Unravelling the Structural Modification (Meso-Nano-) of Cu/ZnO-Al2O3 Catalysts for Methanol Synthesis by the Residual NaNO3 in Hydroxycarbonate Precursors

 , , ,
, , , 

Abstract

1. Introduction
2. Results and Discussion
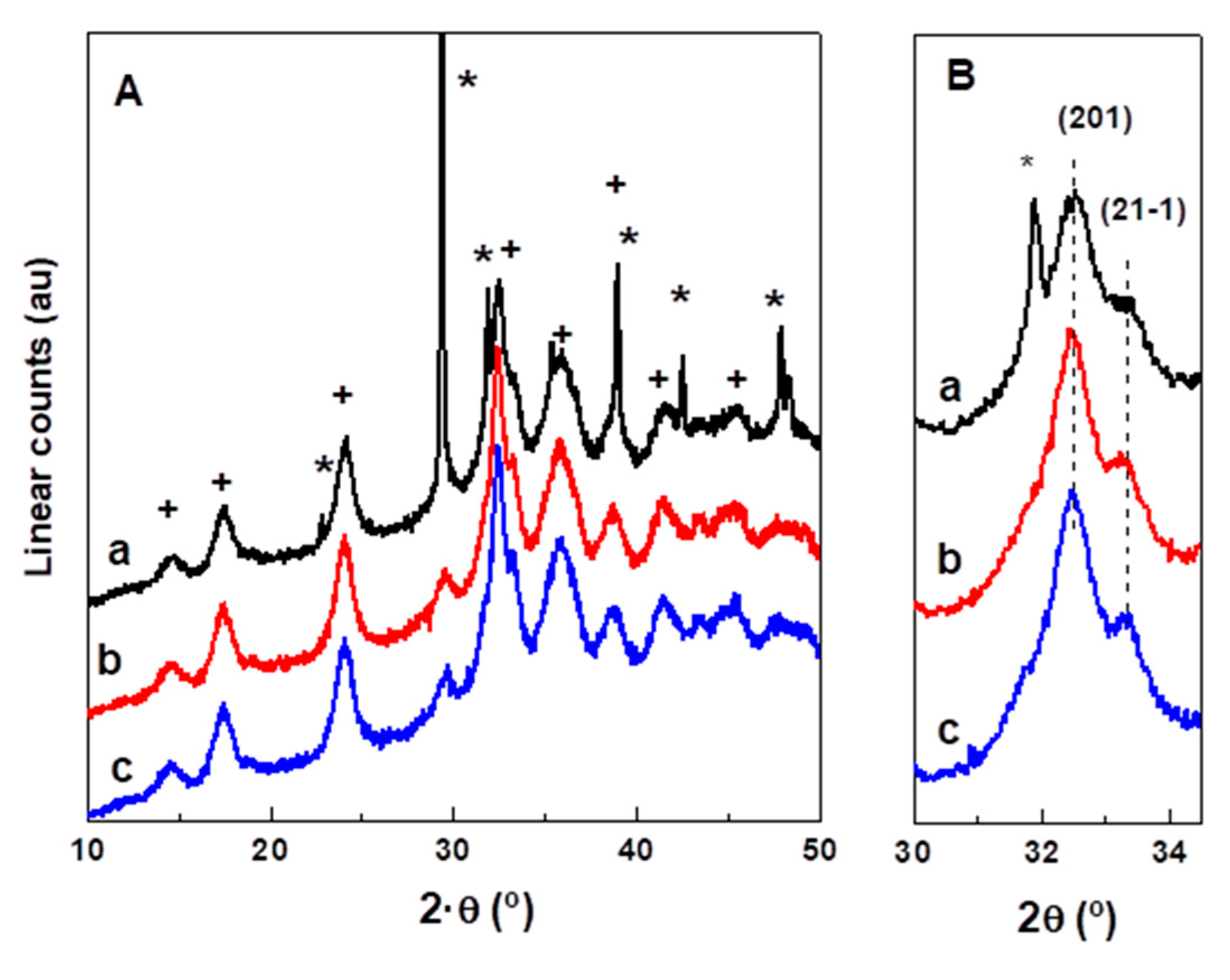
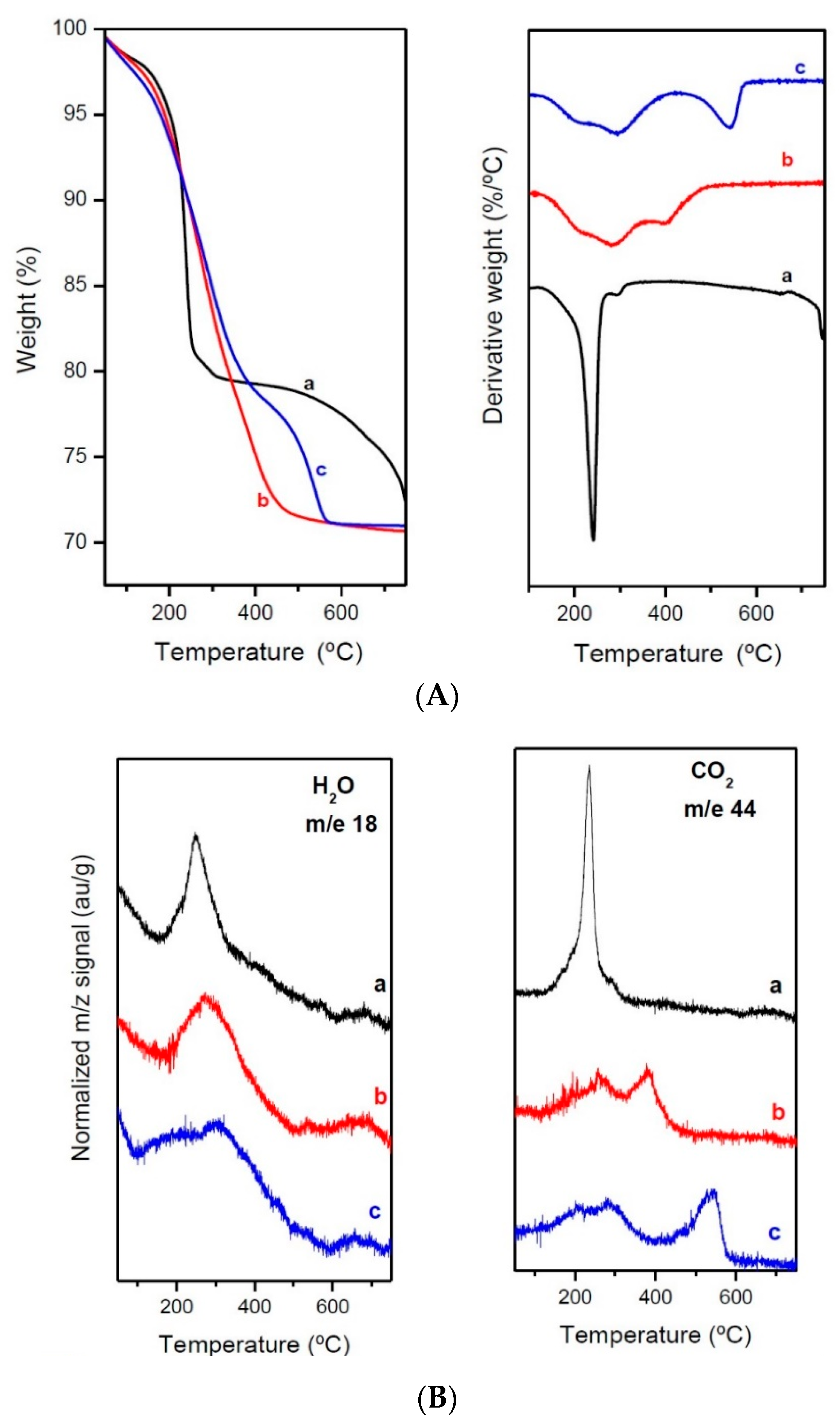
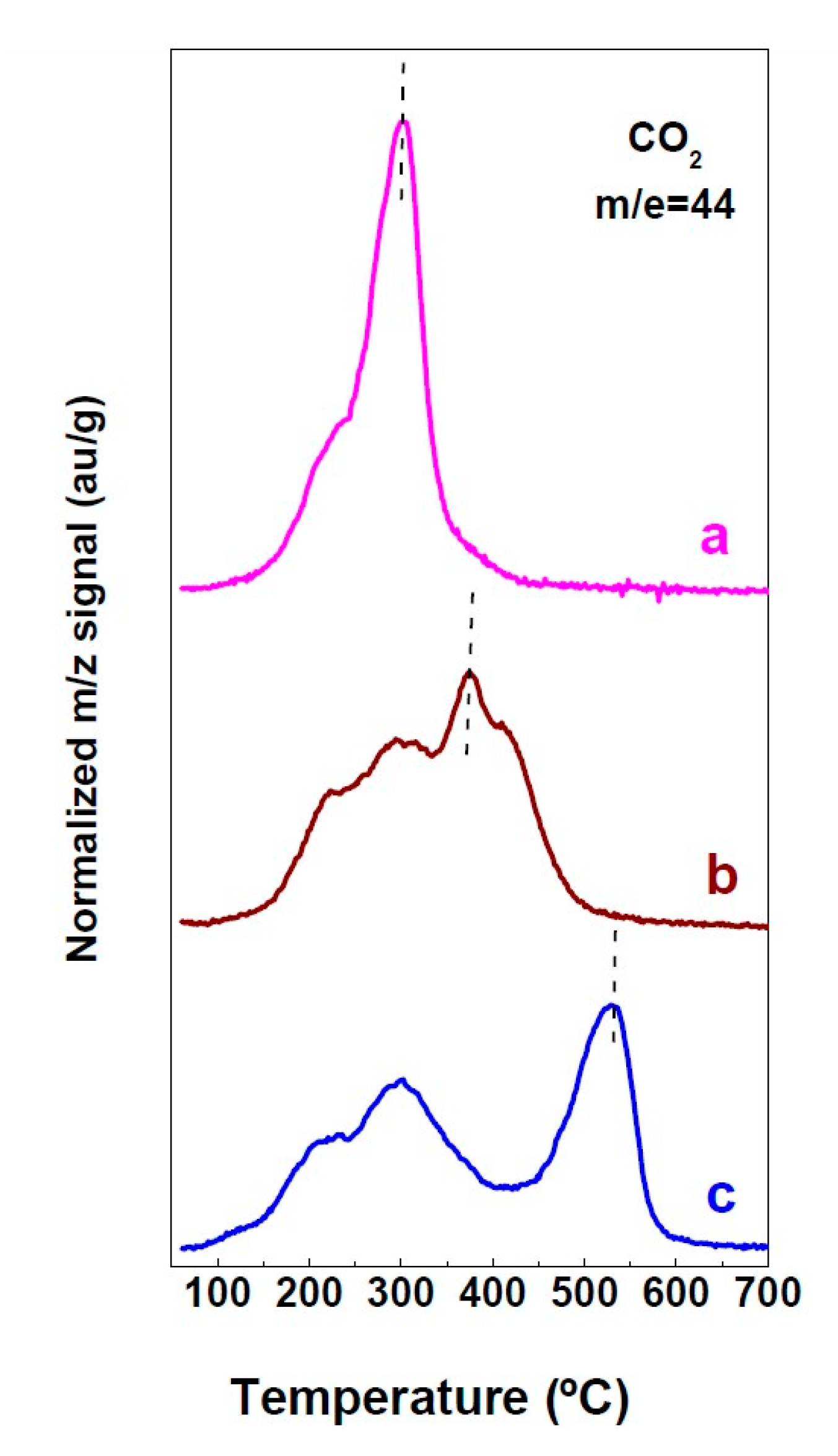
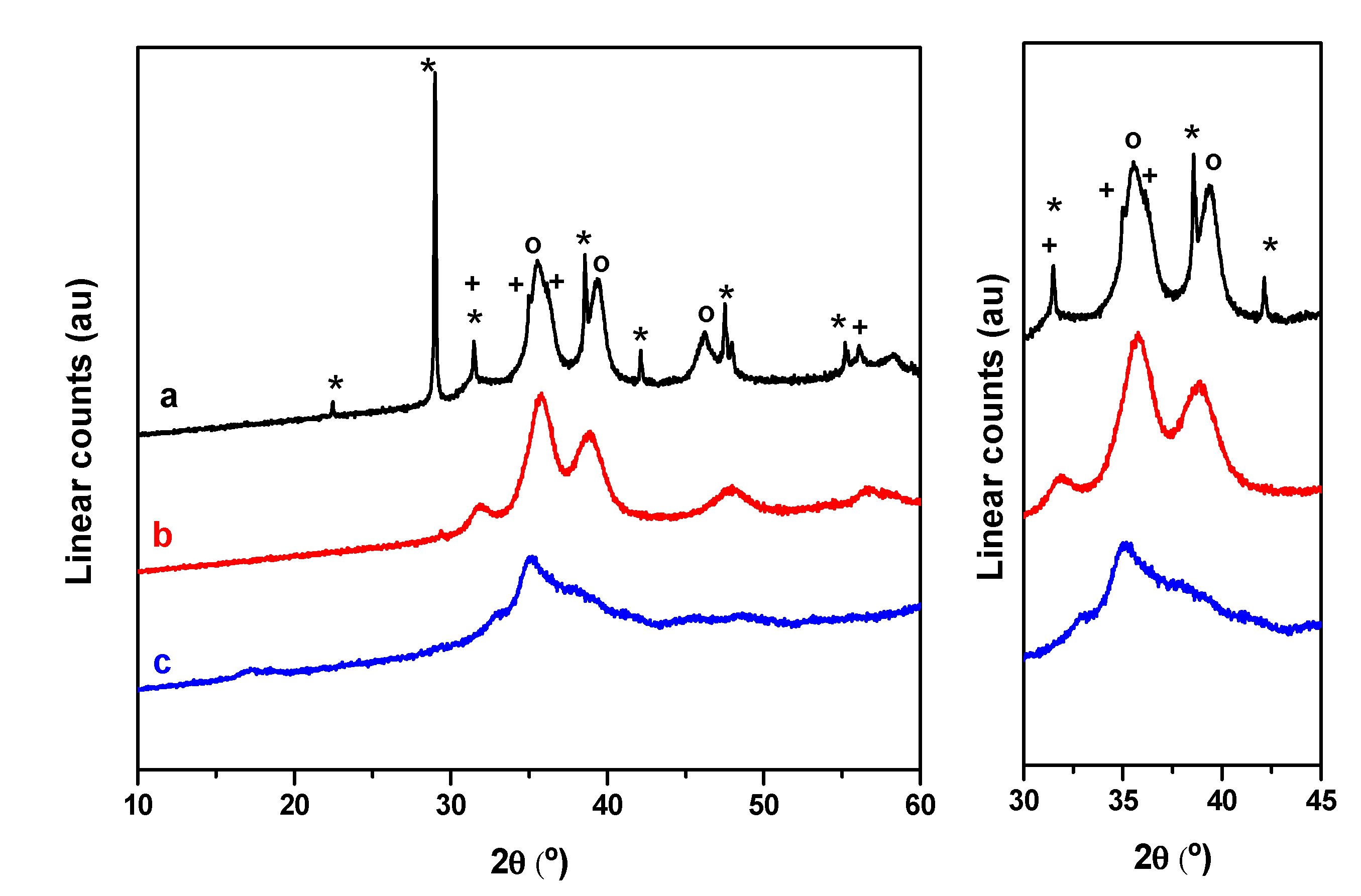
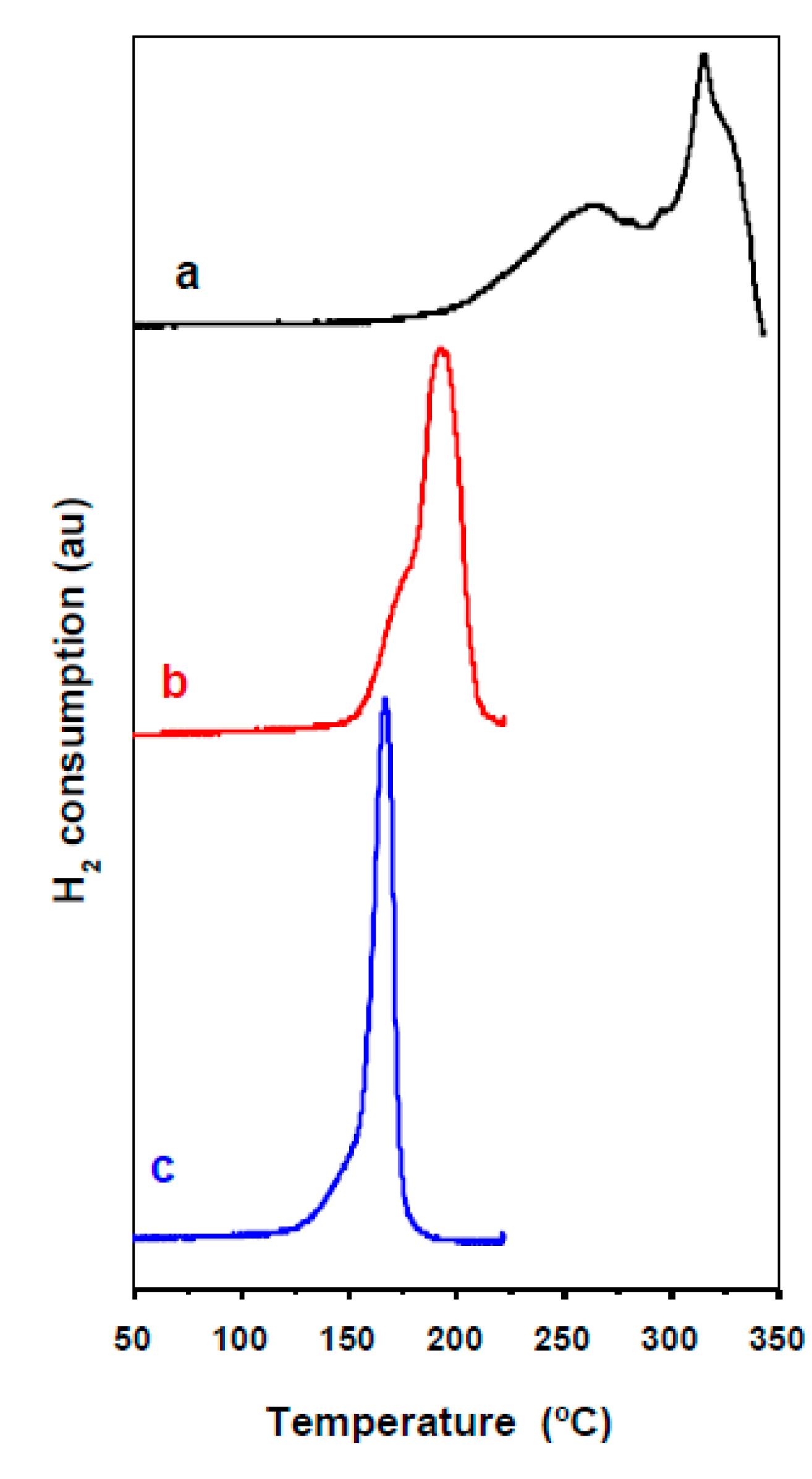
2.1. Characterization of Precursors, Calcined and Reduced Catalysts
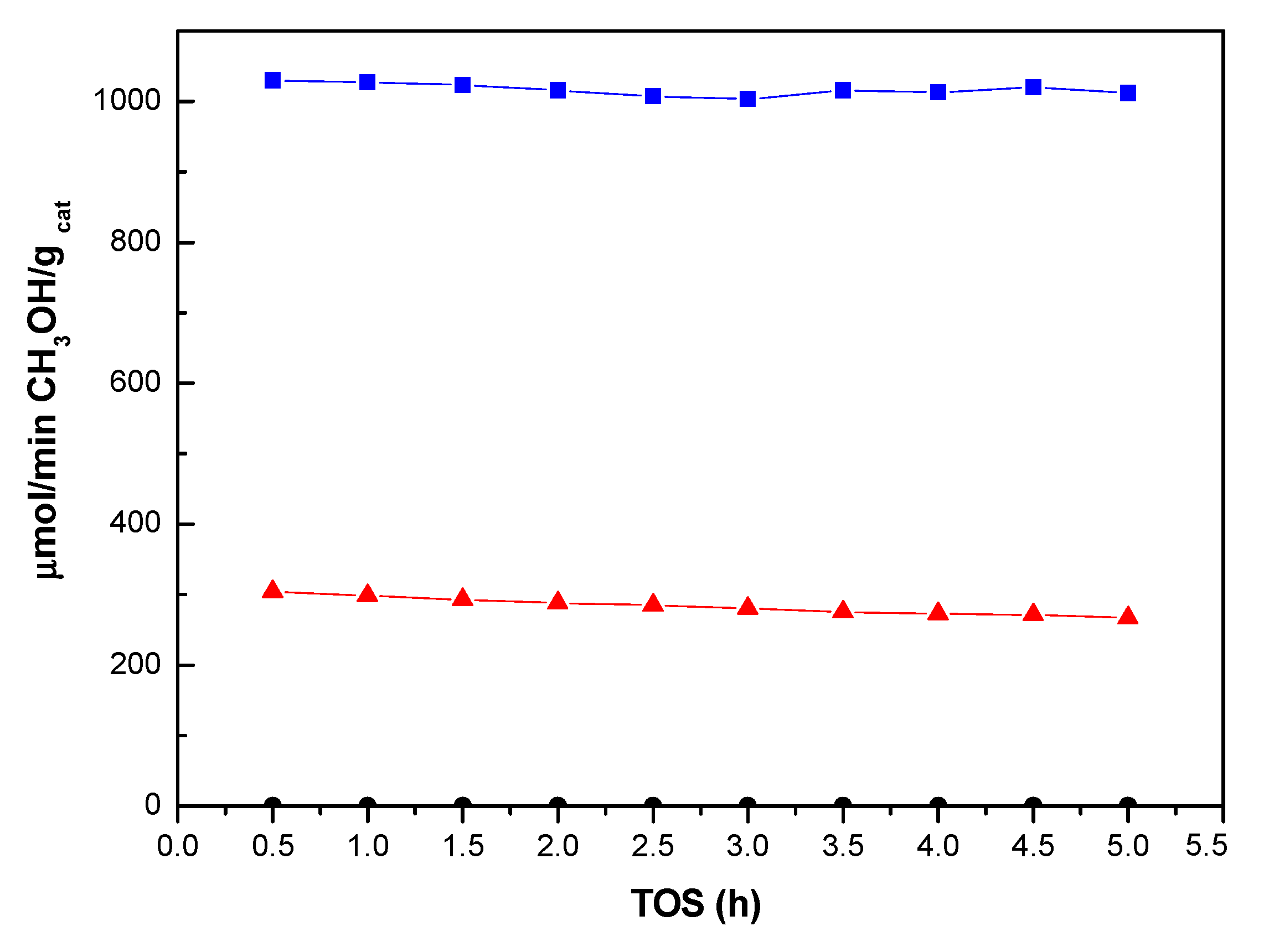
2.2. Activity in Methanol Synthesis
3. Materials and Methods
3.1. Catalysts’ Preparation
3.2. Catalysts Characterization
3.3. Catalytic Activity Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Olah, G.A. Towards oil independence through renewable methanol chemistry. Angew. Chem. Int. Ed. 2013, 52, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Smit, B. Carbon Capture and Storage: Introductory Lecture. Faraday Discuss. 2016, 192, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Melian-Cabrera, I.; Lopez-Granados, M.; Fierro, J.L.G. Pd-Modified Cu–Zn Catalysts for Methanol Synthesis from CO2/H2 Mixtures: Catalytic Structures and Performance. J. Catal. 2002, 210, 285–294. [Google Scholar] [CrossRef]
- Klier, K. Methanol synthesis. Adv. Catal. 1982, 31, 243–313. [Google Scholar] [CrossRef]
- Fichtl, M.B.; Schlereth, D.; Jacobsen, N.; Kasatkin, I.; Schumann, J.; Behrens, M.; Schlögl, R.; Hinrichsen, O. Kinetics of Deactivation on Cu/ZnO/Al2O3 Methanol Synthesis Catalysts. Appl. Catal. A 2015, 502, 262–270. [Google Scholar] [CrossRef]
- Bonura, G.; Khassin, A.A.; Yurieva, T.M.; Cannilla, C.; Frusteri, F.; Frusteri, L. Structure control on kinetics of copper oxides to methanol. Catal. Today 2020, 342, 39–45. [Google Scholar] [CrossRef]
- Kattel, S.; Ramírez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kuhl, S.; Havecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Ghosh, S.; Uday, V.; Giri, A.; Srinivas, S. Biogas to methanol: A comparison of conversion processes involving direct carbon dioxide hydrogenation and via reverse water gas shift reaction. J. Clean. Prod. 2019, 217, 615–626. [Google Scholar] [CrossRef]
- Kondrat, S.A.; Smith, P.J.; Carter, J.H.; Hayward, J.S.; Pudge, G.J.; Shaw, G.; Spencer, M.S.; Bartley, J.K.; Taylor, S.H.; Hutchings, G.J. The effect of sodium species on methanol synthesis and water-gas shift Cu/ZnO catalysts: Utilising high purity zincian georgeite. Faraday Discuss. 2017, 197, 287–307. [Google Scholar] [CrossRef]
- Behrens, M. Meso- and nano-structuring of industrial Cu/ZnO/(Al2O3) catalysts. J. Catal. 2009, 267, 24–29. [Google Scholar] [CrossRef]
- Kühl, S.; Tarasov, A.; Zander, S.; Kasatkin, I.; Behrens, M. Cu-Based Catalyst Resulting from a Cu, Zn, Al Hydrotalcite-Like Compound: A Microstructural, Thermoanalytical, and In Situ XAS Study. Chem. Eur. J. 2014, 20, 3782–3792. [Google Scholar] [CrossRef] [PubMed]
- Mota, N.; Guil-Lopez, R.; Pawelec, B.G.; Fierro, J.L.G.; Navarro, R.M. Highly active Cu/ZnO-Al catalyst for methanol synthesis: Effect of aging on its structure and activity. RSC Adv. 2018, 8, 20619–20629. [Google Scholar] [CrossRef]
- Höppener, R.H.; Doesburg, E.B.M.; Scholten, J.J.F. Preparation and characterization of stable copper/zinc oxide/alumina catalysts for methanol synthesis. Appl. Catal. 1986, 25, 109–119. [Google Scholar] [CrossRef]
- Fujita, S.I.; Satriyo, A.M.; Shen, G.C.; Takezawa, N. Mechanism of the formation of precursors for the Cu/ZnO methanol synthesis catalysts by a co-precipitation method. Catal. Lett. 1995, 34, 85–92. [Google Scholar] [CrossRef]
- Anton, J.; Nebel, J.; Song, H.; Froese, C.; Weide, P.; Ruland, H.; Muhler, M.; Kaluza, S. The effect of sodium on the structure–activity relationships of cobalt-modified Cu/ZnO/Al2O3 catalysts applied in the hydrogenation of carbon monoxide to higher alcohols. J. Catal. 2016, 335, 175–186. [Google Scholar] [CrossRef]
- Prieto, G.; de Jong, K.P.; de Jongh, P.E. Towards “greener” catalyst manufacture: Reduction of wastewater from the preparation of Cu/ZnO/Al2O3 methanol synthesis catalysts. Catal. Today 2013, 215, 142–151. [Google Scholar] [CrossRef]
- Jeong, C.; Kim, T.; Kim, J.; Suh, Y.W. Use of tetraethylammoium bicarbonate as a precipitation agent on the preparation of co-precipitated Cu/ZnO catalysts. Appl. Catal. A Gen. 2017, 541, 35–41. [Google Scholar] [CrossRef]
- Zuo, Y.Z.; An, X.; Han, M.H.; Wang, J.F.; Wang, D.Z.; Jin, Y. Effect of residual sodium on the catalytic activity and stability of Cu-based methanol synthesis catalyst. Chin. J. Catal. 2008, 29, 1266–1270. [Google Scholar]
- Jun, K.W.; Shen, W.J.; Rao, K.S.R.; Lee, K.W. Residual sodium effect on the catalytic activity of Cu/ZnO/Al2O3 in methanol synthesis from CO2 hydrogenation. Appl. Catal. A Gen. 1998, 174, 231–238. [Google Scholar] [CrossRef]
- Suh, Y.W.; Rhee, H.K. Optimum Washing Conditions for the Preparation of Cu/ZnO/ZrO2 for Methanol Synthesis from CO Hydrogenation: Effects of Residual Sodium. Korean J. Chem. Eng. 2002, 19, 17–19. [Google Scholar] [CrossRef]
- Gimenez, P.; Fereres, S. Effect of Heating Rates and Composition on the Thermal Decomposition of Nitrate Based Molten Salts. Energy Procedia 2015, 69, 654–662. [Google Scholar] [CrossRef]
- Millar, G.J.; Holm, I.H.; Uwins, P.J.; Drennan, J.J. Characterization of precursors to methanol synthesis catalysts Cu/ZnO system. Chem. Soc. Faraday Trans. 1998, 94, 593–600. [Google Scholar] [CrossRef]
- Frost, R.L.; Locke, A.J.; Hales, M.C.; Martens, W.N. Thermal stability of synthetic aurichalcite implications for making mixed metal oxides for use as catalysts. J. Therm. Anal. Calorim. 2008, 94, 203–208. [Google Scholar] [CrossRef]
- Schumann, J.; Tarasov, S.; Thomas, N.; Schlögl, R.; Behrens, M. Cu,Zn-based catalysts for methanol synthesis: On the effect of calcination conditions and the part of residual carbonates. Appl. Catal. A Gen. 2015, 516, 117–126. [Google Scholar] [CrossRef]
- Bems, B.; Schur, M.; Dassenoy, A.; Junkes, H.; Herein, D.; Schlögl, R. Relations between Synthesis and Microstructural Properties of Copper/Zinc Hydroxycarbonates. Chem. Europ. J. 2003, 9, 2039–2052. [Google Scholar] [CrossRef]
- Dal Pozzo, A.; Armutlulu, A.; Rekhtina, M.; Abdala, P.M.; Müller, C.R. CO2 Uptake and Cyclic Stability of MgO-Based CO2 Sorbents Promoted with Alkali Metal Nitrates and Their Eutectic Mixtures. ACS Appl. Energy Mater. 2019, 2, 1295–1307. [Google Scholar] [CrossRef]
- Zhang, K.; Li, X.S.; Li, W.Z.; Rohatgi, A.; Duan, Y.; Singh, P.; Li, L.; King, D.L. Phase Transfer-Catalyzed Fast CO2 Absorption by MgO-Based Absorbents with High Cycling Capacity. Adv. Mater. Interfaces 2014, 1, 1400030. [Google Scholar] [CrossRef]
- Rekhtina, M.; Dal Pozzo, A.; Stoian, D.; Armutlulu, A.; Donat, F.; Blanco, M.V.; Wang, Z.-J.; Willinger, M.G.; Fedorov, A.; Abdala, P.M.; et al. Effect of molten sodium nitrate on the decomposition pathways of hydrated magnesium hydroxycarbonate to magnesium oxide probed by in situ total scattering. Nanoscale 2020, 12, 16462–16473. [Google Scholar] [CrossRef]
- Jo, S.-I.; An, Y.-N.; Kim, K.-Y.; Choi, S.-Y.; Kwak, J.-S.; Oh, K.-R.; Kwon, Y.U. Mechanisms of absorption and desorption of CO2 by molten NaNO3-promoted MgO. Phys. Chem. Chem. Phys. 2017, 19, 6224–6232. [Google Scholar] [CrossRef]
- Harada, T.; Simeon, F.; Hamad, E.Z. Hatton, Alkali Metal Nitrate-Promoted High-Capacity MgO Adsorbents for Regenerable CO2 Capture at Moderate Temperatures. Chem. Mater. 2015, 27, 1943–1947. [Google Scholar] [CrossRef]
- Tarasov, A.; Schumann, J.; Thomas, N.; Behrens, M. Thermokinetic investigation of binary Cu/Zn hydroxycarbonates as precursors for Cu/ZnO catalysts. Termochim. Acta 2014, 591, 1–9. [Google Scholar] [CrossRef]
- Fujita, S.; Moribe, S.; Kanamori, Y.; Kakudate, M.; Tazekawa, N. Preparation of a coprecipitated Cu/ZnO catalyst for the methanol synthesis from CO2–Effects of the calcination and reduction conditions on the catalytic performance. Appl. Catal. A Gen. 2001, 207, 121–128. [Google Scholar] [CrossRef]
- Sloczynski, J.; Grabowski, R.; Kozlowska, A.; Olszewski, P.K.; Stoch, J. Reduction kinetics of CuO in CuO/ZnO/ZrO2 systems. Phys. Chem. Chem. Phys. 2003, 5, 4631–4640. [Google Scholar] [CrossRef]
- Gao, P.; Li, F.; Xiao, F.K.; Zhao, N.; Sun, N.N.; Wei, W.; Zhong, L.S.; Sun, Y.H. Preparation and activity of Cu/Zn/Al/Zr catalysts via hydrotalcite-containing precursors for methanol synthesis from CO2 hydrogenation. Catal. Sci. Technol. 2012, 2, 1447–1454. [Google Scholar] [CrossRef]
- Shimokawabe, M.; Asakawa, H.; Takezawa, N. Characterization of copper/zirconia catalysts prepared by an impregnation method. Appl. Catal. 1990, 59, 45–58. [Google Scholar] [CrossRef]
- Breen, J.P.; Ross, J.R. Methanol reforming for fuel-cell applications: Development of zirconia-containing Cu–Zn–Al catalysts. Catal. Today 1999, 51, 521–533. [Google Scholar] [CrossRef]
- Yuvaraj, S.; Fan-Yuan, L.; Tsong-Huei, C.; Chuin-Tih, Y. Thermal Decomposition of Metal Nitrates in Air and Hydrogen Environments. J. Phys. Chem. B 2003, 107, 1044–1047. [Google Scholar] [CrossRef]
- Ressler, T.; Kneip, B.L.; Kasatkin, I.; Schlögl, R. The Microstructure of Copper Zinc Oxide Catalysts: Bridging the Materials Gap. Angew. Chem. Int. Ed. 2003, 44, 4704–4707. [Google Scholar] [CrossRef]
- Prašnikar, A.; Pavlišič, A.; Ruiz-Zepeda, F.; Kovač, J.; Likozar, B. Mechanisms of Copper-Based Catalyst Deactivation during CO2 Reduction to Methanol. Ind. Eng. Chem. Res. 2019, 58, 13021–13029. [Google Scholar] [CrossRef]
- Fichtl, M.B.; Schumann, J.; Jacobsen, N.C.; Behrens, M.; Schlögl, R.; Muhler, M.; Hinrichsen, O. Counting of Oxygen Defects versus Metal Surface Sites in Methanol Synthesis Catalysts by Different Probe Molecules. Angew. Chem. Int. Ed. 2014, 53, 7043–7047. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Choi, Y.; Fujitani, T. On the Issue of the Active Site and the Role of ZnO in Cu/ZnO Methanol Synthesis Catalysts. Top. Catal. 2003, 22, 277–285. [Google Scholar] [CrossRef]
- Lunkenbein, T.; Schuman, J.; Behrens, M.; Schlögl, R.; Willinger, M.G. Formation of a ZnO Overlayer in Industrial Cu/ZnO/Al2O3 Catalysts Induced by Strong Metal–Support Interactions. Angew. Chem. 2015, 127, 4627–4631. [Google Scholar] [CrossRef]
- Schuman, J.; Lunkebein, T.; Tarasov, A.; Thomas, N.; Schlögl, R.; Behrens, M. Synthesis and Characterisation of a Highly Active Cu/ZnO:Al Catalyst. ChemCatChem 2014, 6, 2889–2897. [Google Scholar] [CrossRef]
- Behrens, M.; Zander, S.; Kurr, P.; Jacobsen, N.; Senker, J.; Koch, G.; Ressler, T.; Fischer, R.W.; Schlögl, R. Performance Improvement of Nanocatalysts by Promoter-Induced Defects in the Support Material: Methanol Synthesis over Cu/ZnO:Al. J. Am. Chem. Soc. 2013, 135, 6061–6068. [Google Scholar] [CrossRef]
- Bond, G.C.; Namijo, S.N. An improved procedure for estimating the metal surface area of supported copper catalysts. J. Catal. 1989, 118, 507–510. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Washing (mL H2O/g)/Time (min) | Na/NaNO3 (wt %) | SBET (m2·g−1) a | Vpore (cm3·g−1) a |
|---|---|---|---|---|
| W/O | 00 | 7.339.4 | 107 | 0.461 |
| W1 | 1005 | 0.502.8 | 128 | 0.558 |
| W3 | 30,015 | 0.010.05 | 128 | 0.578 |
| W6 | 60,030 | 0.010.05 | 127 | 0.579 |
| Catalyst | dp CuO (nm) | HT-CO3 T (°C)-wt% | SBET (a) (m2/g) | TPR T (°C) |
|---|---|---|---|---|
| CuO/ZnO-Al-W/O | 9 | - | 67.1 | 265 |
| CuO/ZnO-Al W1 | 5 | 386–2.6 | 87.5 | 195 |
| CuO/ZnO-Al W3 | <2 | 528–8.5 | 107 | 165 |
| Catalyst | Effective Cu Surface Area (m2·g−1) | Initial CH3OH Time Yield a (μmol min−1·g−1) | TOF b (s−1) | Deactivation Rate (μmol min−1·g−1) |
|---|---|---|---|---|
| Cu/ZnO-Al W/O | 0 | 0 | - | - |
| Cu/ZnO-Al W1 | 29.1 | 300 | 7.0 | 0.05 |
| Cu/ZnO-Al W3 | 41.3 | 1050 | 17.3 | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guil-López, R.; Mota, N.; Llorente, J.; Millan, E.; Pawelec, B.G.; Fierro, J.L.G.; Navarro, R.M. Unravelling the Structural Modification (Meso-Nano-) of Cu/ZnO-Al2O3 Catalysts for Methanol Synthesis by the Residual NaNO3 in Hydroxycarbonate Precursors. Catalysts 2020, 10, 1346. https://doi.org/10.3390/catal10111346
Guil-López R, Mota N, Llorente J, Millan E, Pawelec BG, Fierro JLG, Navarro RM. Unravelling the Structural Modification (Meso-Nano-) of Cu/ZnO-Al2O3 Catalysts for Methanol Synthesis by the Residual NaNO3 in Hydroxycarbonate Precursors. Catalysts. 2020; 10(11):1346. https://doi.org/10.3390/catal10111346
Chicago/Turabian StyleGuil-López, Rut, Noelia Mota, Jorge Llorente, Elena Millan, Bárbara G. Pawelec, Jose Luis G. Fierro, and Rufino M. Navarro. 2020. "Unravelling the Structural Modification (Meso-Nano-) of Cu/ZnO-Al2O3 Catalysts for Methanol Synthesis by the Residual NaNO3 in Hydroxycarbonate Precursors" Catalysts 10, no. 11: 1346. https://doi.org/10.3390/catal10111346
APA StyleGuil-López, R., Mota, N., Llorente, J., Millan, E., Pawelec, B. G., Fierro, J. L. G., & Navarro, R. M. (2020). Unravelling the Structural Modification (Meso-Nano-) of Cu/ZnO-Al2O3 Catalysts for Methanol Synthesis by the Residual NaNO3 in Hydroxycarbonate Precursors. Catalysts, 10(11), 1346. https://doi.org/10.3390/catal10111346