaction of chalcone electrophiles under atmospheric CO2 pressure at ambient reaction temperature. This is a unique system considering the reversibility of the reaction and competition reaction pathways. We ascribed the observed selectivity to the active role of non-inert gas CO2, which can interact with substrates (cyanide and chalcones), intermediates, transition states and products. Further applications of CO2-mediated organic synthesis are underway in our laboratory to expand the concept to various organic transformation.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/12/1481/s1, Synthetic procedures for substrates, reaction conditions and 1H and 13C NMR spectra.
Author Contributions
Conceptualization, J.-W.L. and S.D.; investigation, S.D., G.B.H., F.S.K.; Crystallography A.K.; writing—original draft preparation, J.-W.L.; writing—review and editing, S.D., J.-W.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Novo Nordisk Fonden, grant number NNF17OC0027598 and Villum Fonden (00019062). S.D. was funded by ERASMUS Program.
Acknowledgments
The generous support from the Department of Chemistry, University of Copenhagen and the analytic department are gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lee, J.-W. Toward ideal carbon dioxide functionalization. Chem. Sci. 2019, 10, 3905–3926. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Chand-Thakuri, P.; Young, M.C. Carbon Dioxide-Mediated C(sp2)–H Arylation of Primary and Secondary Benzylamines. J. Am. Chem. Soc. 2019, 141, 7980–7989. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Liu, D.; Young, M.C. Carbon Dioxide-Mediated C(sp3)–H Arylation of Amine Substrates. J. Am. Chem. Soc. 2018, 140, 6818–6822. [Google Scholar] [CrossRef]
- White, D.A. Cyanocarboxylation of activated olefins. J. Chem. Soc. Perkin Trans. 1976, 1, 1926–1930. [Google Scholar] [CrossRef]
- Leow, D.; Li, G.; Mei, T.-S.; Yu, J.-Q. Activation of remote meta-C–H bonds assisted by an end-on template. Nature 2012, 486, 518–522. [Google Scholar] [CrossRef]
- Dai, H.-X.; Li, G.; Zhang, X.-G.; Stepan, A.F.; Yu, J.-Q. Pd(II)-Catalyzed ortho- or meta-C–H Olefination of Phenol Derivatives. J. Am. Chem. Soc. 2013, 135, 7567–7571. [Google Scholar] [CrossRef]
- Bornschein, C.; Werkmeister, S.; Wendt, B.; Jiao, H.; Alberico, E.; Baumann, W.; Junge, H.; Junge, K.; Beller, M. Mild and selective hydrogenation of aromatic and aliphatic (di)nitriles with a well-defined iron pincer complex. Nat. Commun. 2014, 5, 4111. [Google Scholar] [CrossRef]
- Ahmed, T.J.; Knapp, S.M.M.; Tyler, D.R. Frontiers in catalytic nitrile hydration: Nitrile and cyanohydrin hydration catalyzed by homogeneous organometallic complexes. Coord. Chem. Rev. 2011, 255, 949–974. [Google Scholar] [CrossRef]
- Chu, X.-Q.; Ge, D.; Shen, Z.-L.; Loh, T.-P. Recent Advances in Radical-Initiated C(sp3)–H Bond Oxidative Functionalization of Alkyl Nitriles. ACS Catal. 2018, 8, 258–271. [Google Scholar] [CrossRef]
- Mattalia, J.-M.R. The reductive decyanation reaction: An overview and recent developments. Beilstein J. Org. Chem. 2017, 13, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Seebach, D. Methods of Reactivity Umpolung. Angew. Chem. Int. Ed. 1979, 18, 239–258. [Google Scholar] [CrossRef]
- Tolman, C.A.; McKinney, R.J.; Seidel, W.C.; Druliner, J.D.; Stevens, W.R. Homogeneous Nickel-Catalyzed Olefin Hydrocyanation. In Advances in Catalysis; Eley, D.D., Pines, H., Weisz, P.B., Eds.; Academic Press Inc.: Cambridge, MA, USA, 1985; Volume 33, pp. 1–46. [Google Scholar]
- Friedman, L.; Shechter, H. Preparation of Nitriles from Halides and Sodium Cyanide. An Advantageous Nucleophilic Displacement in Dimethyl Sulfoxide1a. J. Org. Chem. 1960, 25, 877–879. [Google Scholar] [CrossRef]
- Roy, T.; Kim, M.J.; Yang, Y.; Kim, S.; Kang, G.; Ren, X.; Kadziola, A.; Lee, H.-Y.; Baik, M.-H.; Lee, J.-W. Carbon Dioxide-Catalyzed Stereoselective Cyanation Reaction. ACS Catal. 2019, 9, 6006–6011. [Google Scholar] [CrossRef]
- Li, Z.-F.; Li, Q.; Ren, L.-Q.; Li, Q.-H.; Peng, Y.-G.; Liu, T.-L. Cyano-borrowing reaction: Nickel-catalyzed direct conversion of cyanohydrins and aldehydes/ketones to β-cyano ketone. Chem. Sci. 2019, 10, 5787–5792. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Fujii, A.; Nakano, Y.; Sakaguchi, S.; Ishii, Y. Acetylcyanation of Aldehydes with Acetone Cyanohydrin and Isopropenyl Acetate by Cp*2Sm(thf)2. J. Org. Chem. 1999, 64, 4214–4216. [Google Scholar] [CrossRef]
- Ramesh, S.; Lalitha, A. Scandium(III) Triflate Catalyzed 1,4-Addition of Cyano Group to Enones Using Tetraethylammonium Cyanide as the Cyanide Source. Acta Chim. Slov. 2013, 60, 689–694. [Google Scholar]
- Guo, S.; Mi, X. Tetraarylphosphonium inner-salts (TAPIS) as both Lewis base catalyst and phase tag. Tetrahedron Lett. 2017, 58, 2881–2884. [Google Scholar] [CrossRef]
- Yang, J.; Shen, Y.; Chen, F.-X. Highly Efficient Cs2CO3-Catalyzed 1,4-Addition of Me3SiCN to Enones with Water as the Additive. Synthesis 2010, 2010, 1325–1333. [Google Scholar] [CrossRef]
- Ciller, J.A.; Seoane, C.; Soto, J.L. Synthesis of Heterocyclic Compounds, XXXVIII. Five-membered Heterocycles by Cyclization of 3-Benzoyl-4-oxobutanenitriles. Liebigs Ann. Chem. 1985, 1985, 51–57. [Google Scholar] [CrossRef]
- Allen, C.F.H.; Kimball, R.K. α-Phenyl-β-Benzoylpropionitrile. Org. Synth. 1930, 10, 80. [Google Scholar]
- Bellinger, T.J.; Harvin, T.; Pickens-Flynn, T.B.; Austin, N.; Whitaker, S.H.; Tang Yuk Tutein, M.L.C.; Hukins, D.T.; Deese, N.; Guo, F. Conjugate Addition of Grignard Reagents to Thiochromones Catalyzed by Copper Salts: A Unified Approach to Both 2-Alkylthiochroman-4-One and Thioflavanone. Molecules 2020, 25, 2128. [Google Scholar] [CrossRef] [PubMed]
- Davis, R. Notes: Condensation of Aromatic Aldehydes with Methyl Aryl Ketones and Sodium Cyanide. J. Org. Chem. 1959, 24, 880–882. [Google Scholar] [CrossRef]
- Vorona, S.; Artamonova, T.; Zevatskii, Y.; Myznikov, L. An Improved Protocol for the Preparation of 5-Substituted Tetrazoles from Organic Thiocyanates and Nitriles. Synthesis 2014, 46, 781–786. [Google Scholar] [CrossRef]
- Schilling, W.; Das, S. CO2-catalyzed/promoted transformation of organic functional groups. Tetrahedron Lett. 2018, 59, 3821–3828. [Google Scholar] [CrossRef]
- Lee, J.-W.; Juhl, M.; Petersen, A.R. CO2-Enabled Cyanohydrin Synthesis and Facile Iterative Homologation Reactions. Chem. Eur. J. 2020. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, J.; Lee, J.-W. A CO2-Catalyzed Transamidation Reaction. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Chiang, P.-C.; Bode, J.W. On the Role of CO2 in NHC-Catalyzed Oxidation of Aldehydes. Org. Lett. 2011, 13, 2422–2425. [Google Scholar] [CrossRef]
- Li, H.; Wu, H.; Yu, Z.; Zhang, H.; Yang, S. CO(2)-Enabled Biomass Fractionation/Depolymerization: A Highly Versatile Pre-Step for Downstream Processing. ChemSusChem 2020, 13, 3565–3582. [Google Scholar] [CrossRef]





























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).