Ruthenium-Catalyzed C–H Activations for the Synthesis of Indole Derivatives


{kind=link}
{kind=link}
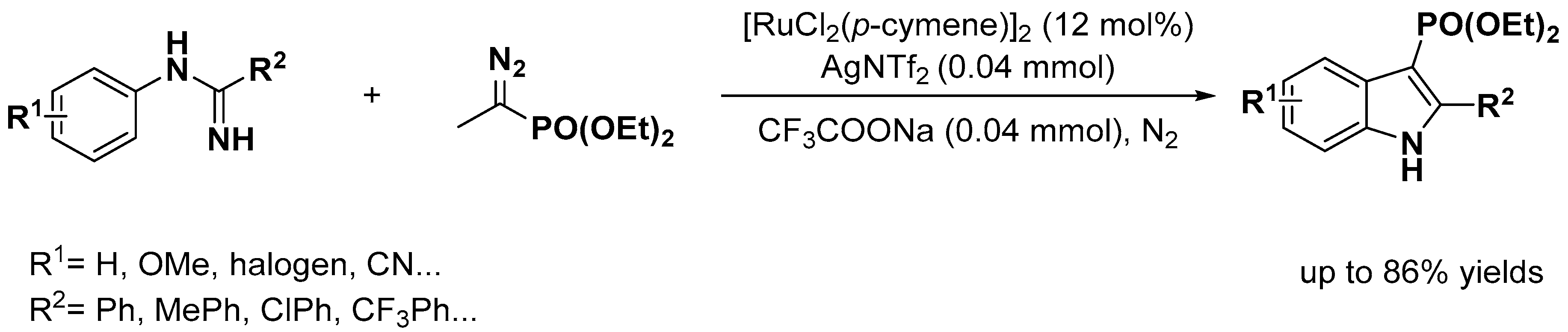{kind=link}
{kind=link}
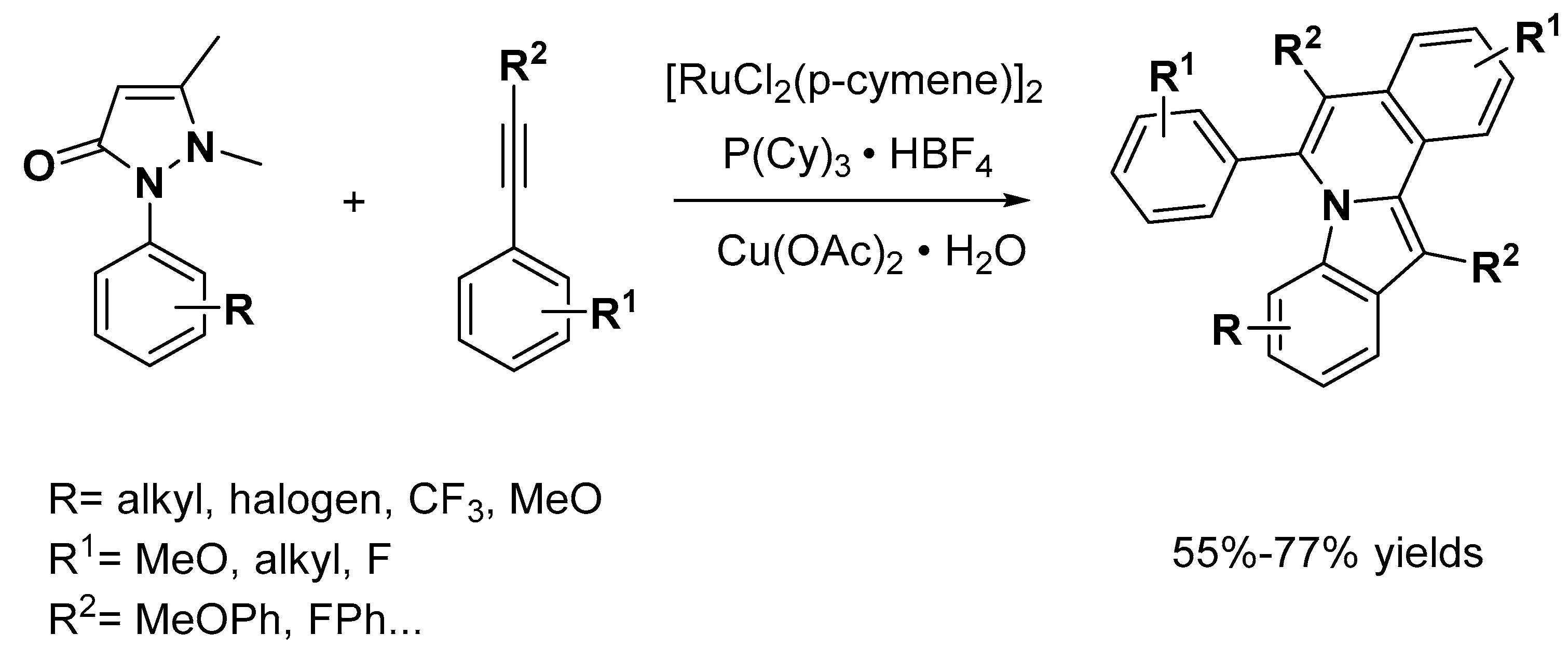{kind=link}
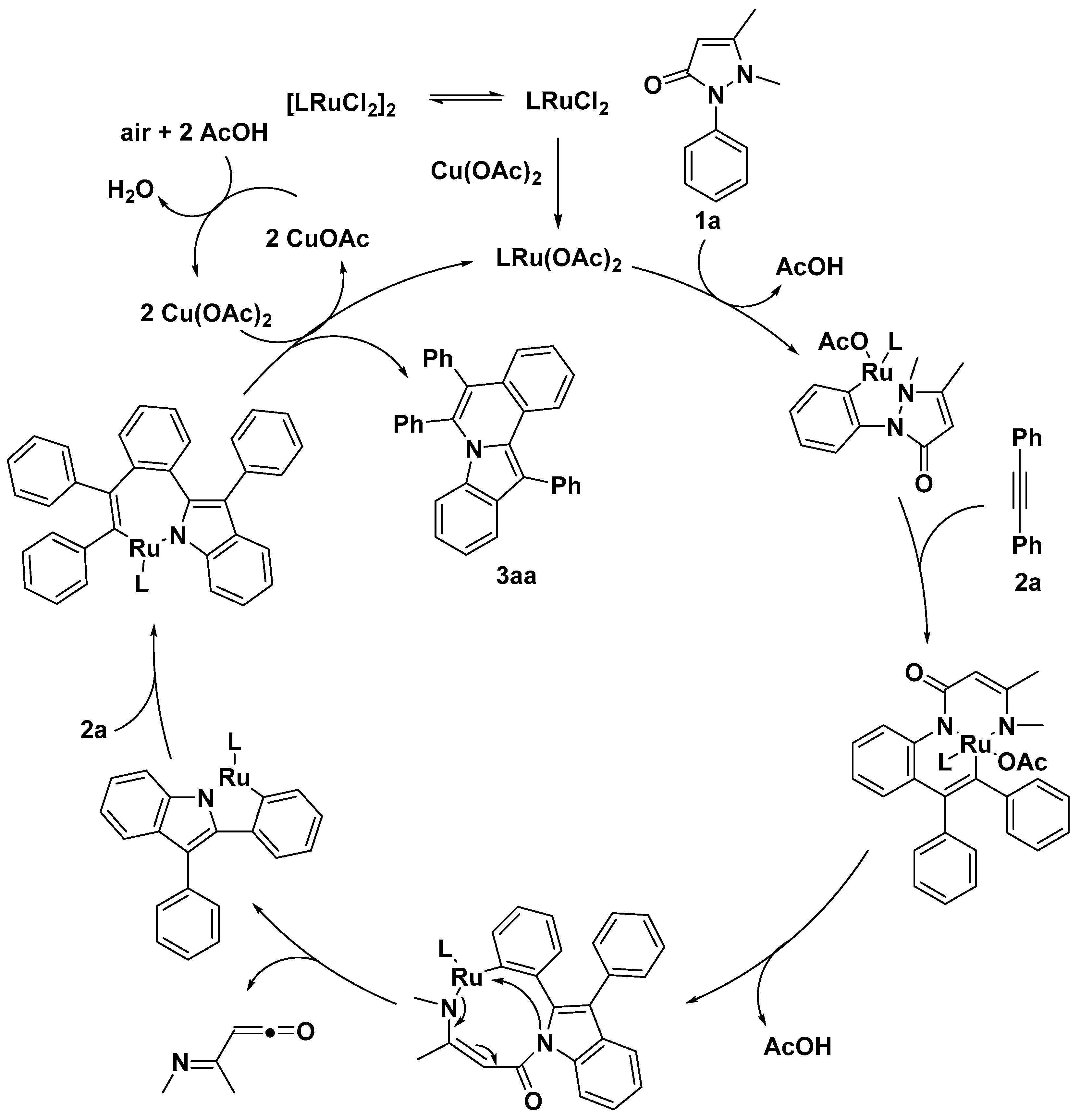{kind=link}
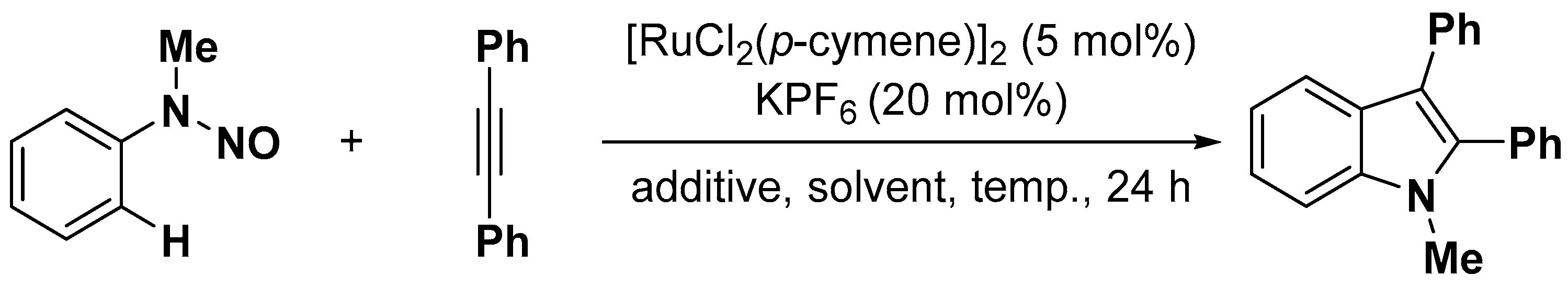{kind=link}
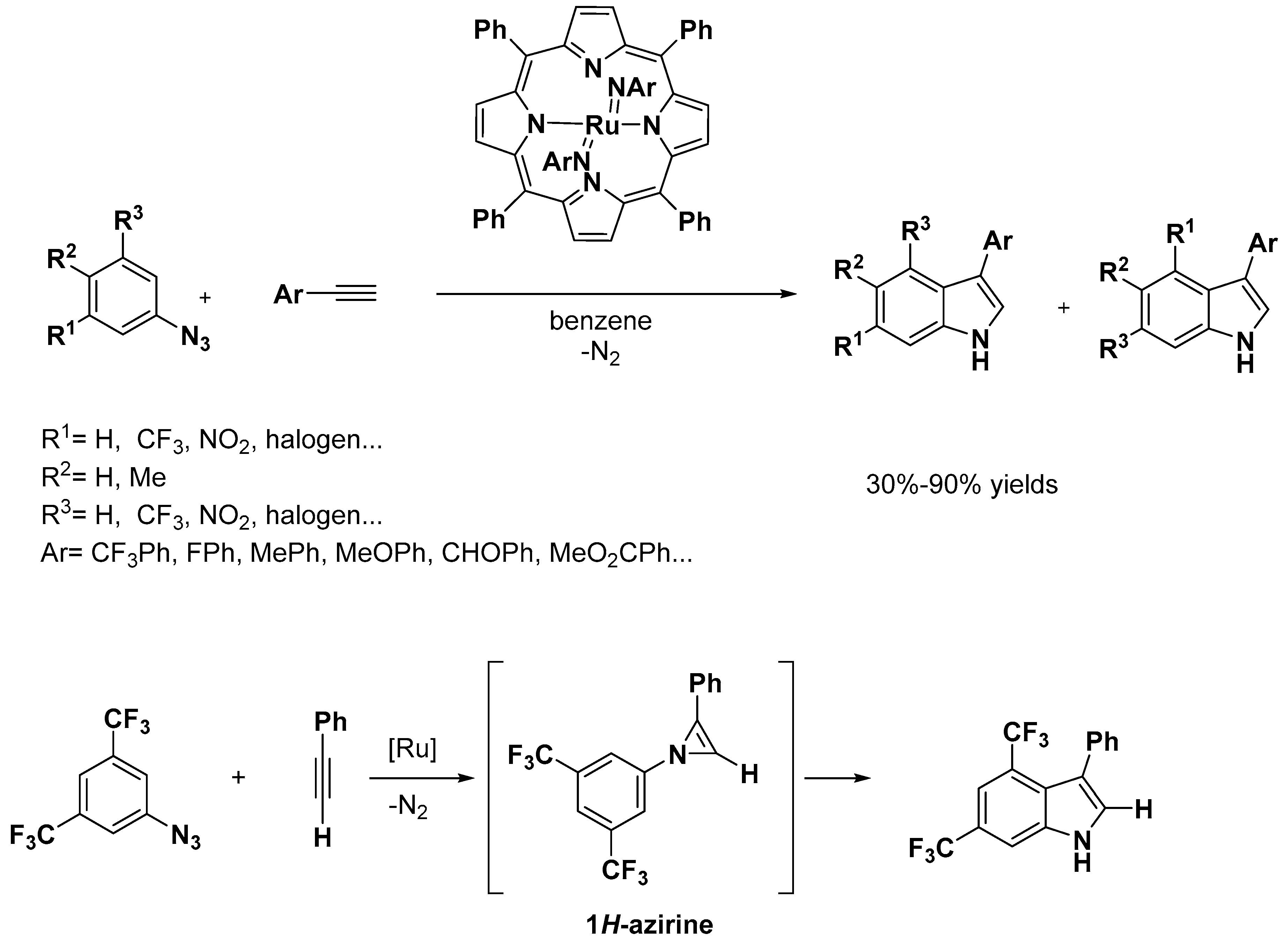{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
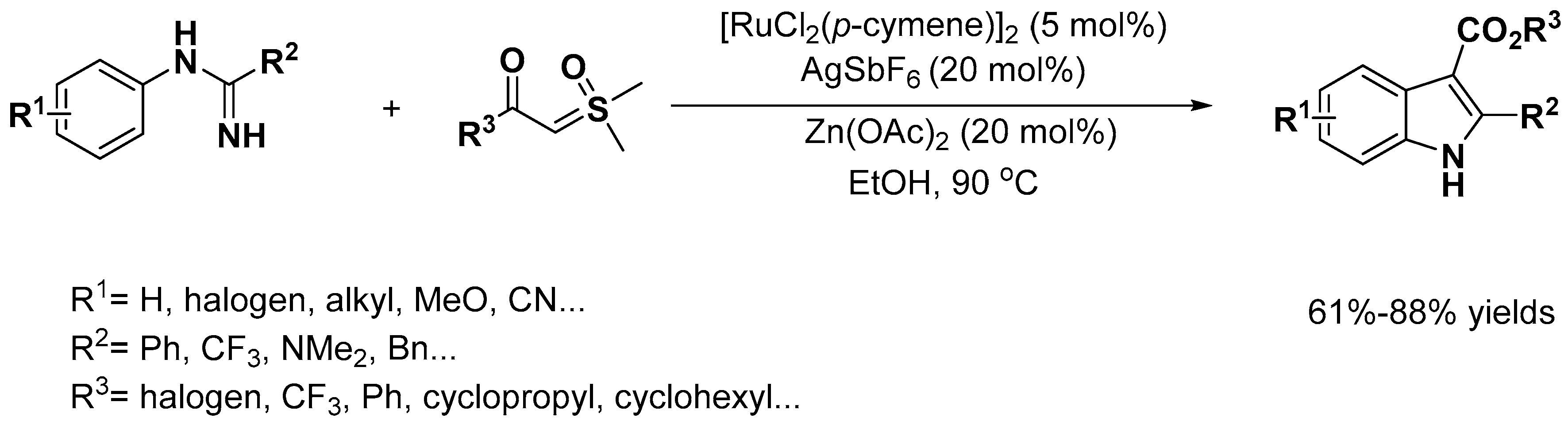{kind=link}
{kind=link}
{kind=link}
{kind=link}
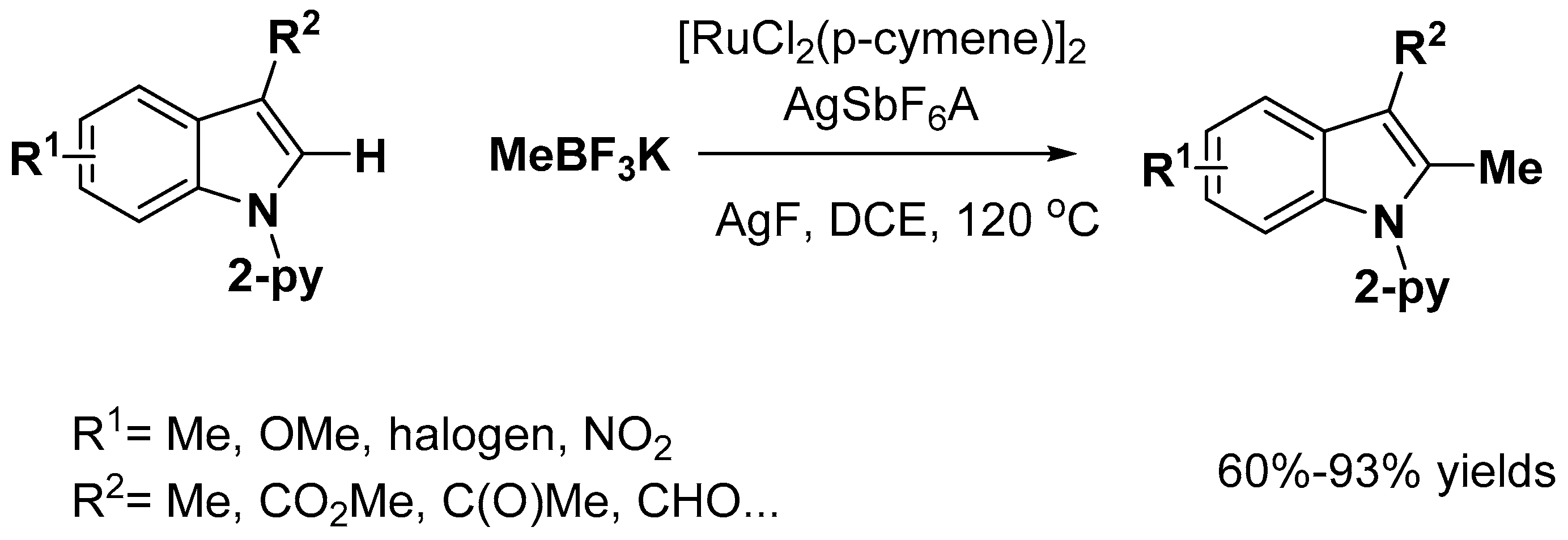{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
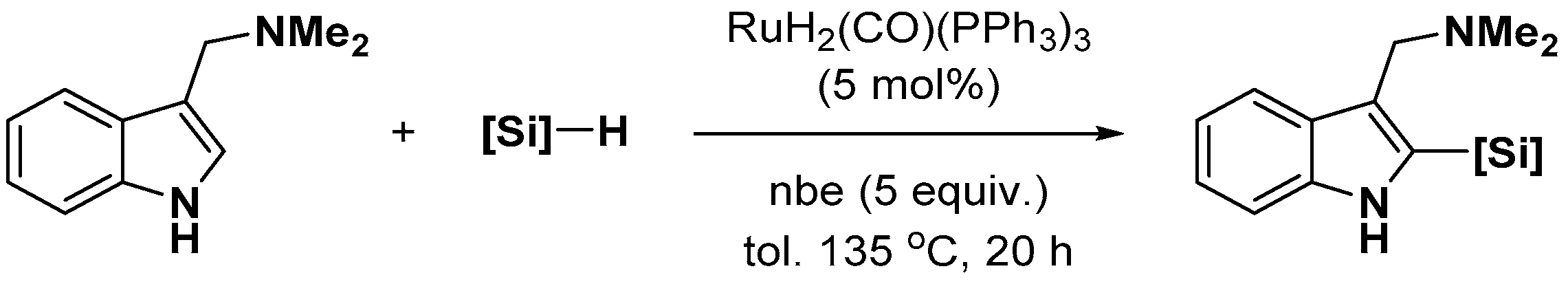{kind=link}
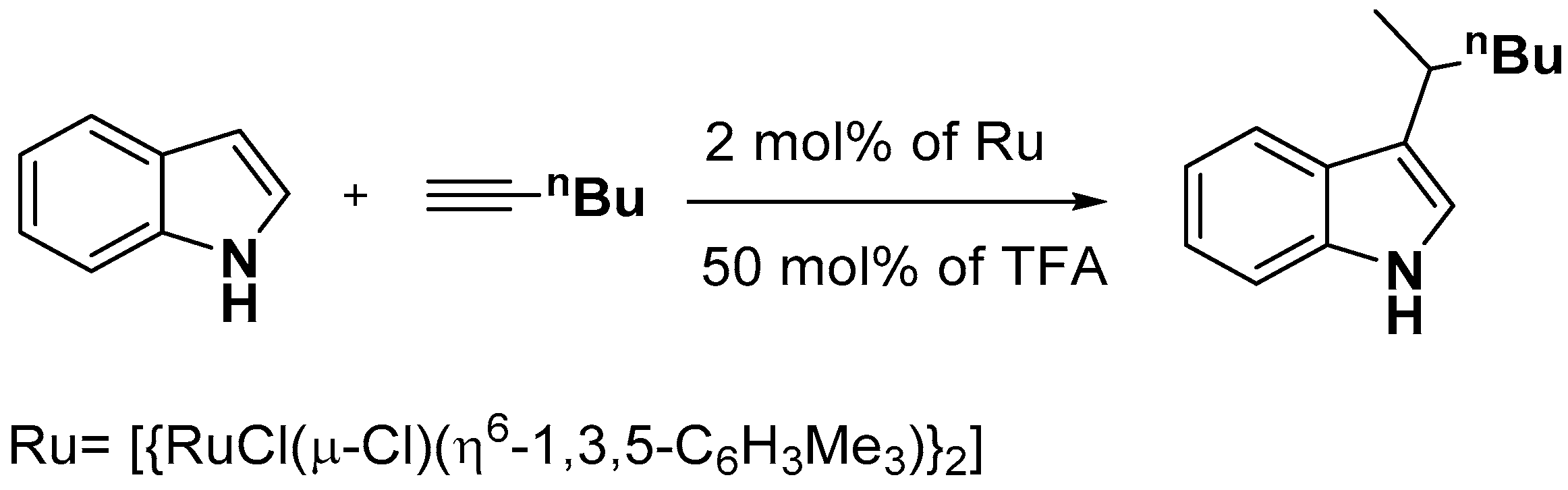{kind=link}
{kind=link}
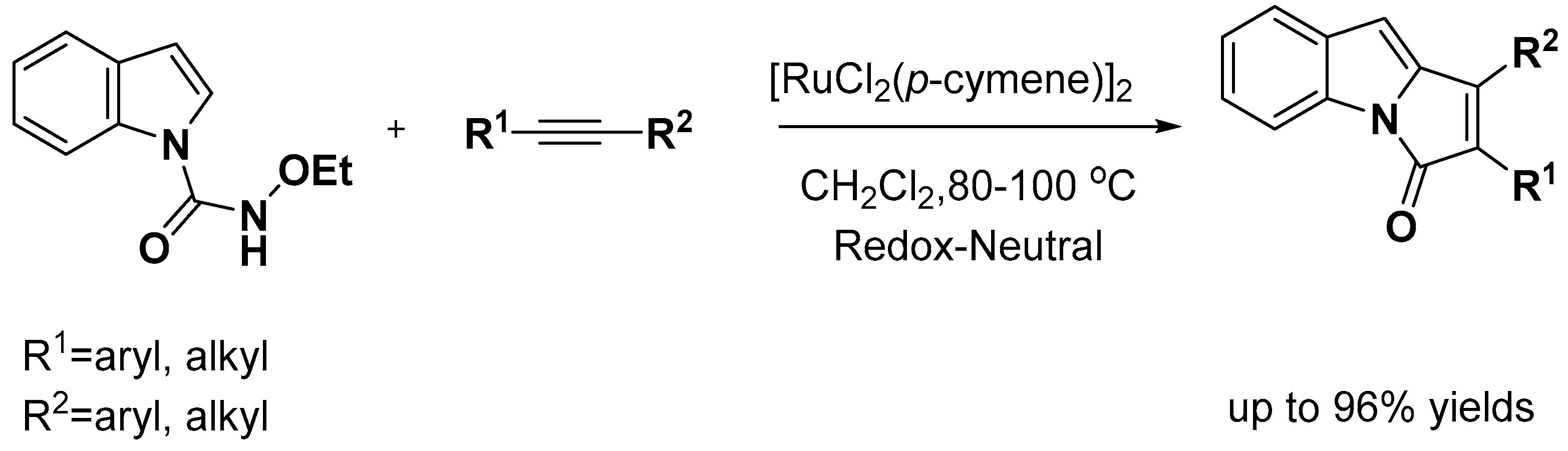{kind=link}
{kind=link}
{kind=link}
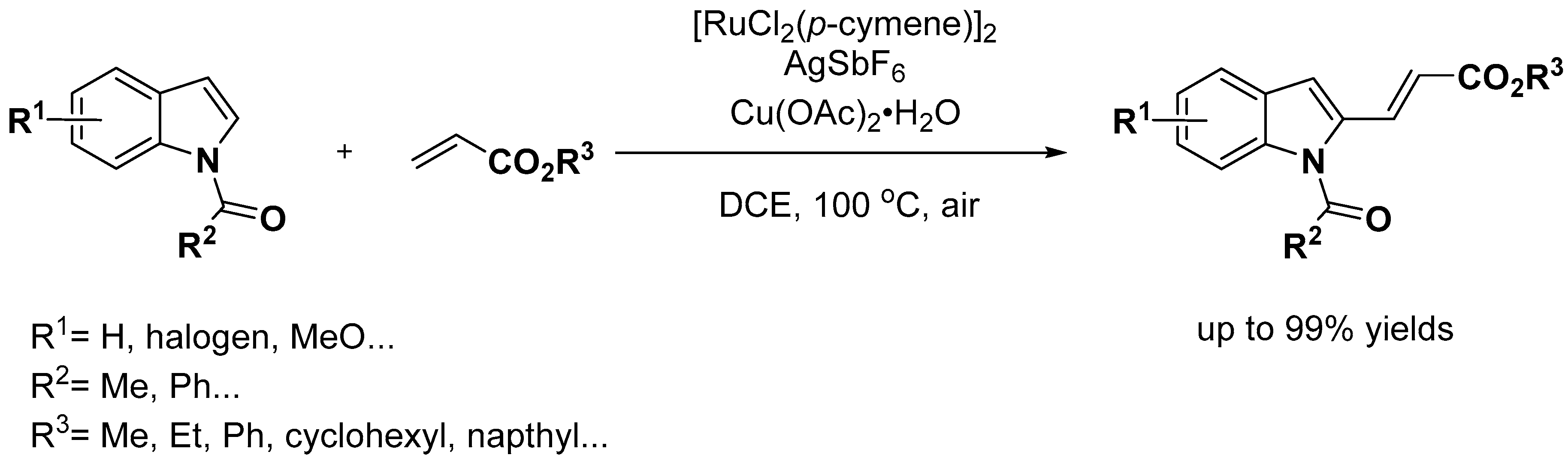{kind=link}
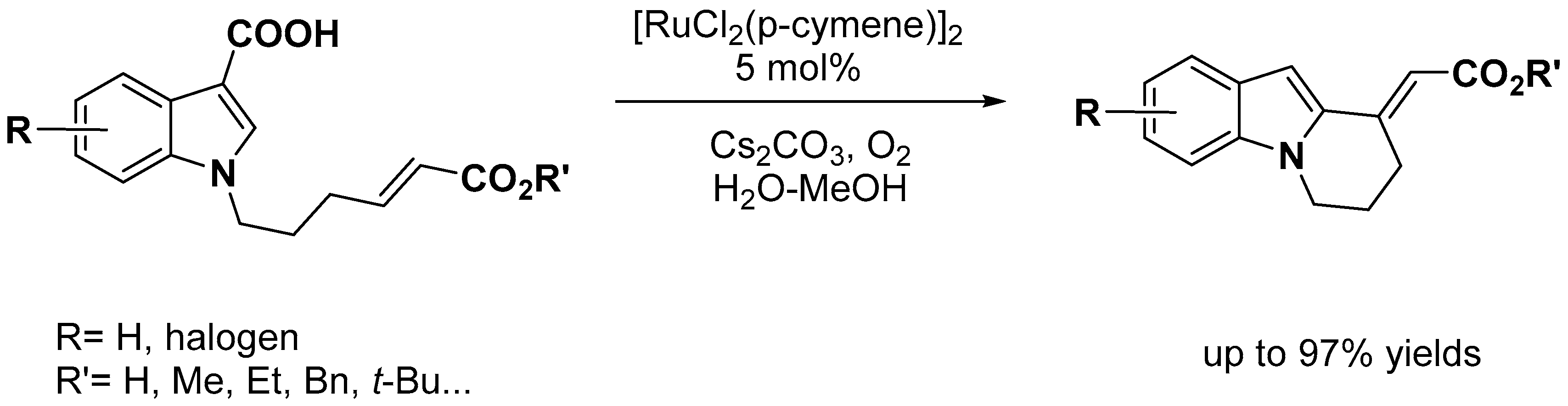{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
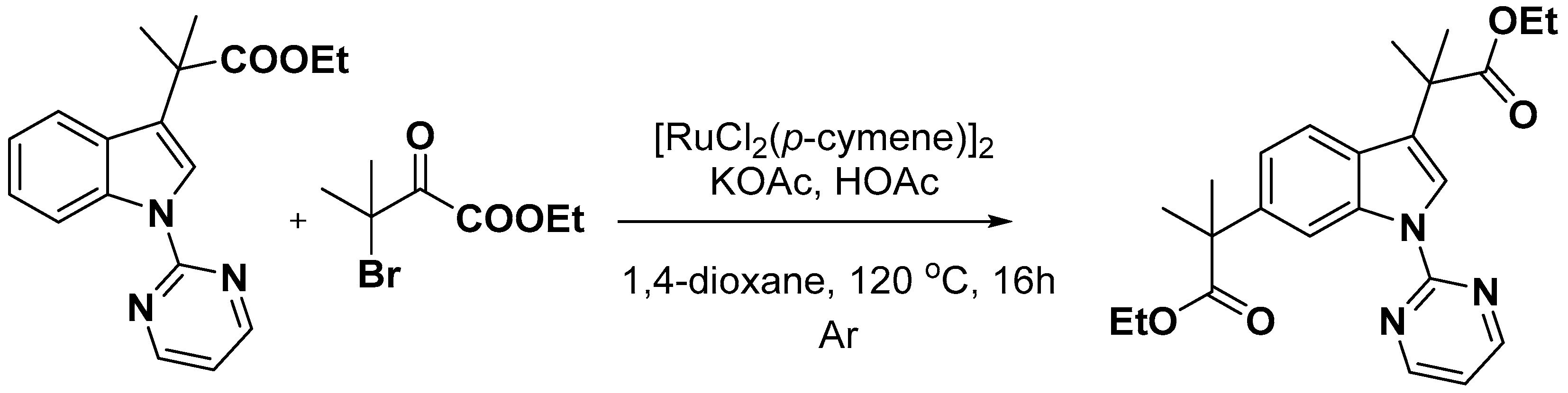{kind=link}
{kind=link}
{kind=link}
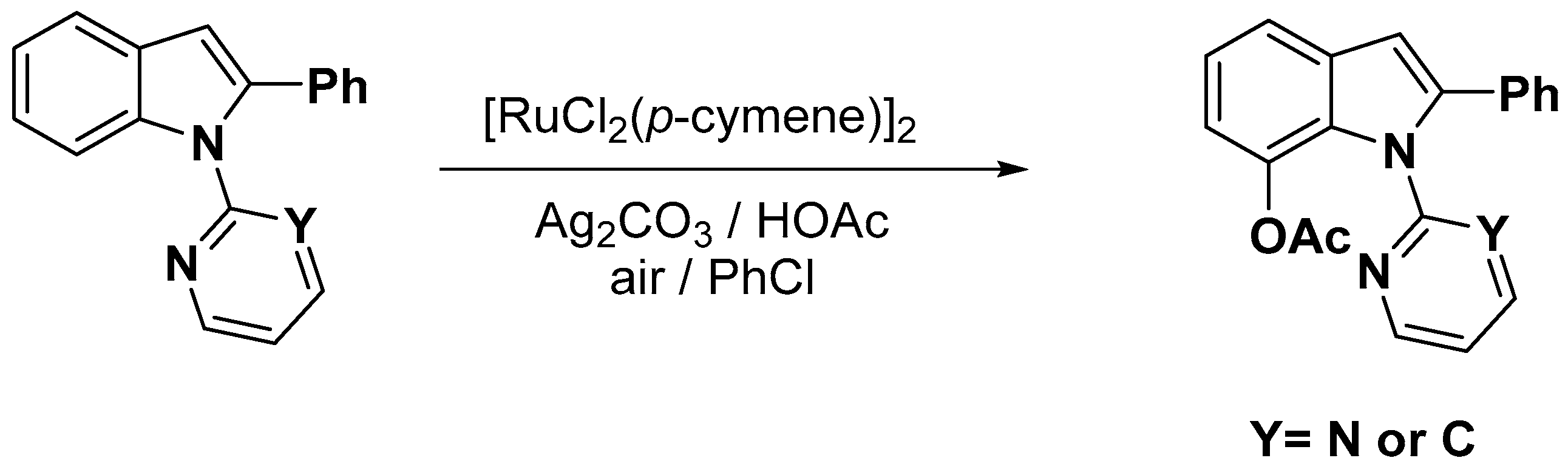{kind=link}
{kind=link}
Abstract
1. Introduction
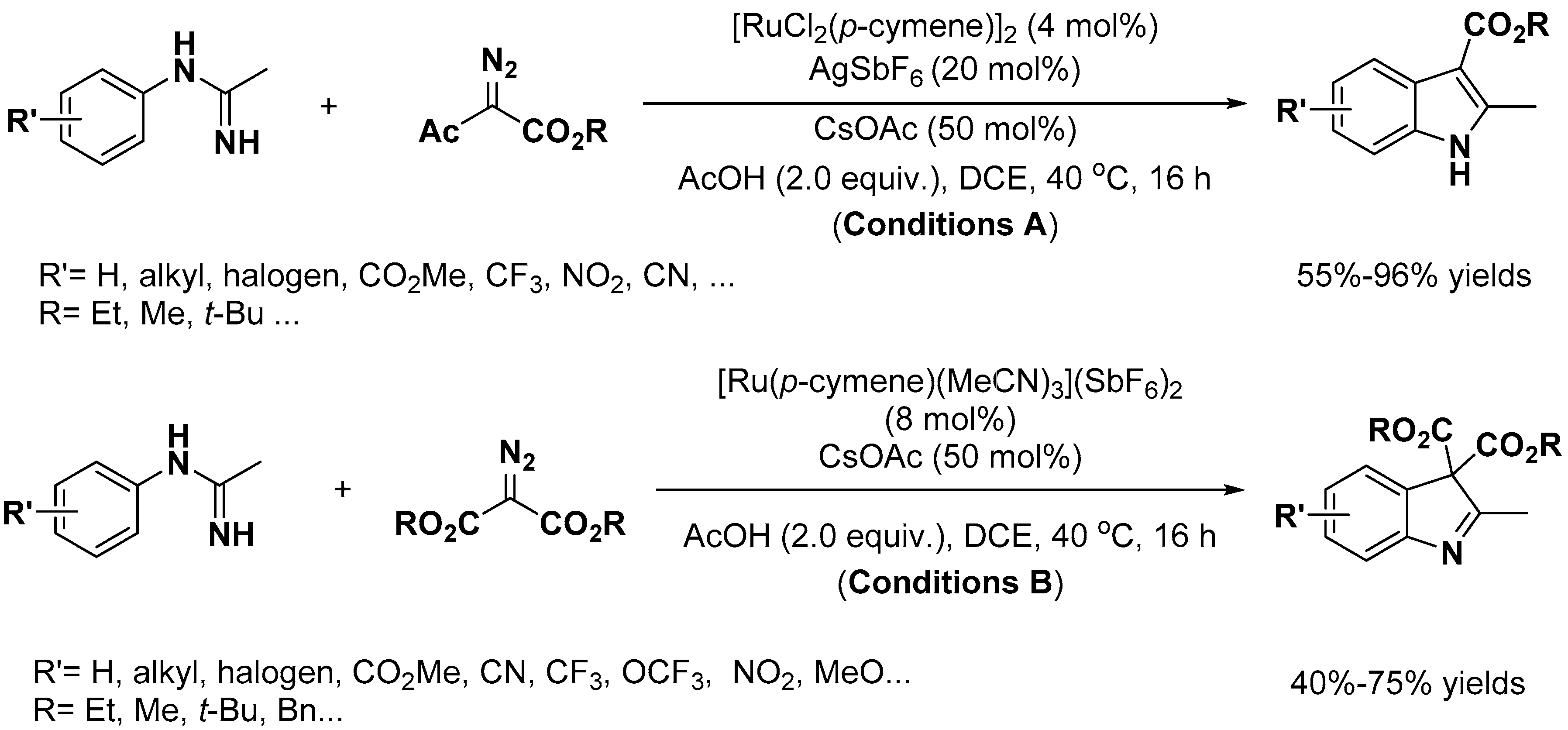
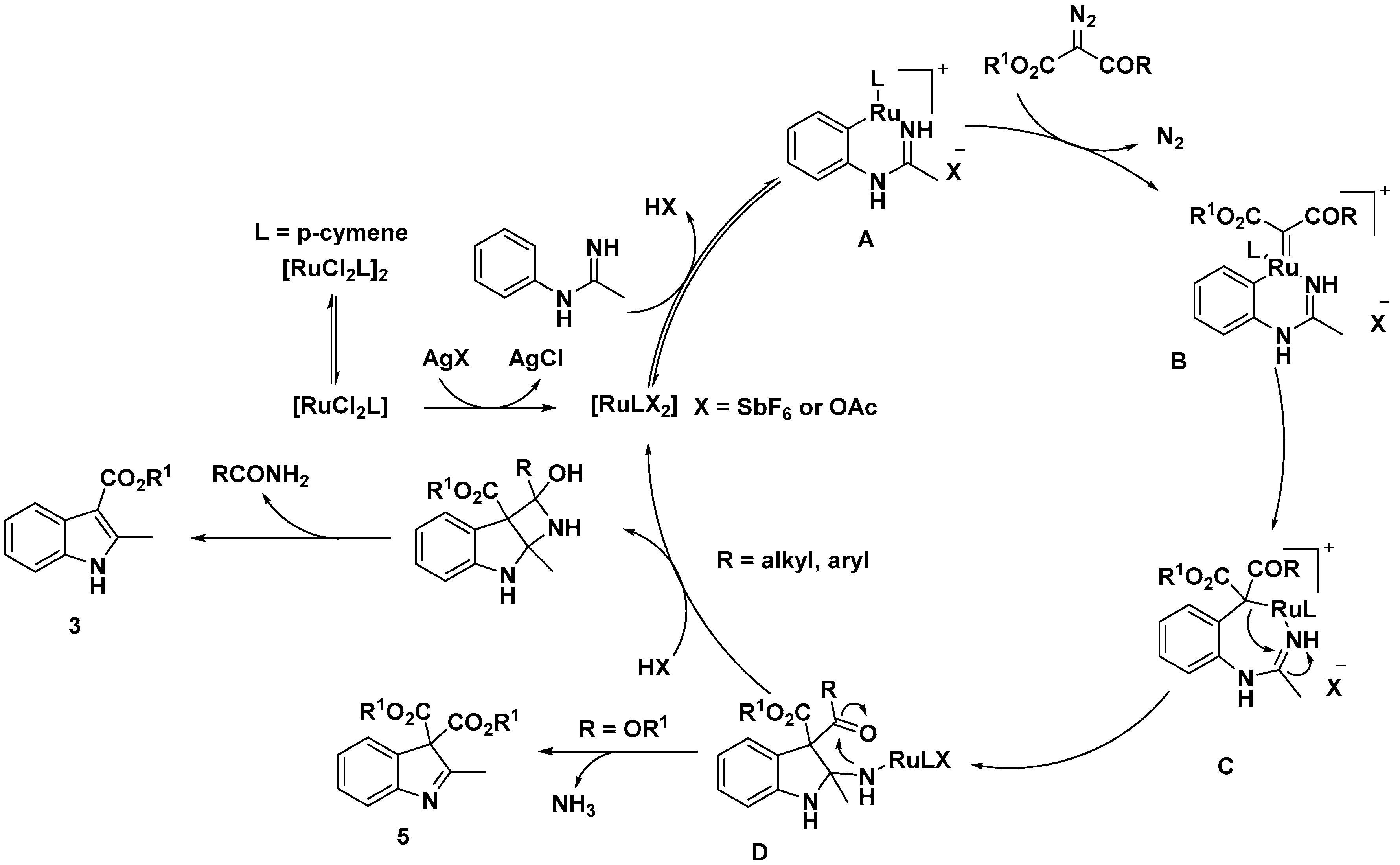
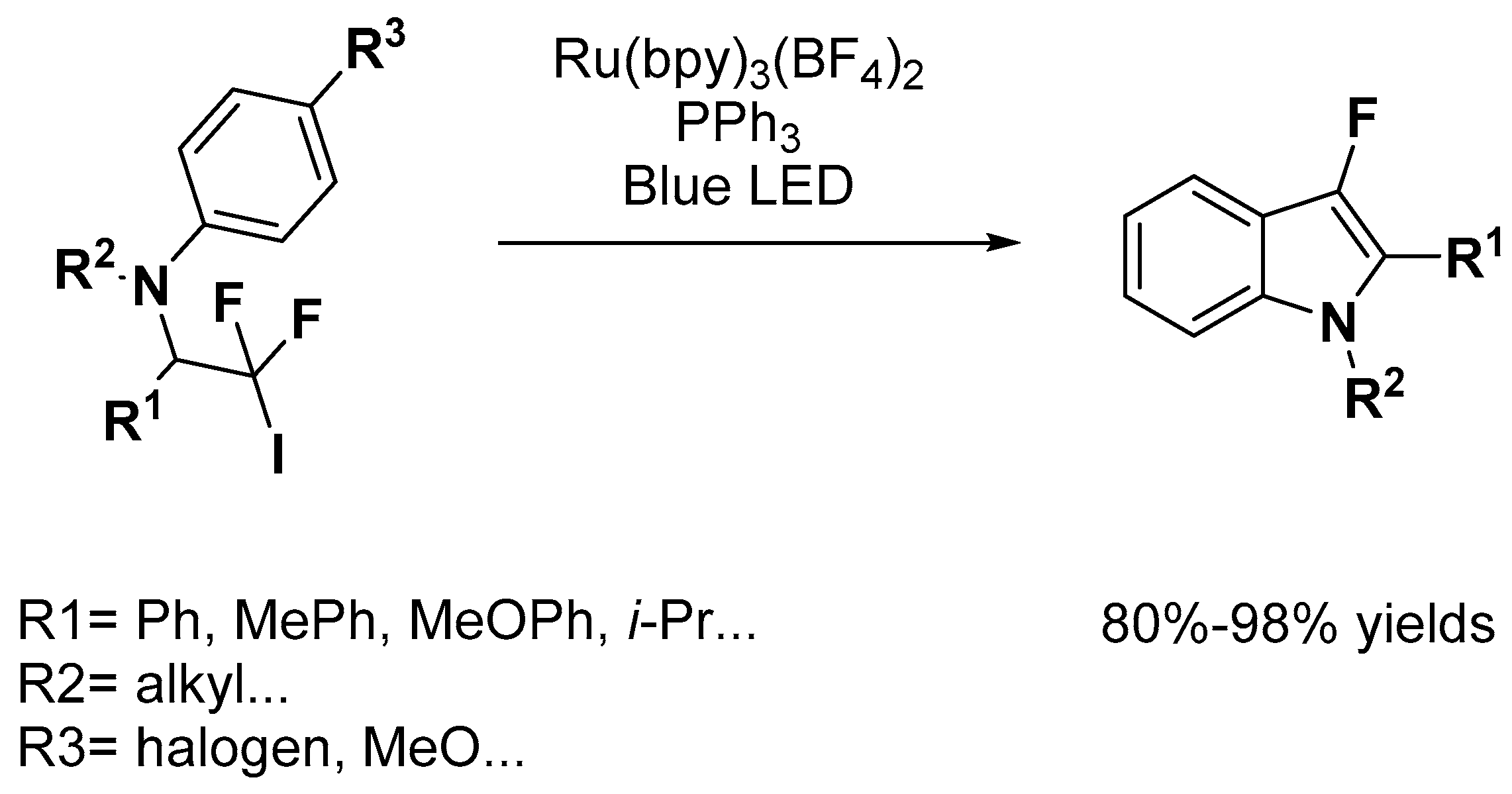
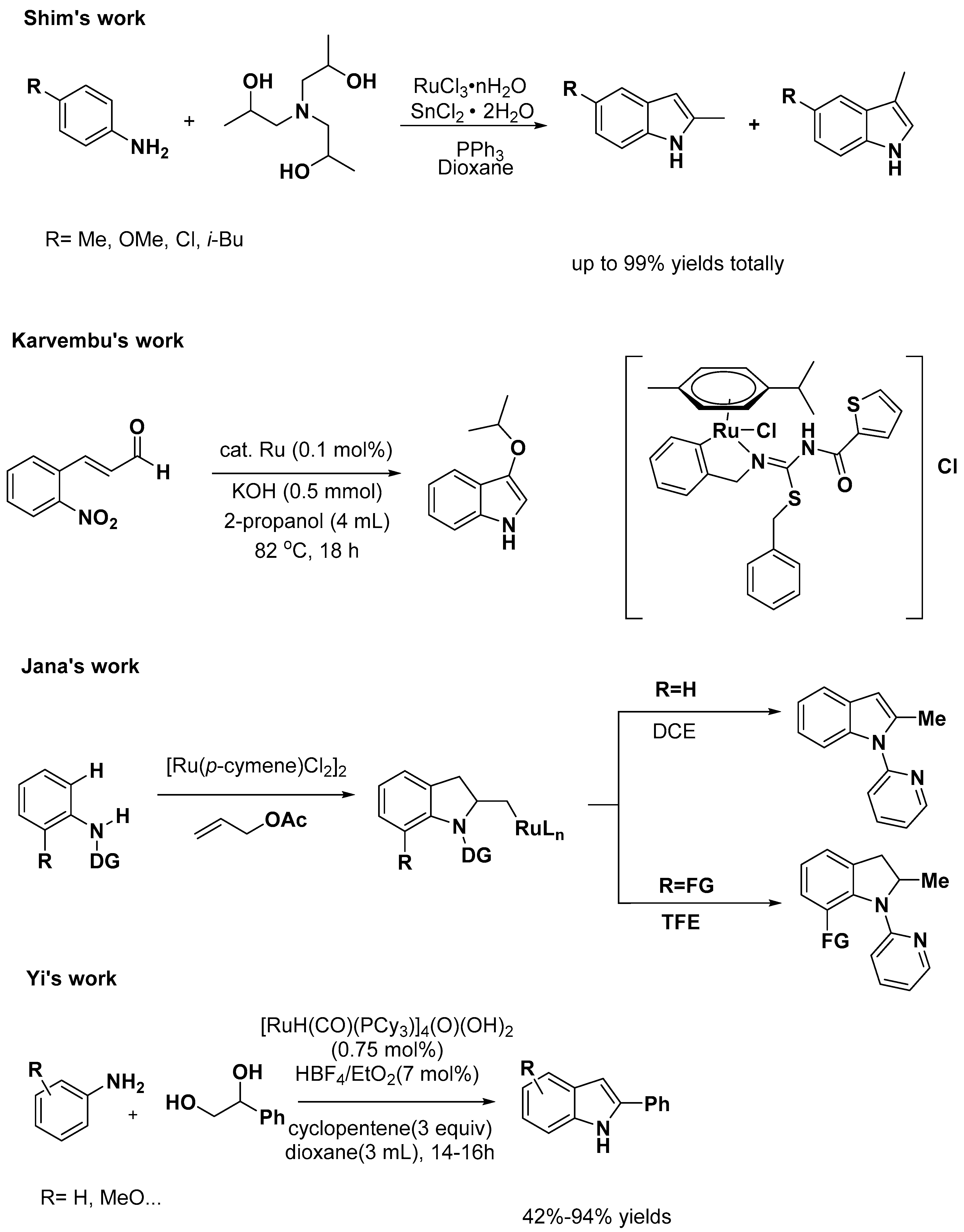
Indole Scaffold Construction via Ruthenium-Catalyzed C–H Activation
2. Site-Specific Modification for Indole Scaffold via Ruthenium-Catalyzed C–H Activation
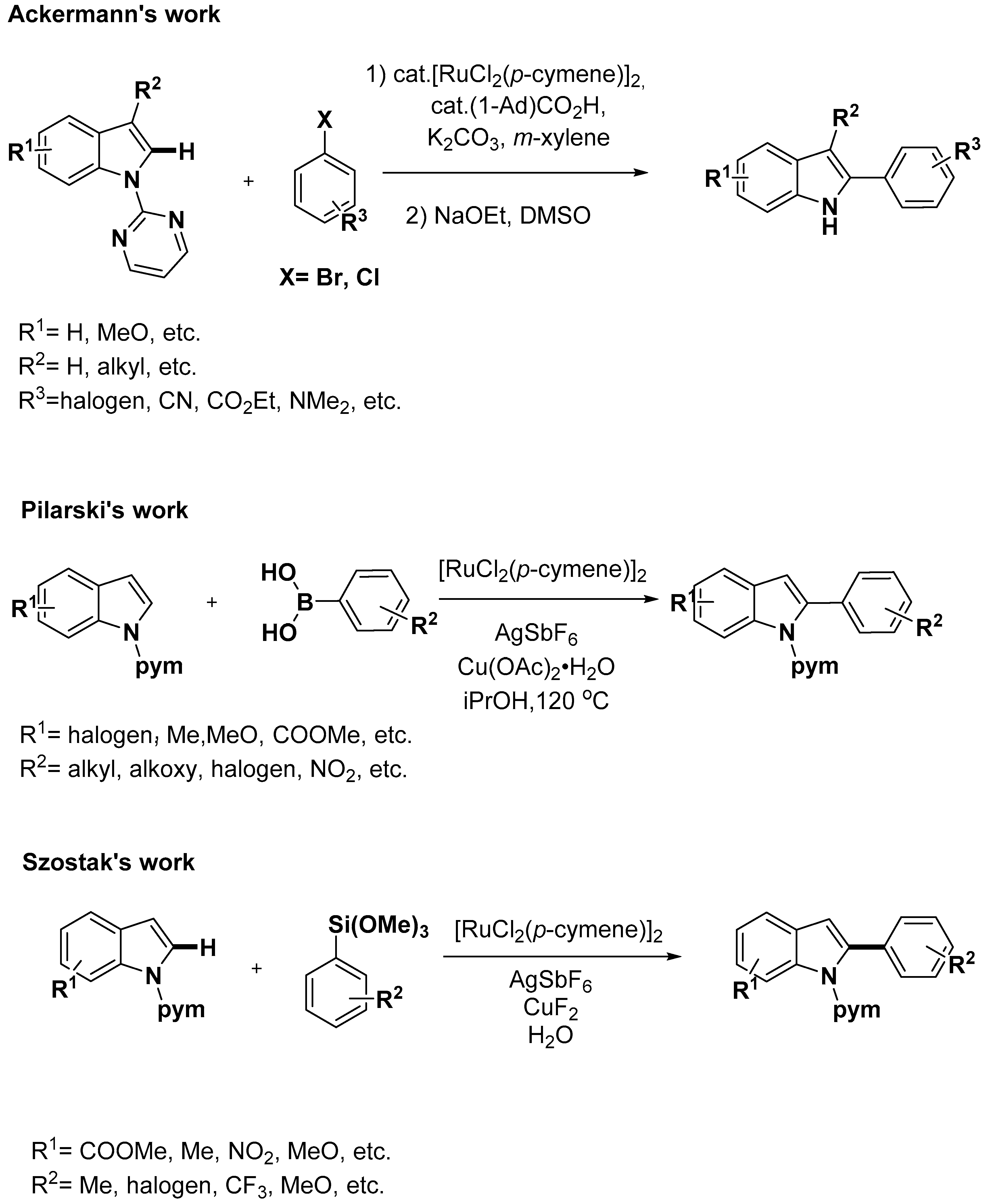
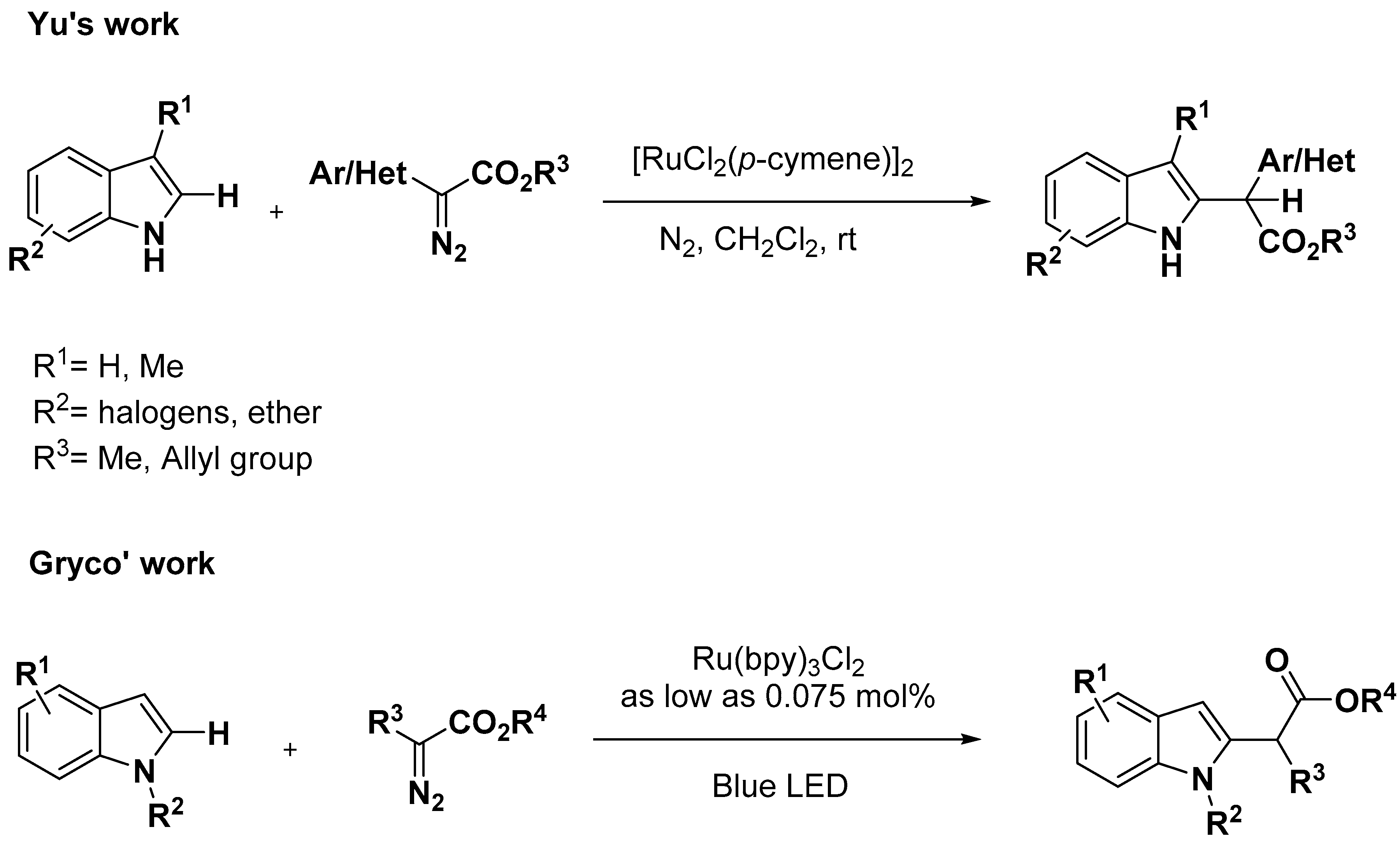
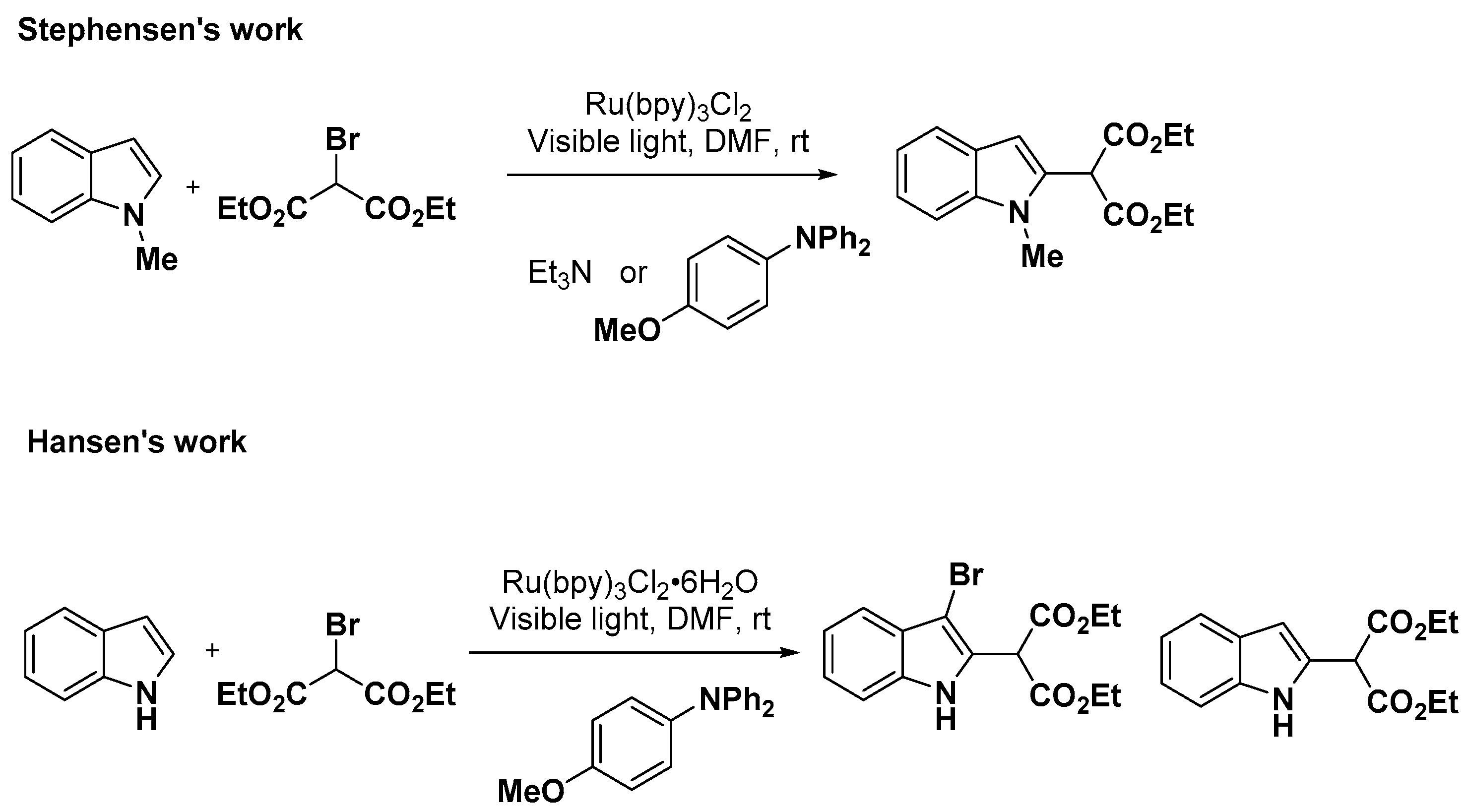
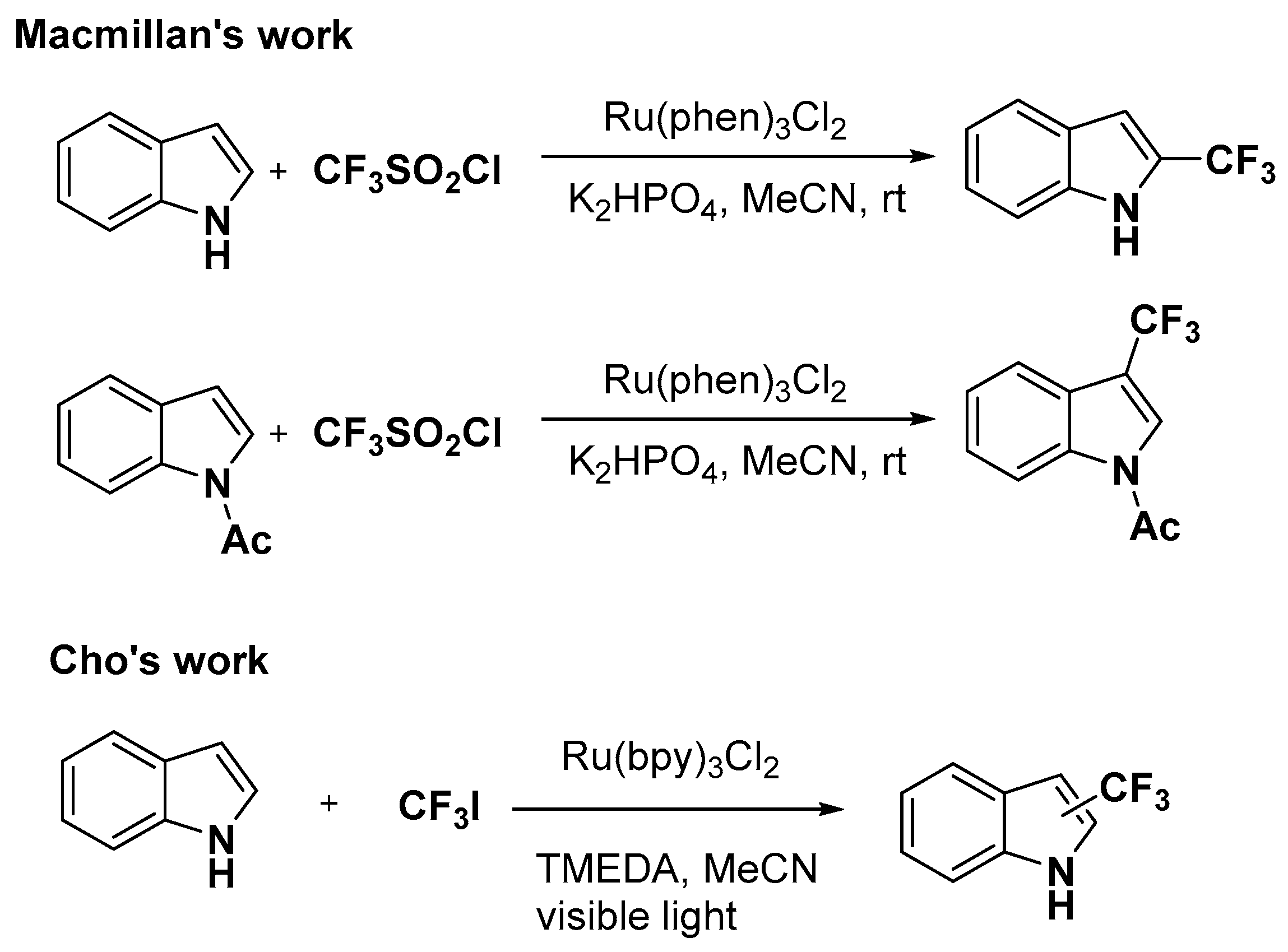
2.1. C–H Activation on C2 and C3
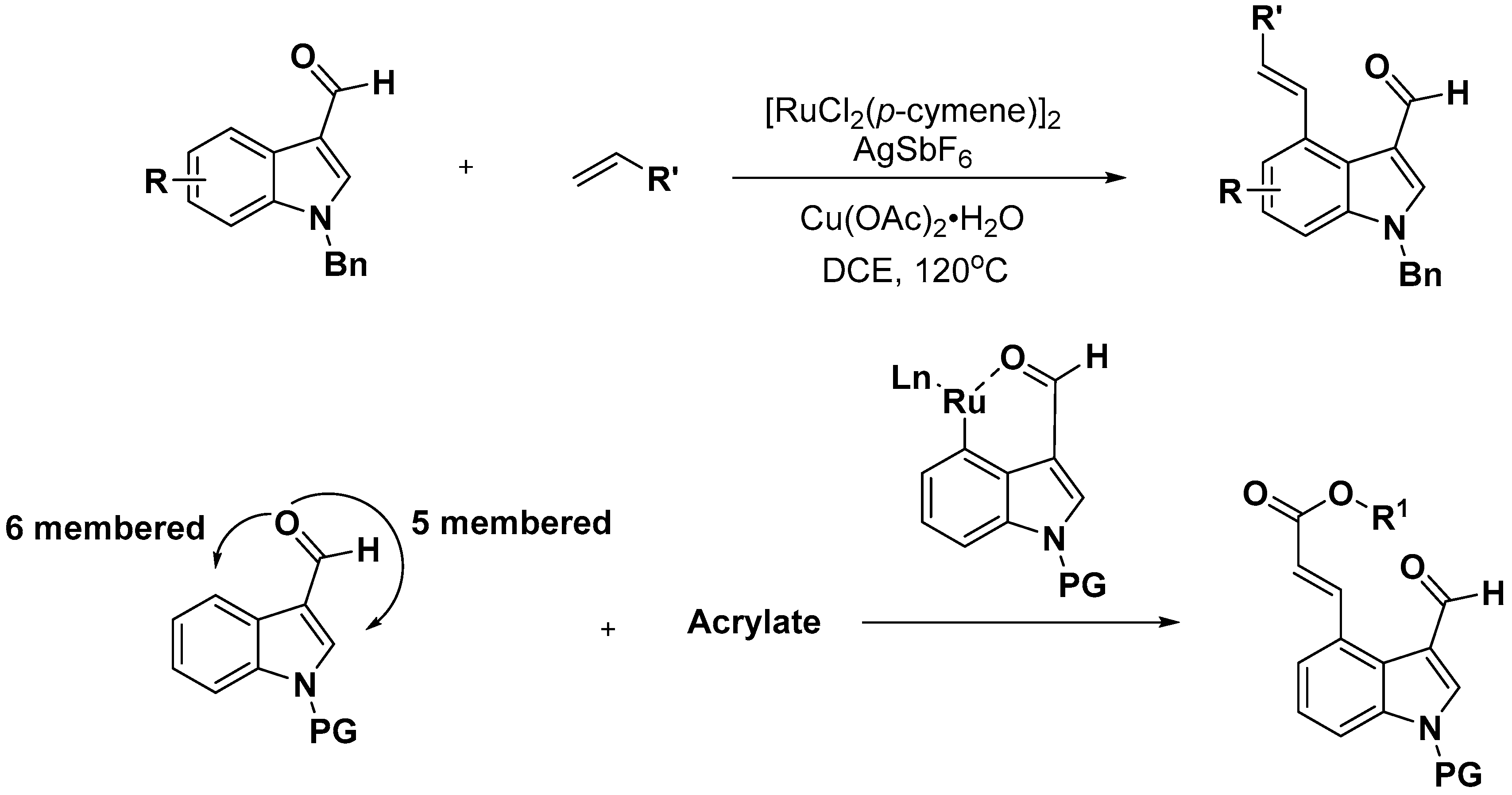
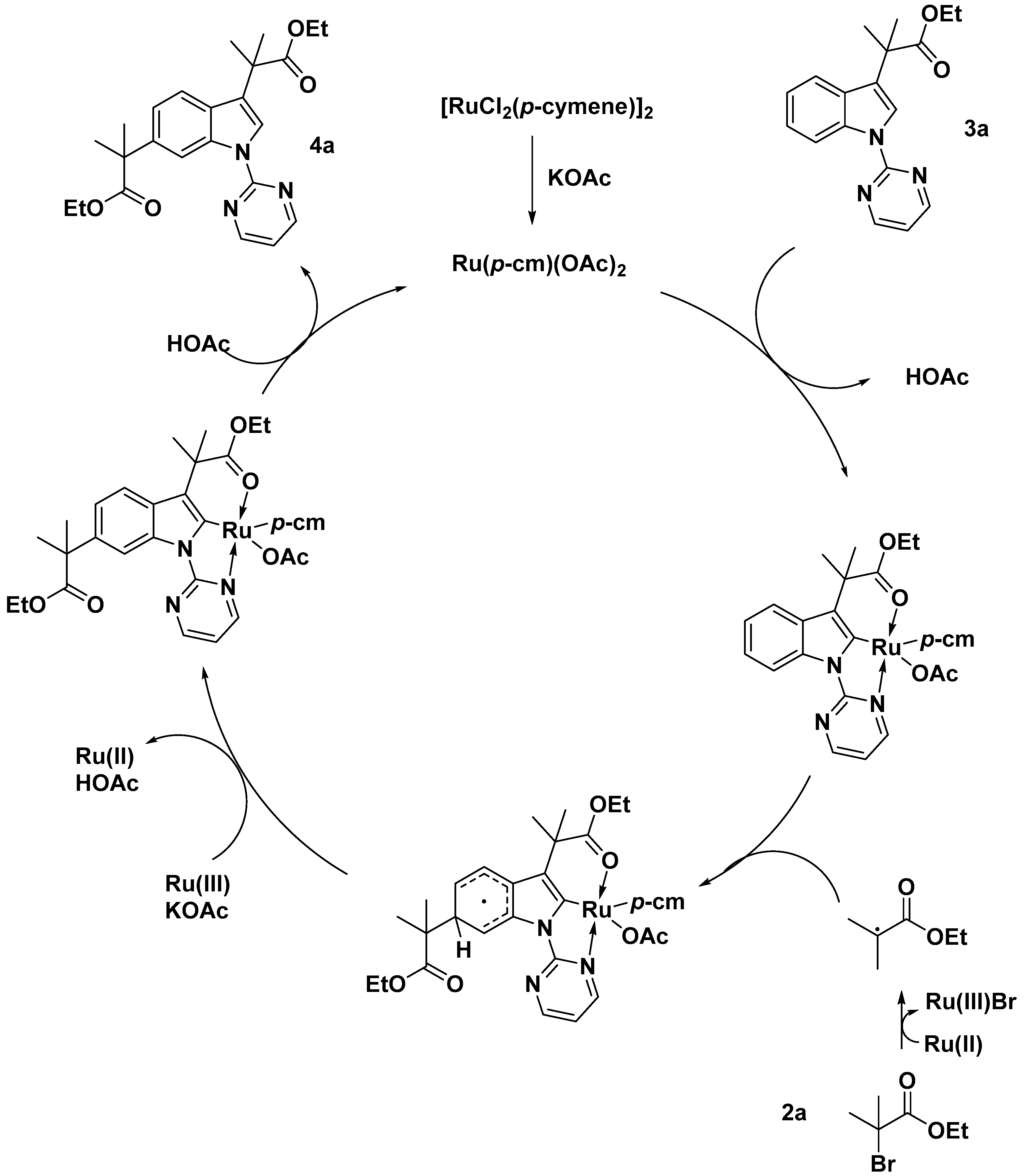
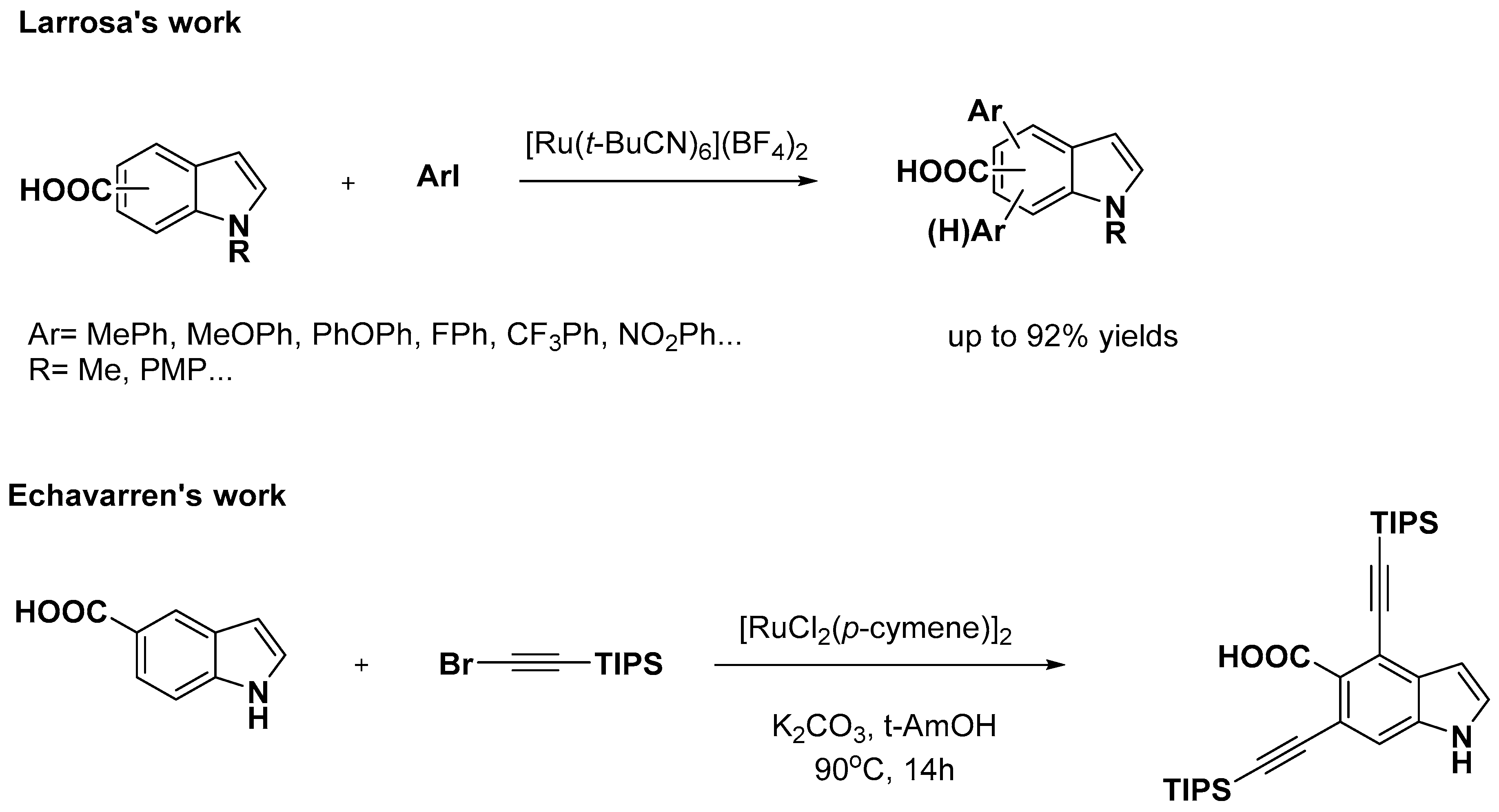
2.2. C–H Activation on C4–C7
3. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Gadelha, M.N.R.; Bronstein, M.D.; Brue, T.; Coculescu, M.; Fleseriu, M.; Guitelman, M.; Pronin, V.; Raverot, G.; Shimon, I.; Lievre, K.K.J.L.D.; et al. Pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA): A randomised, phase 3 trial. Lancet Diabetes Endocrinol. 2014, 2, 875–884. [Google Scholar] [CrossRef]
- Modlin, I.M.; Pavel, M.; Kidd, M.; Gustafsson, B.I. Review article: Somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment. Pharmacol. Ther. 2010, 31, 169–188. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cang, S.; Liu, D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L. Osimertinib: First Global Approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef]
- Das, A.; Durrant, D.; Salloum, F.N.; Xi, L.; Kukreja, R.C. PDE5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol. Therapeut. 2015, 147, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Elexacaftor/Ivacaftor/Tezacaftor: First Approval. Drugs 2019, 79, 2001–2007. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331. [Google Scholar] [CrossRef]
- Tan, D.-X.; Zhou, J.; Liu, C.-Y.; Han, F.-S. Enantioselective Total Synthesis and Absolute Configuration Assignment of (+)-Tronocarpine Enabled by an Asymmetric Michael/Aldol Reaction. Angew. Chem. Int. Ed. 2020, 59, 3834–3839. [Google Scholar] [CrossRef]
- Eicher, T.; Hauptmann, S. The Chemistry of Heterocycles: Structures, Reactions, Synthesis, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef]
- Li, Y.; Qi, Z.; Wang, H.; Yang, X.; Li, X. Ruthenium(II)-Catalyzed C-H Activation of Imidamides and Divergent Couplings with Diazo Compounds: Substrate-Controlled Synthesis of Indoles and 3H-Indoles. Angew. Chem. Int. Ed. 2016, 55, 11877–11881. [Google Scholar] [CrossRef]
- Yang, Q.; Wu, C.; Zhou, J.; He, G.; Liu, H.; Zhou, Y. Highly selective C–H bond activation of N-arylbenzimidamide and divergent couplings with diazophosphonate compounds: A catalyst-controlled selective synthetic strategy for 3-phosphorylindoles and 4-phosphorylisoquinolines. Org. Chem. Front. 2019, 6, 393–398. [Google Scholar] [CrossRef]
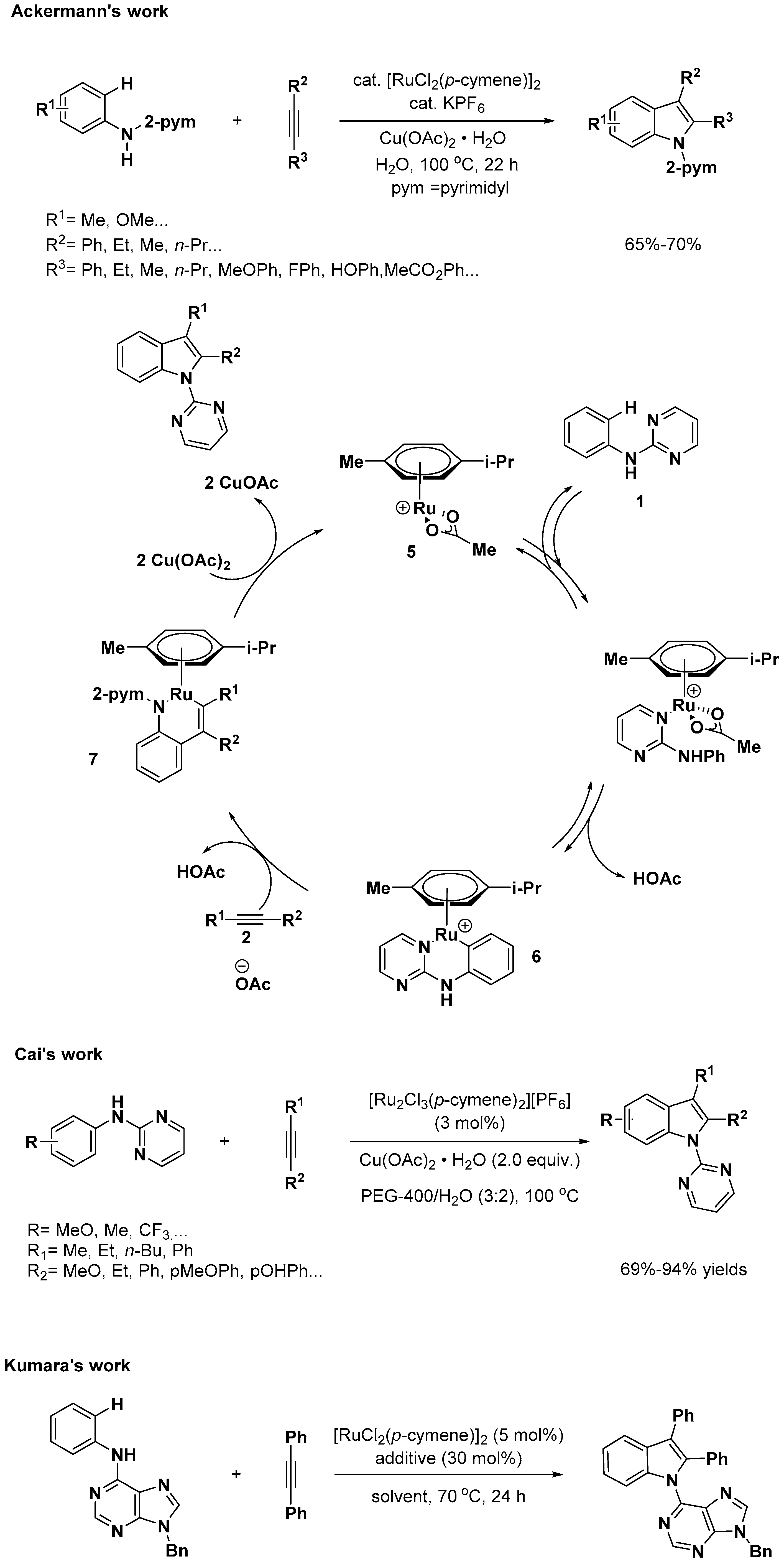
- Ackermann, L.; Lygin, A.V. Cationic Ruthenium(II) Catalysts for Oxidative C–H/N–H Bond Functionalizations of Anilines with Removable Directing Group: Synthesis of Indoles in Water. Org. Lett. 2012, 14, 764–767. [Google Scholar] [CrossRef] [PubMed]
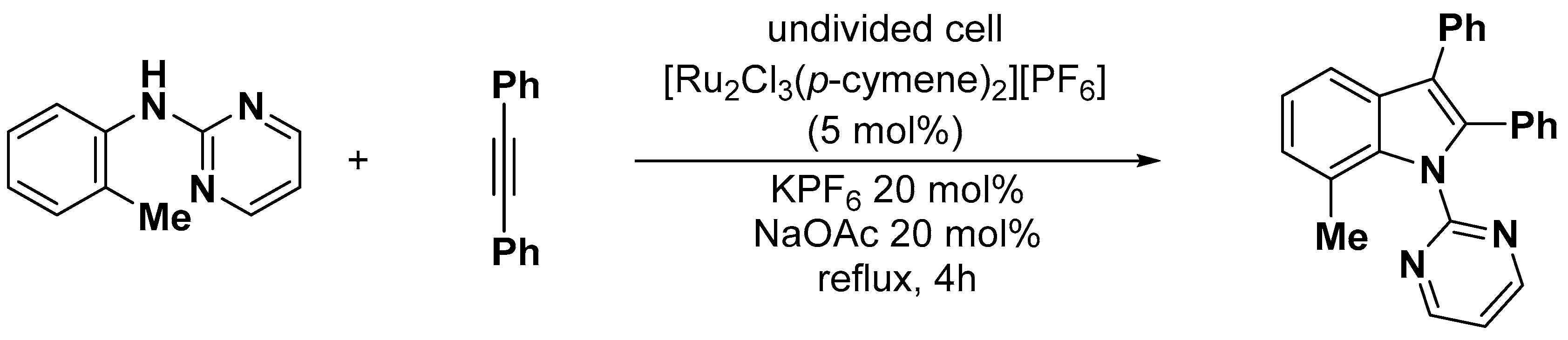
- Liao, Y.; Wei, T.; Yan, T.; Cai, M. Recyclable [Ru2Cl3(p-cymene)2][PF6]/Cu(OAc)2/PEG-400/H2O system for oxidative annulation of alkynes by aniline derivatives: Green synthesis of indoles. Tetrahedron 2017, 73, 1238–1246. [Google Scholar] [CrossRef]
- Allu, S.; Kumara Swamy, K.C. Ruthenium-Catalyzed Oxidative Annulation of 6-Anilinopurines with Alkynes via C-H Activation: Synthesis of Indole-Substituted Purines/Purine Nucleosides. Adv. Syn. Catal. 2015, 357, 2665–2680. [Google Scholar] [CrossRef]
- Borthakur, S.; Sarma, B.; Gogoi, S. Ruthenium(II)-Catalyzed Oxidative Double C-H Activation and Annulation Reaction: Synthesis of Indolo[2,1-a]isoquinolines. Org. Lett. 2019, 21, 7878–7882. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cui, X.; Bai, L.; Wang, Y.; Xie, Y.; Wang, S.; Zhai, R.; Zhao, K.; Kong, D.; Li, Y. Ruthenium(II)-Catalyzed C−H Bond [3+2] Annulation of N-Nitrosoanilines with Alkynes in Water. Asian J. Org. Chem. 2019, 8, 2209–2212. [Google Scholar] [CrossRef]
- Intrieri, D.; Carminati, D.M.; Zardi, P.; Damiano, C.; Manca, G.; Gallo, E.; Mealli, C. Indoles from Alkynes and Aryl Azides: Scope and Theoretical Assessment of Ruthenium Porphyrin-Catalyzed Reactions. Chem. Eur. J. 2019, 25, 16591–16605. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, Y.-J.; Huang, C.; Xu, H.-C. Ruthenium-Catalyzed Electrochemical Dehydrogenative Alkyne Annulation. ACS Catal. 2018, 8, 3820–3824. [Google Scholar] [CrossRef]
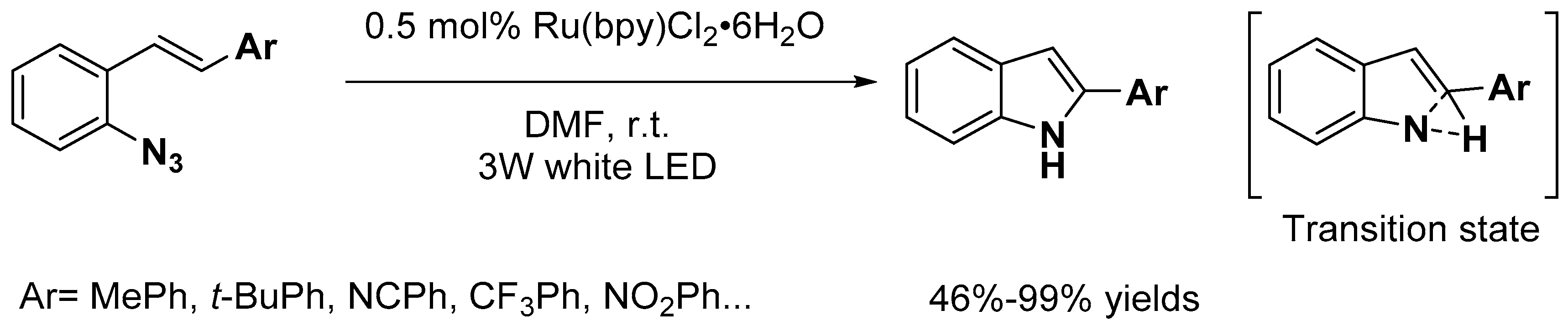
- Xia, X.-D.; Xuan, J.; Wang, Q.; Lu, L.-Q.; Chen, J.-R.; Xiao, W.-J. Synthesis of 2-Substituted Indoles through Visible Light-Induced Photocatalytic Cyclizations of Styryl Azides. Adv. Syn. Catal. 2014, 356, 2807–2812. [Google Scholar] [CrossRef]
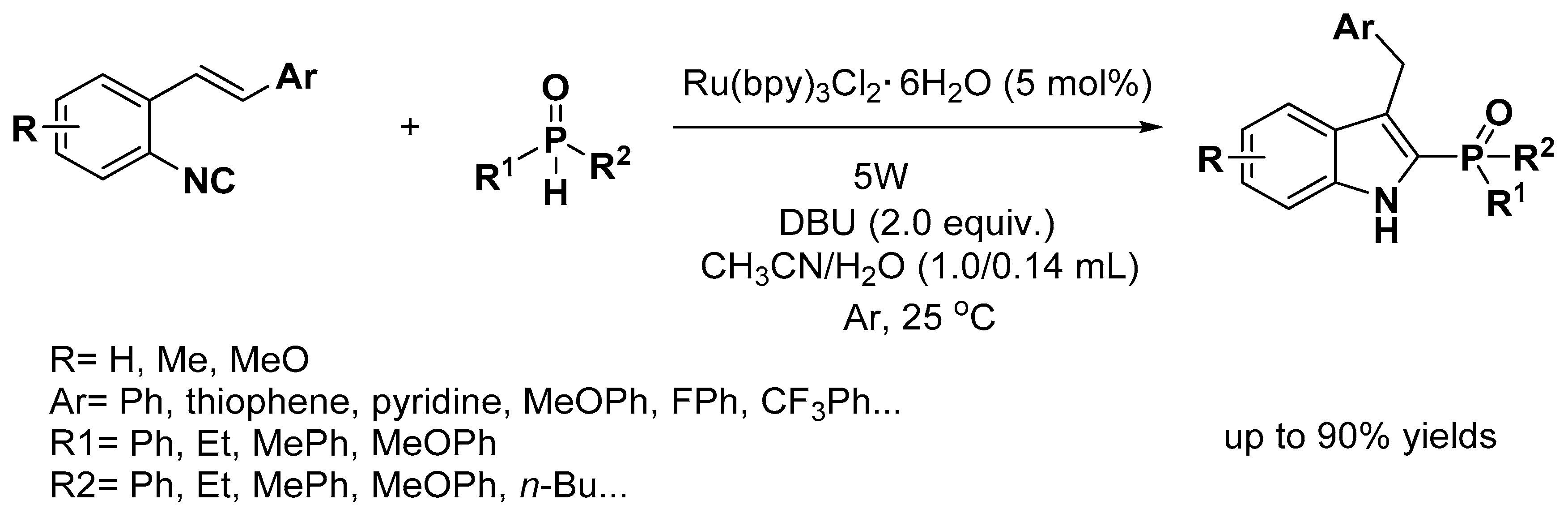
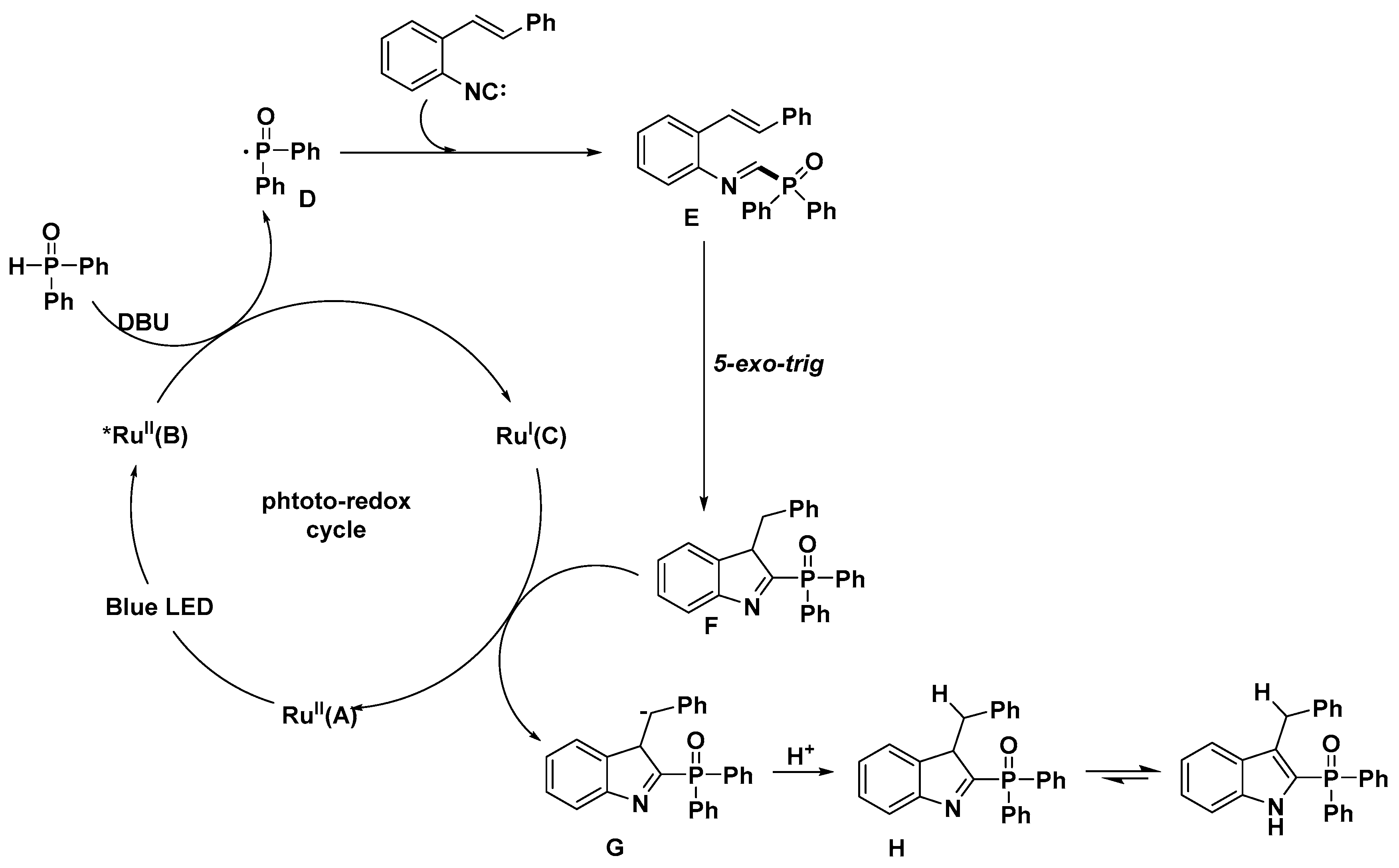
- Wang, C.H.; Li, Y.H.; Yang, S.D. Autoxidation Photoredox Catalysis for the Synthesis of 2-Phosphinoylindoles. Org. Lett. 2018, 20, 2382–2385. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, J.; He, G.; Li, H.; Yang, Q.; Wang, R.; Zhou, Y.; Liu, H. Ruthenium(ii)-catalyzed selective C–H bond activation of imidamides and coupling with sulfoxonium ylides: An efficient approach for the synthesis of highly functional 3-ketoindoles. Org. Chem. Front. 2019, 6, 1183–1188. [Google Scholar] [CrossRef]
- Panferova, L.I.; Smirnov, V.O.; Levin, V.V.; Kokorekin, V.A.; Struchkova, M.I.; Dilman, A.D. Synthesis of 3-Fluoroindoles via Photoredox Catalysis. J. Org. Chem. 2017, 82, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.S.; Kim, J.H.; Kim, T.-J.; Shim, S.C. Ruthenium-catalyzed heteroannulation of anilines with alkanolammonium chlorides leading to indoles. Tetrahedron 2001, 57, 3321–3329. [Google Scholar] [CrossRef]
- Sathishkumar, P.N.; Prabha, P.S.; Bhuvanesh, N.S.P.; Karvembu, R. Tuning acylthiourea ligands in Ru(II) catalysts for altering the reactivity and chemoselectivity of transfer hydrogenation reactions, and synthesis of 3-isopropoxy-1H-indole through a new synthetic approach. J. Organomet. Chem. 2020, 908, 121087. [Google Scholar] [CrossRef]
- Manna, M.K.; Bairy, G.; Jana, R. Sterically Controlled Ru(II)-Catalyzed Divergent Synthesis of 2-Methylindoles and Indolines through a C-H Allylation/Cyclization Cascade. J. Org. Chem. 2018, 83, 8390–8400. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yi, C.S. Catalytic Synthesis of Substituted Indoles and Quinolines from the Dehydrative C-H Coupling of Arylamines with 1,2- and 1,3-Diols. Organometallics 2016, 35, 1973–1977. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.A.; Bergman, R.G.; Ellman, J.A. Rhodium-Catalyzed C−C Bond Formation via Heteroatom-Directed C−H Bond Activation. Chem. Rev. 2010, 110, 624–655. [Google Scholar] [CrossRef]
- Ackermann, L.; Lygin, A.V. Ruthenium-catalyzed direct C-H bond arylations of heteroarenes. Org. Lett. 2011, 13, 3332–3335. [Google Scholar] [CrossRef]
- Sollert, C.; Devaraj, K.; Orthaber, A.; Gates, P.J.; Pilarski, L.T. Ru-Catalysed C H Arylation of Indoles and Pyrroles with Boronic Acids: Scope and Mechanistic Studies. Chem. Eur. J. 2015, 21, 5380–5386. [Google Scholar] [CrossRef]
- Nareddy, P.; Jordan, F.; Szostak, M. Ruthenium(II)-Catalyzed Direct C-H Arylation of Indoles with Arylsilanes in Water. Org. Lett. 2018, 20, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Tonin, M.; Zell, D.; Müller, V. Ruthenium(II)-Catalyzed C−H Methylation with Trifluoroborates. Synthesis 2016, 49, 127–134. [Google Scholar] [CrossRef]
- Chan, W.-W.; Yeung, S.-H.; Zhou, Z.; Chan, A.S.C.; Yu, W.-Y. Ruthenium Catalyzed Directing Group-Free C2-Selective Carbenoid Functionalization of Indoles by α-Aryldiazoesters. Org. Lett. 2010, 12, 604–607. [Google Scholar] [CrossRef]
- Ciszewski, Ł.W.; Durka, J.; Gryko, D. Photocatalytic Alkylation of Pyrroles and Indoles with α-Diazo Esters. Org. Lett. 2019, 21, 7028–7032. [Google Scholar] [CrossRef] [PubMed]
- Furst, L.; Matsuura, B.S.; Narayanam, J.M.R.; Tucker, J.W.; Stephenson, C.R.J. Visible Light-Mediated Intermolecular C−H Functionalization of Electron-Rich Heterocycles with Malonates. Org. Lett. 2010, 12, 3104–3107. [Google Scholar] [CrossRef] [PubMed]
- Erdenebileg, U.; Demissie, T.B.; Hansen, J.H. Visible-Light Photocatalytic Double C–H Functionalization of Indoles: A Synergistic Experimental and Computational Study. Synlett 2017, 28, 907–912. [Google Scholar] [CrossRef]
- De Montellano, P.R.O. Cytochrome P450: Structure, Mechanism, and Biochemistry; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Acena, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II-III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Nagib, D.A.; MacMillan, D.W. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224–228. [Google Scholar] [CrossRef]
- Iqbal, N.; Choi, S.; Ko, E.; Cho, E.J. Trifluoromethylation of heterocycles via visible light photoredox catalysis. Tetrahedron Lett. 2012, 53, 2005–2008. [Google Scholar] [CrossRef]
- Straathof, N.J.; Gemoets, H.P.; Wang, X.; Schouten, J.C.; Hessel, V.; Noel, T. Rapid trifluoromethylation and perfluoroalkylation of five-membered heterocycles by photoredox catalysis in continuous flow. ChemSusChem 2014, 7, 1612–1617. [Google Scholar] [CrossRef]
- Nakao, Y.; Hiyama, T. Silicon-based cross-coupling reaction: An environmentally benign version. Chem. Soc. Rev. 2011, 40, 4893–4901. [Google Scholar] [CrossRef]
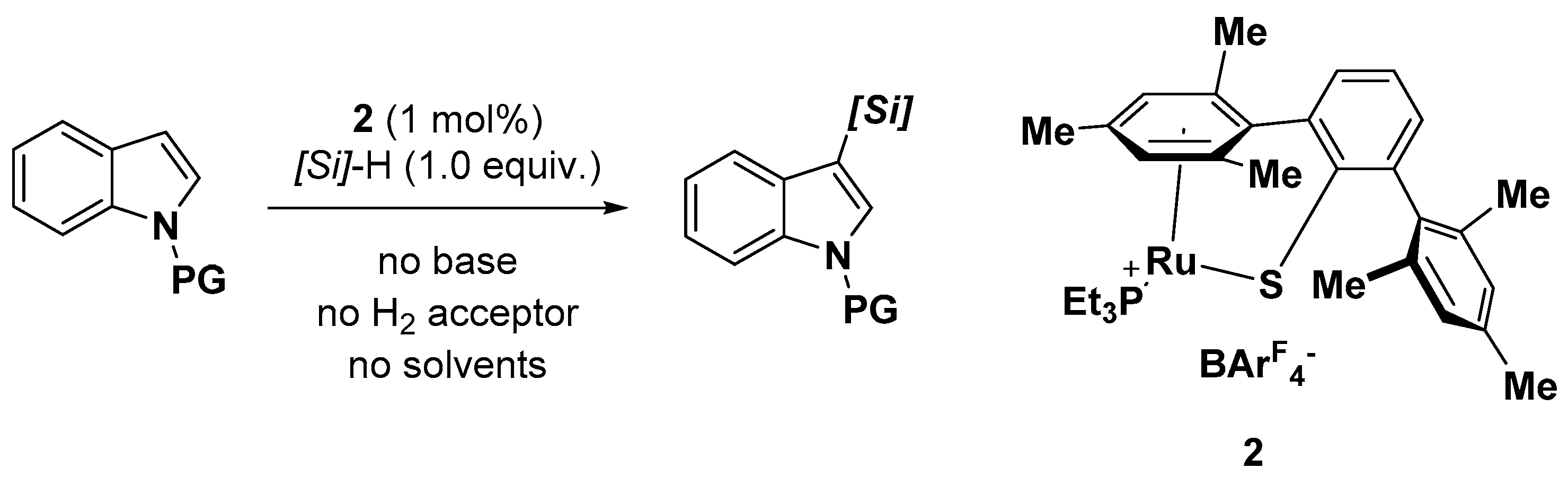
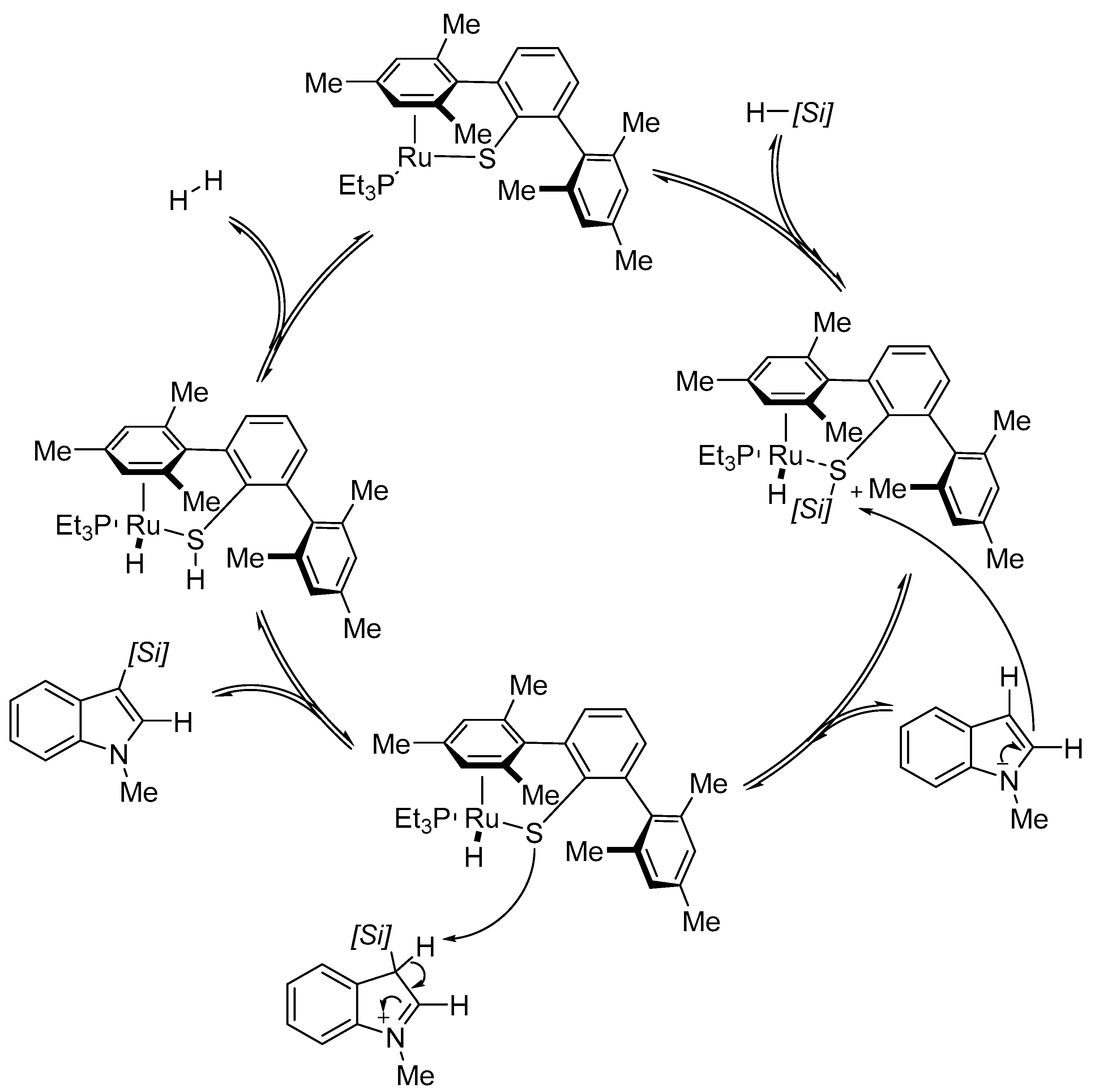
- Klare, H.F.; Oestreich, M.; Ito, J.; Nishiyama, H.; Ohki, Y.; Tatsumi, K. Cooperative catalytic activation of Si-H bonds by a polar Ru-S bond: Regioselective low-temperature C-H silylation of indoles under neutral conditions by a Friedel-Crafts mechanism. J. Am. Chem. Soc. 2011, 133, 3312–3315. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, K.; Sollert, C.; Juds, C.; Gates, P.J.; Pilarski, L.T. Ru-catalysed C-H silylation of unprotected gramines, tryptamines and their congeners. Chem. Commun. 2016, 52, 5868–5871. [Google Scholar] [CrossRef]
- Cadierno, V.; Francos, J.; Gimeno, J. Ruthenium/TFA-catalyzed regioselective C-3-alkylation of indoles with terminal alkynes in water: Efficient and unprecedented access to 3-(1-methylalkyl)-1H-indoles. Chem. Commun. 2010, 46, 4175–4177. [Google Scholar] [CrossRef]
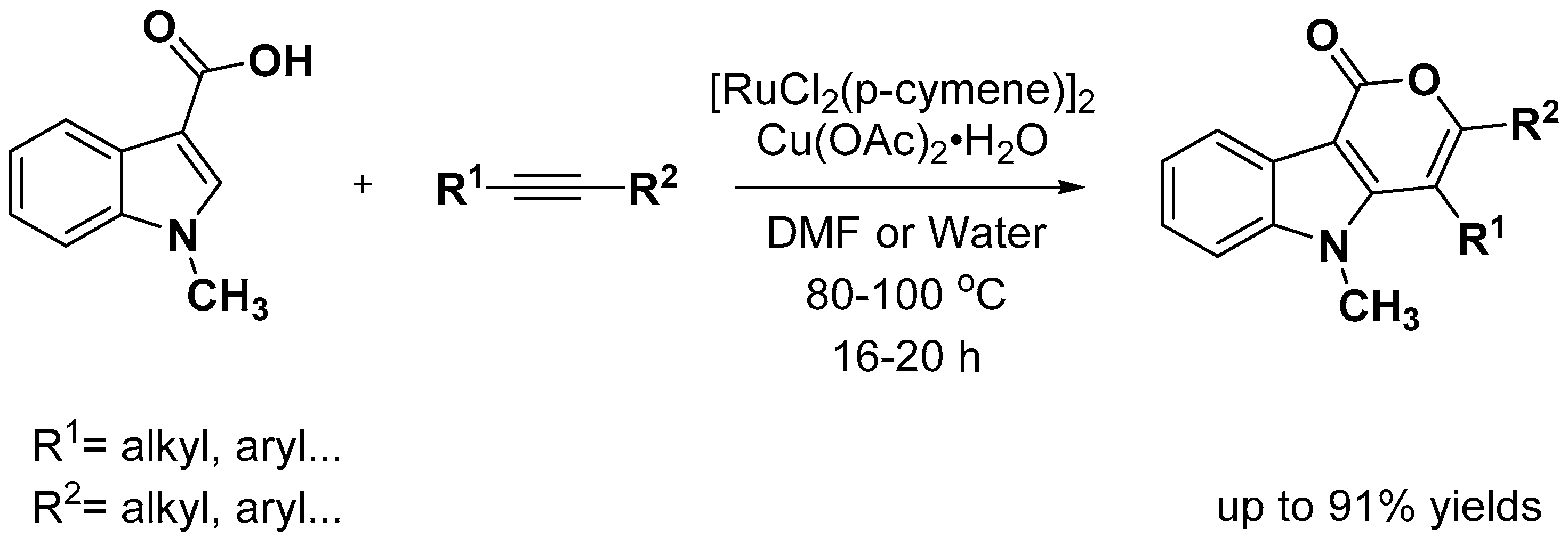
- Singh, K.S.; Sawant, S.G.; Dixneuf, P.H. Ruthenium(II)-Catalyzed Synthesis of Pyrrole- and Indole-Fused Isocoumarins by C−H Bond Activation in DMF and Water. ChemCatChem 2016, 8, 1046–1050. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, X.; Li, C.; Wang, J.; Li, J.; Liu, H. Ruthenium(II)-Catalyzed Redox-Neutral [3+2] Annulation of Indoles with Internal Alkynes via C-H Bond Activation: Accessing a Pyrroloindolone Scaffold. J. Org. Chem. 2017, 82, 5263–5273. [Google Scholar] [CrossRef] [PubMed]
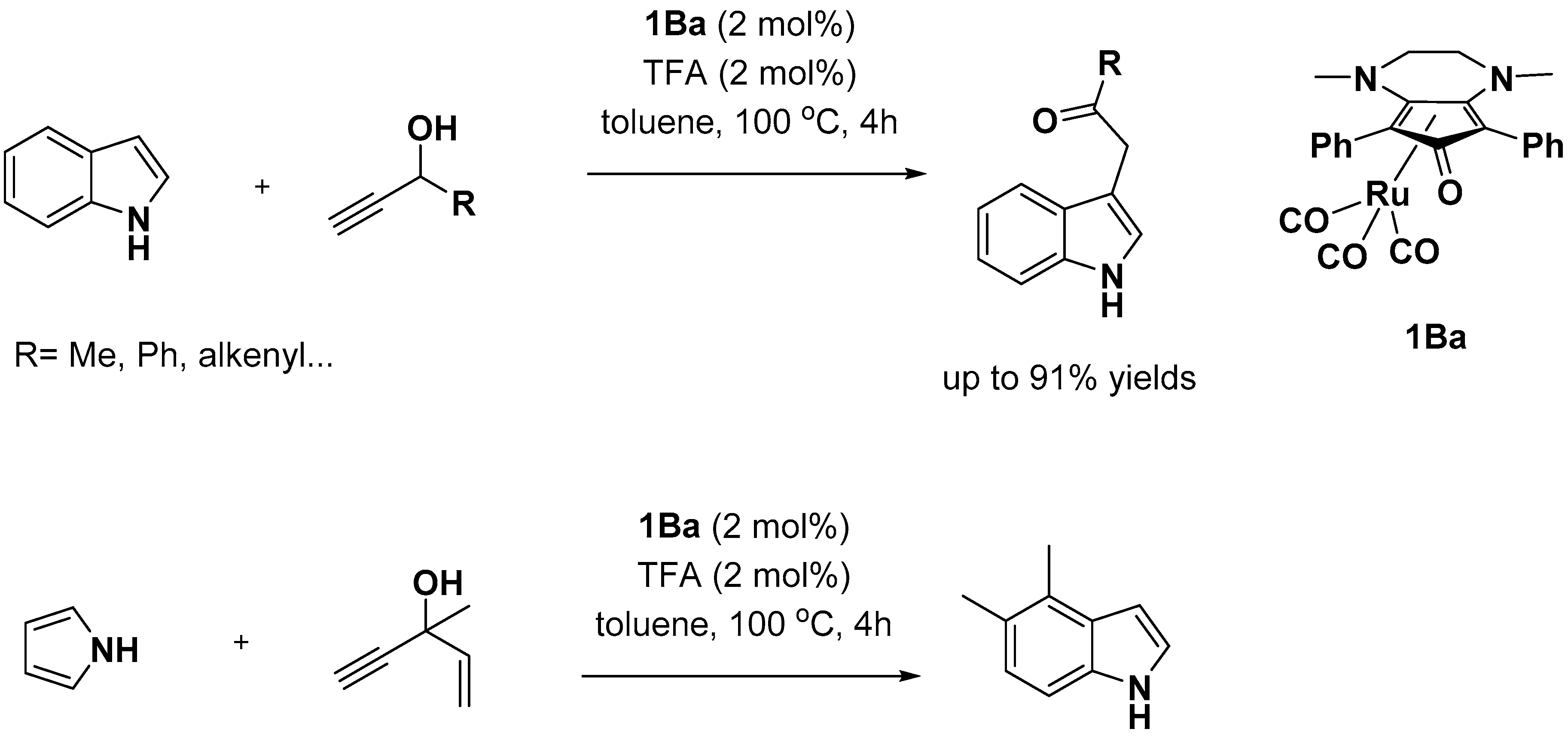
- Thies, N.; Hrib, C.G.; Haak, E. Ruthenium-Catalyzed Functionalization of Pyrroles and Indoles with Propargyl Alcohols. Chem. Eur. J. 2012, 18, 6302–6308. [Google Scholar] [CrossRef] [PubMed]
- Haak, E. Modern Annulation Strategies for the Synthesis of Cyclo[b]fused Indoles. Synlett 2018, 30, 245–251. [Google Scholar] [CrossRef]
- Li, B.; Ma, J.; Xie, W.; Song, H.; Xu, S.; Wang, B. Ruthenium-catalyzed regioselective C2 alkenylation of indoles and pyrroles via C-H bond functionalization. J. Org. Chem. 2013, 78, 9345–9353. [Google Scholar] [CrossRef]
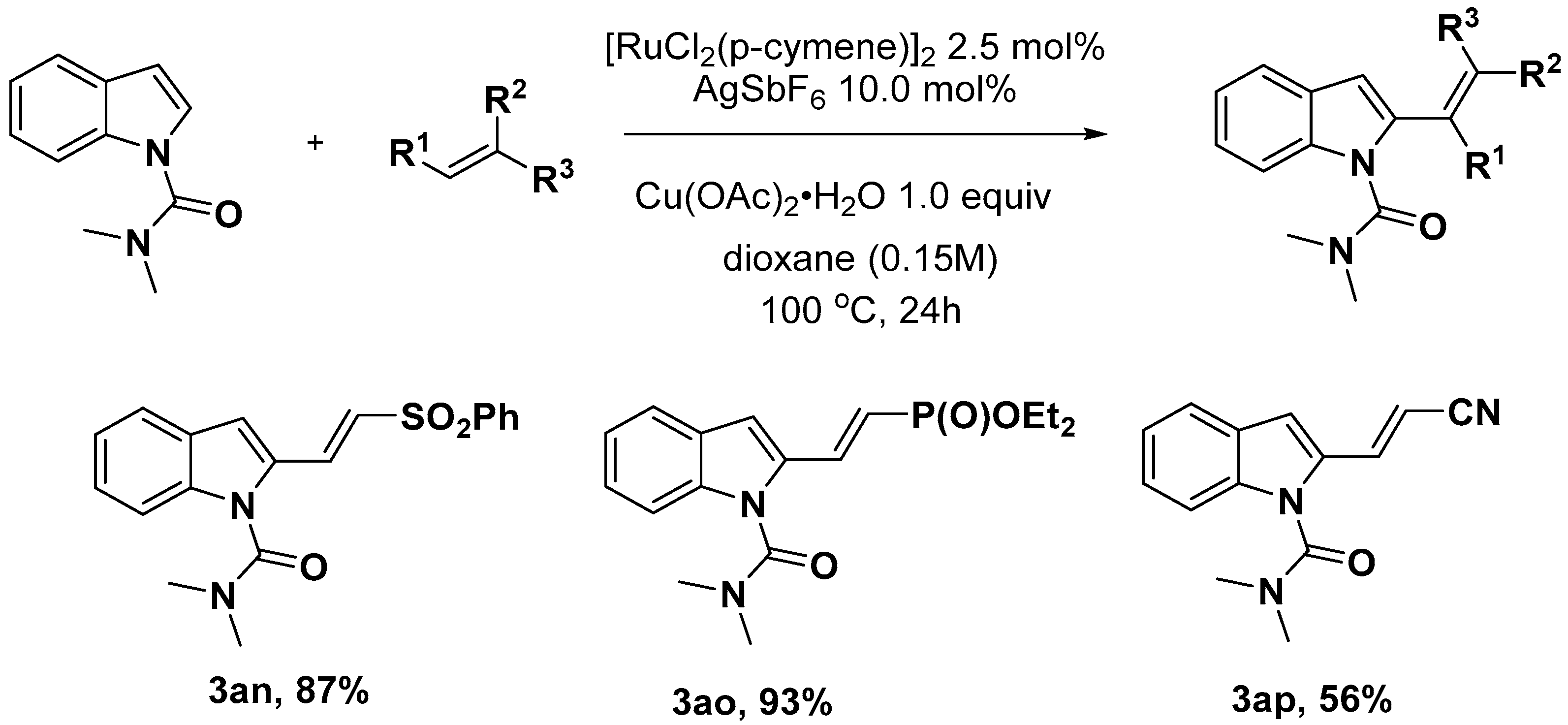
- Zhang, L.Q.; Yang, S.; Huang, X.; You, J.; Song, F. Aerobic Ru-catalyzed direct C2-olefination of N-heteroarenes with alkenes directed by a removable N-dimethylcarbamoyl group. Chem. Commun. 2013, 49, 8830–8832. [Google Scholar] [CrossRef]
- Lanke, V.; Prabhu, K.R. Highly Regioselective C2-Alkenylation of Indoles Using the N-Benzoyl Directing Group: An Efficient Ru-Catalyzed Coupling Reaction. Org. Lett. 2013, 15, 2818–2821. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, H.J.; Li, Q.Y.; Wu, Y.C. Direct oxidative coupling of N-acyl pyrroles with alkenes by ruthenium(ii)-catalyzed regioselective C2-alkenylation. Org. Biomol. Chem. 2020, 18, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.Y.; Xie, L.J.; Cheng, H.P.; Liu, A.D.; Li, X.D.; Wang, D.; Cheng, L.; Liu, L. Ruthenium-Catalyzed Decarboxylative C-H Alkenylation in Aqueous Media: Synthesis of Tetrahydropyridoindoles. J. Org. Chem. 2018, 83, 7514–7522. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Lin, H.; Zhang, X.M.; Dong, L. Ruthenium-catalyzed direct C3 alkylation of indoles with alpha, beta-unsaturated ketones. Org. Biomol. Chem. 2015, 13, 1254–1263. [Google Scholar] [CrossRef]
- Xia, Y.-Q.; Li, C.; Liu, M.; Dong, L. Ruthenium-Catalyzed Selective C−C Coupling of Allylic Alcohols with Free Indoles: Influence of the Metal Catalyst. Chem. Eur. J. 2018, 24, 5474–5478. [Google Scholar] [CrossRef]
- Sundararaju, B.; Achard, M.; Demerseman, B.; Toupet, L.; Sharma, G.V.; Bruneau, C. Ruthenium(IV) complexes featuring P, O-chelating ligands: Regioselective substitution directly from allylic alcohols. Angew. Chem. Int. Ed. 2010, 49, 2782–2785. [Google Scholar] [CrossRef]
- Zaitsev, A.B.; Gruber, S.; Pregosin, P.S. Fast, efficient Ru(IV)-catalysed regioselective allylation of indoles using allyl alcohol (without additives) under mild conditions. Chem. Commun. 2007, 4692–4693. [Google Scholar] [CrossRef]
- Imm, S.; Bähn, S.; Tillack, A.; Mevius, K.; Neubert, L.; Beller, M. Selective Ruthenium-Catalyzed Alkylation of Indoles by Using Amines. Chem. Eur. J. 2010, 16, 2705–2709. [Google Scholar] [CrossRef]
- Wang, M.-Z.; Zhou, C.-Y.; Wong, M.-K.; Che, C.-M. Ruthenium-Catalyzed Alkylation of Indoles with Tertiary Amines by Oxidation of a sp3 C-H Bond and Lewis Acid Catalysis. Chem. Eur. J. 2010, 16, 5723–5735. [Google Scholar] [CrossRef]
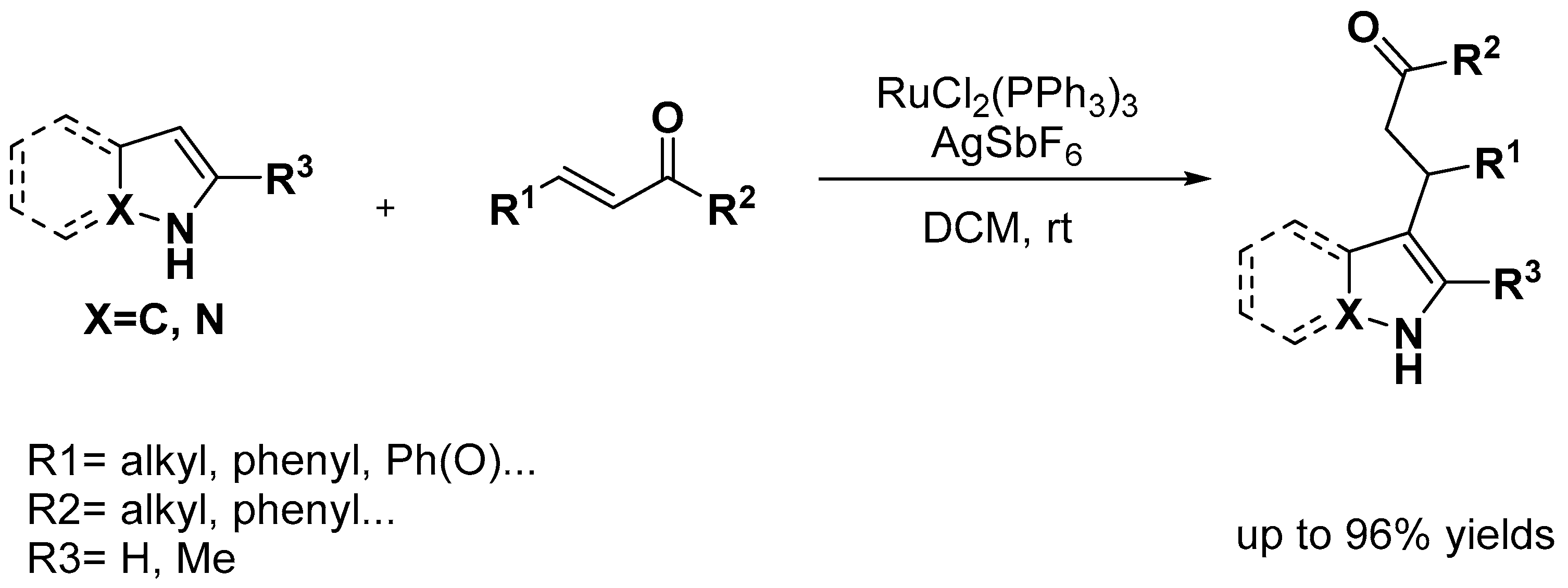
- Lanke, V.; Bettadapur, K.R.; Prabhu, K.R. Site-Selective Addition of Maleimide to Indole at the C-2 Position: Ru(II)-Catalyzed C-H Activation. Org. Lett. 2015, 17, 4662–4665. [Google Scholar] [CrossRef]
- Wang, Z.; Yin, Z.; Wu, X.F. 3-Acylindoles Synthesis: Ruthenium-Catalyzed Carbonylative Coupling of Indoles and Aryl Iodides. Org. Lett. 2017, 19, 4680–4683. [Google Scholar] [CrossRef]
- Lanke, V.; Ramaiah Prabhu, K. Regioselective Synthesis of 4-Substituted Indoles via C–H Activation: A Ruthenium Catalyzed Novel Directing Group Strategy. Org. Lett. 2013, 15, 6262–6265. [Google Scholar] [CrossRef]
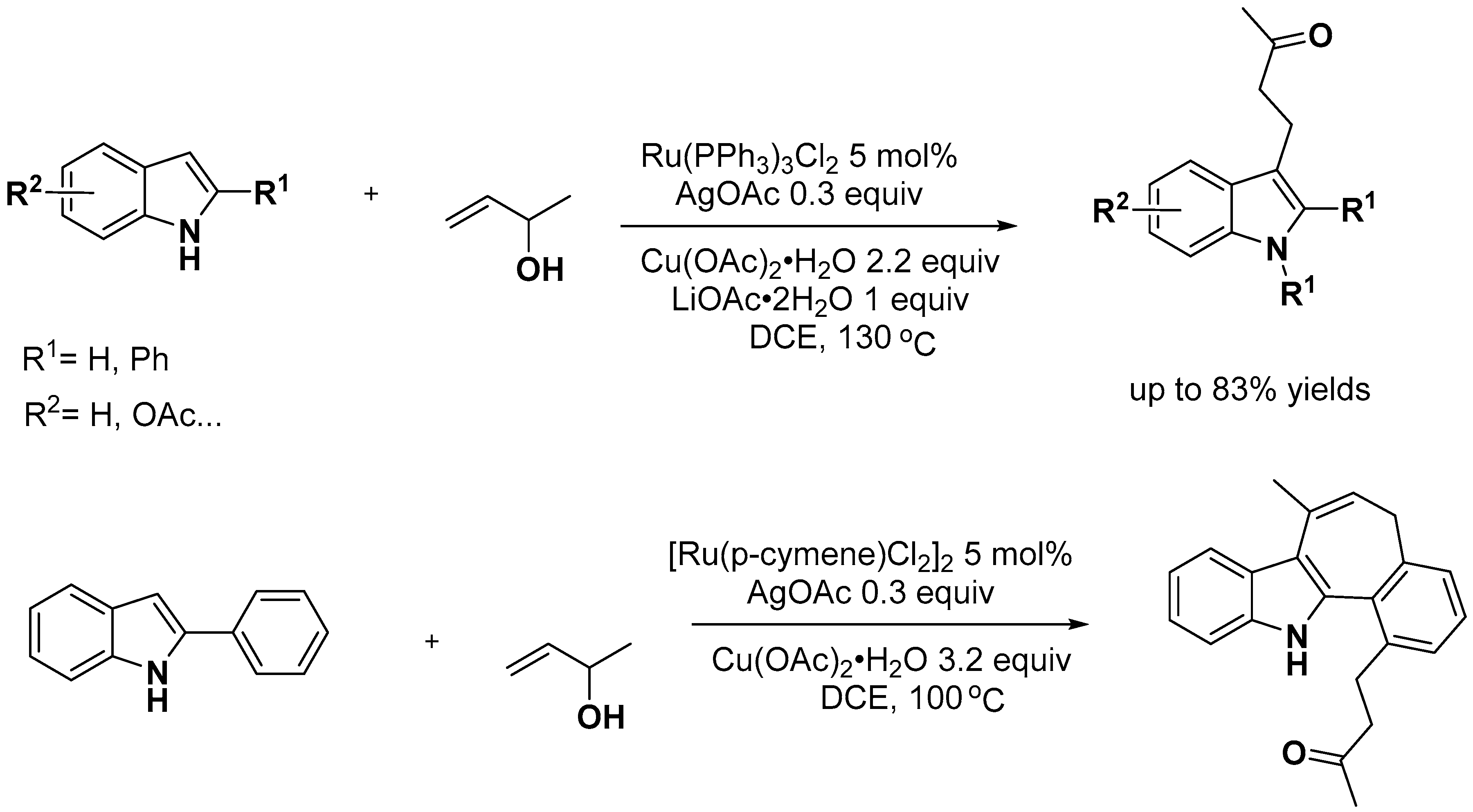
- Leitch, J.A.; McMullin, C.L.; Mahon, M.F.; Bhonoah, Y.; Frost, C.G. Remote C6-Selective Ruthenium-Catalyzed C–H Alkylation of Indole Derivatives via σ-Activation. ACS Catal. 2017, 7, 2616–2623. [Google Scholar] [CrossRef]
- Simonetti, M.; Cannas, D.M.; Panigrahi, A.; Kujawa, S.; Kryjewski, M.; Xie, P.; Larrosa, I. Ruthenium-Catalyzed C−H Arylation of Benzoic Acids and Indole Carboxylic Acids with Aryl Halides. Chem. Eur. J. 2017, 23, 549–553. [Google Scholar] [CrossRef]
- Tan, E.; Konovalov, A.I.; Fernandez, G.A.; Dorel, R.; Echavarren, A.M. Ruthenium-Catalyzed Peri- and Ortho-Alkynylation with Bromoalkynes via Insertion and Elimination. Org. Lett. 2017, 19, 5561–5564. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Nobushige, K.; Satoh, T.; Miura, M. Ruthenium-Catalyzed Regioselective C-H Bond Acetoxylation on Carbazole and Indole Frameworks. Org. Lett. 2016, 18, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
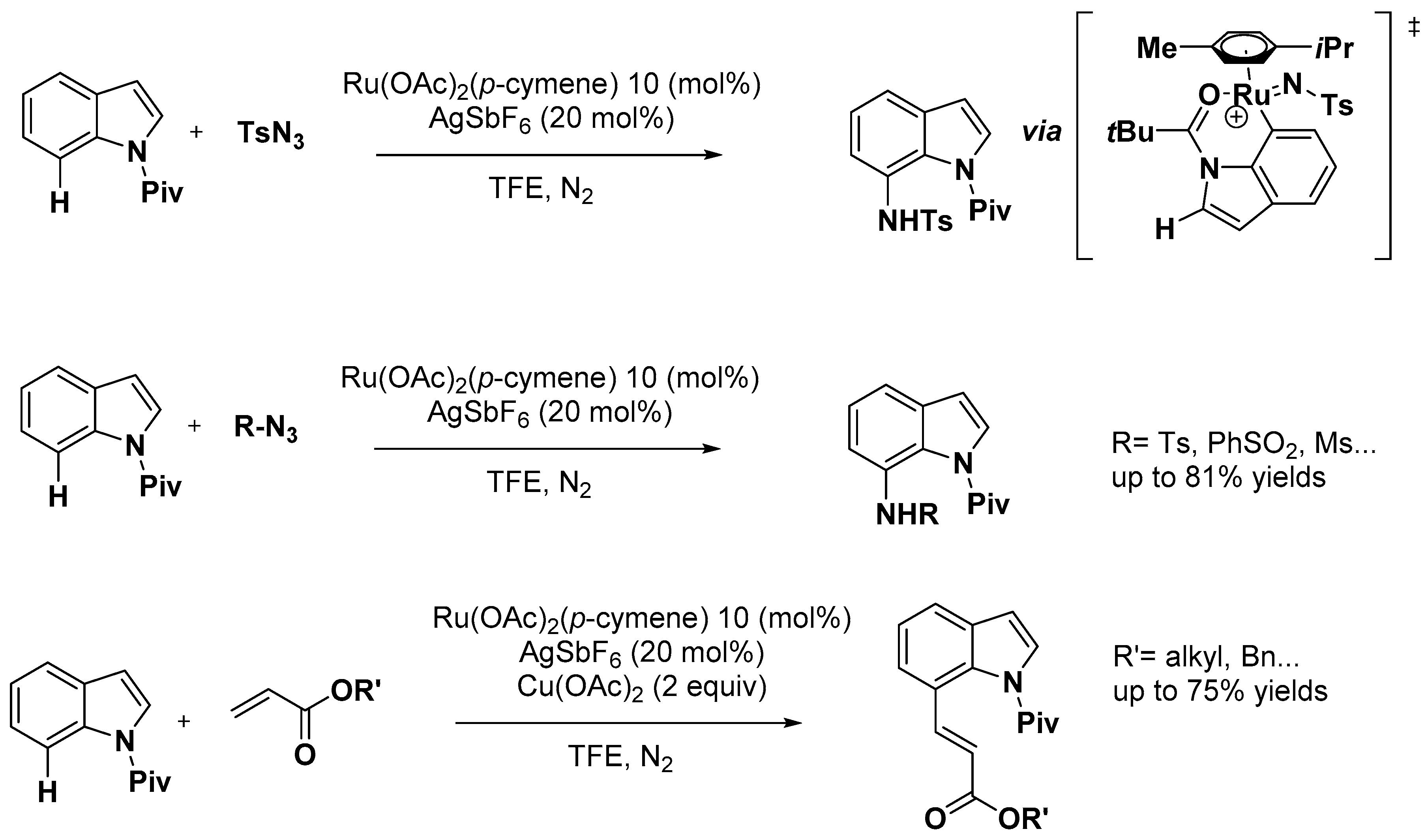
- Choi, I.; Messinis, A.M.; Ackermann, L. C7-Indole Amidations and Alkenylations by Ruthenium(II) Catalysis. Angew. Chem. Int. Ed. 2020, 59, 12534–12540. [Google Scholar] [CrossRef]










































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Zhao, S.; Zhou, Y.; Li, C.; Liu, H. Ruthenium-Catalyzed C–H Activations for the Synthesis of Indole Derivatives. Catalysts 2020, 10, 1253. https://doi.org/10.3390/catal10111253
Zhu H, Zhao S, Zhou Y, Li C, Liu H. Ruthenium-Catalyzed C–H Activations for the Synthesis of Indole Derivatives. Catalysts. 2020; 10(11):1253. https://doi.org/10.3390/catal10111253
Chicago/Turabian StyleZhu, Haoran, Sen Zhao, Yu Zhou, Chunpu Li, and Hong Liu. 2020. "Ruthenium-Catalyzed C–H Activations for the Synthesis of Indole Derivatives" Catalysts 10, no. 11: 1253. https://doi.org/10.3390/catal10111253
APA StyleZhu, H., Zhao, S., Zhou, Y., Li, C., & Liu, H. (2020). Ruthenium-Catalyzed C–H Activations for the Synthesis of Indole Derivatives. Catalysts, 10(11), 1253. https://doi.org/10.3390/catal10111253