Enhancement of CO2 Reforming of CH4 Reaction Using Ni,Pd,Pt/Mg1−xCex4+O and Ni/Mg1−xCex4+O Catalysts


Abstract
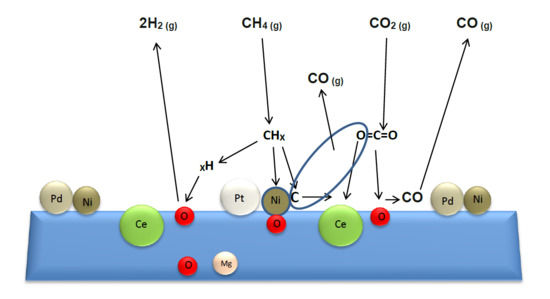
1. Introduction
2. Results and Discussion
2.1. Characterization of Catalysts
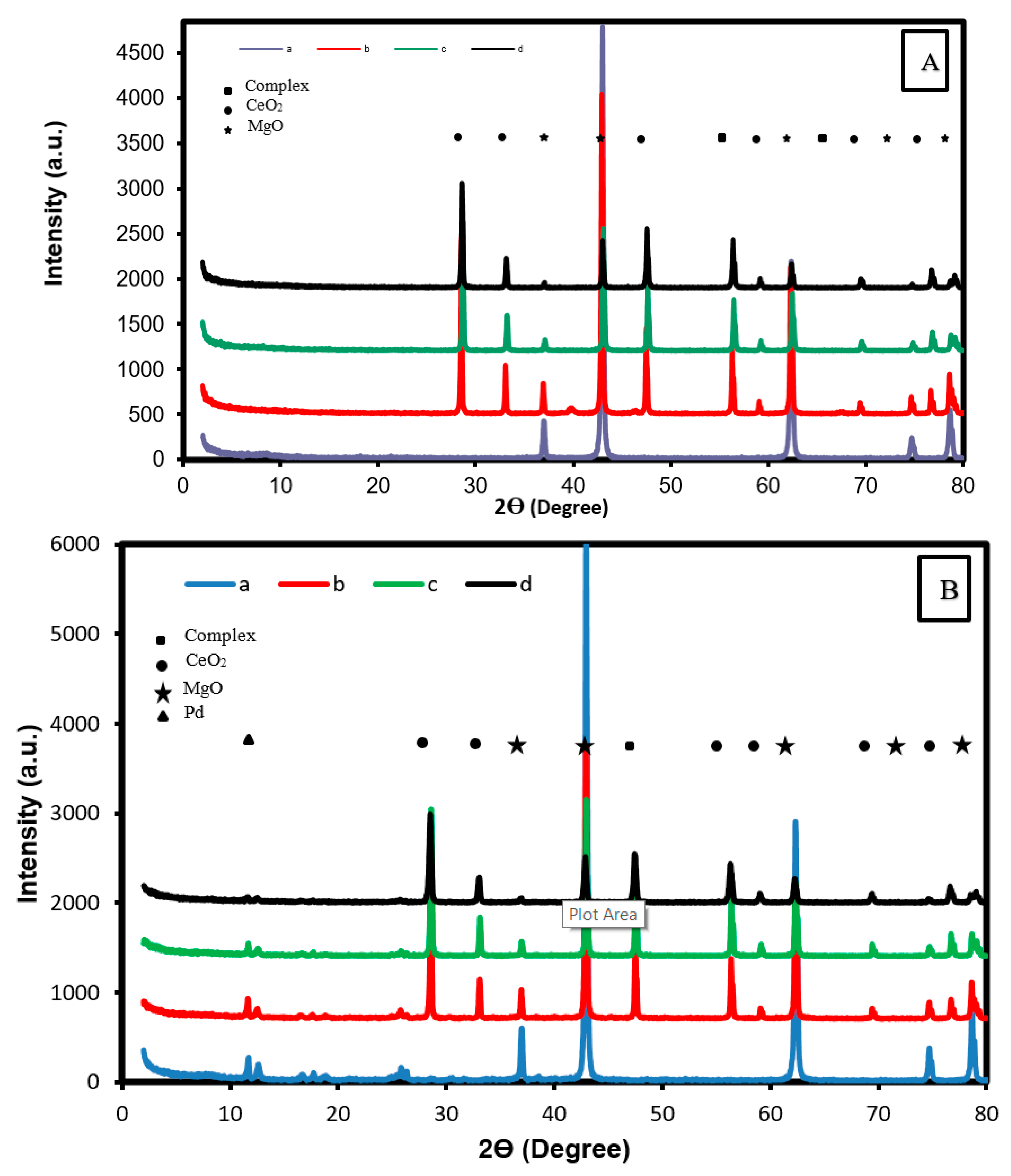
2.1.1. Patterns of XRD
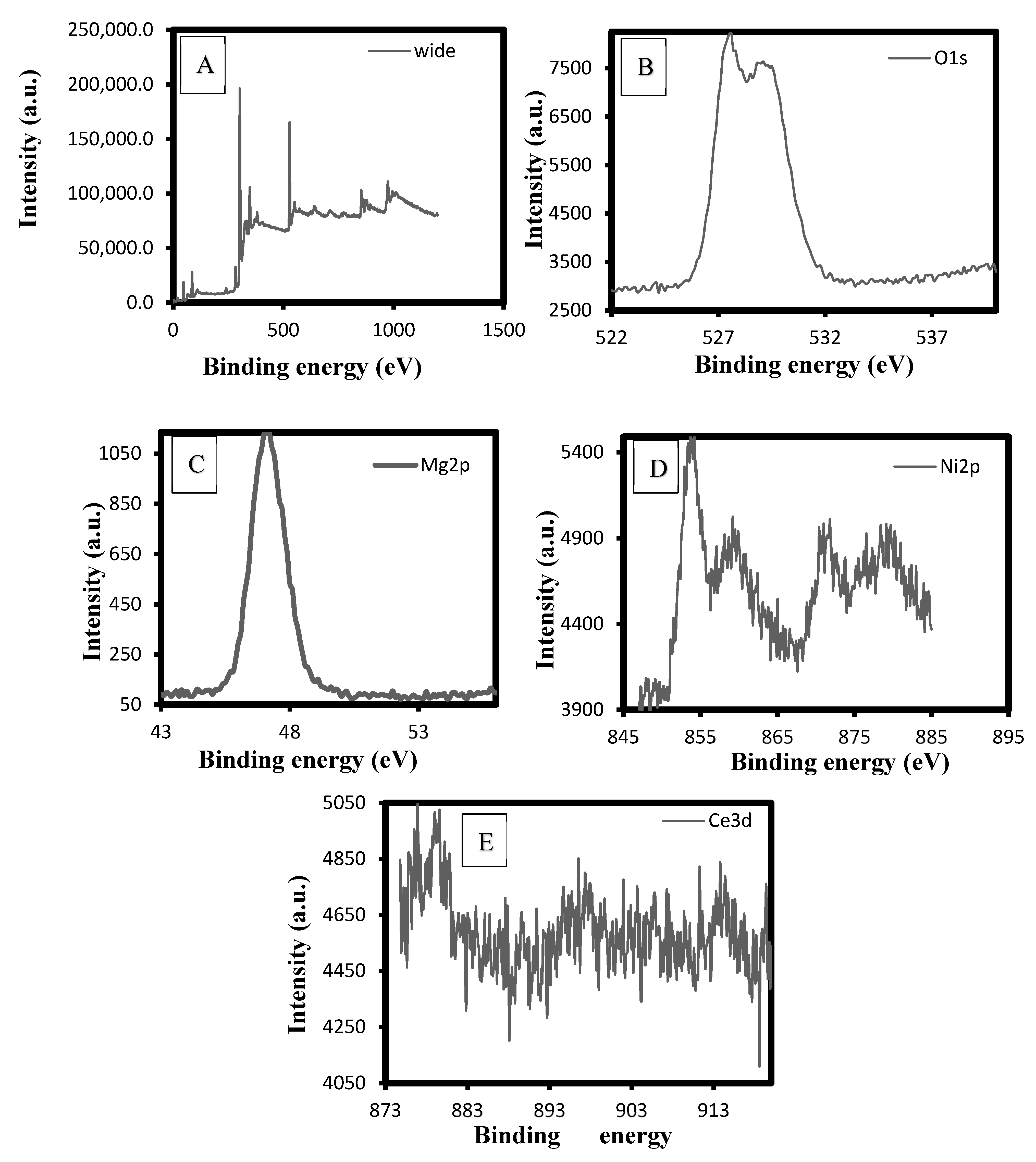
2.1.2. XPS Analysis
2.1.3. TEM and FE-SEM Characterization
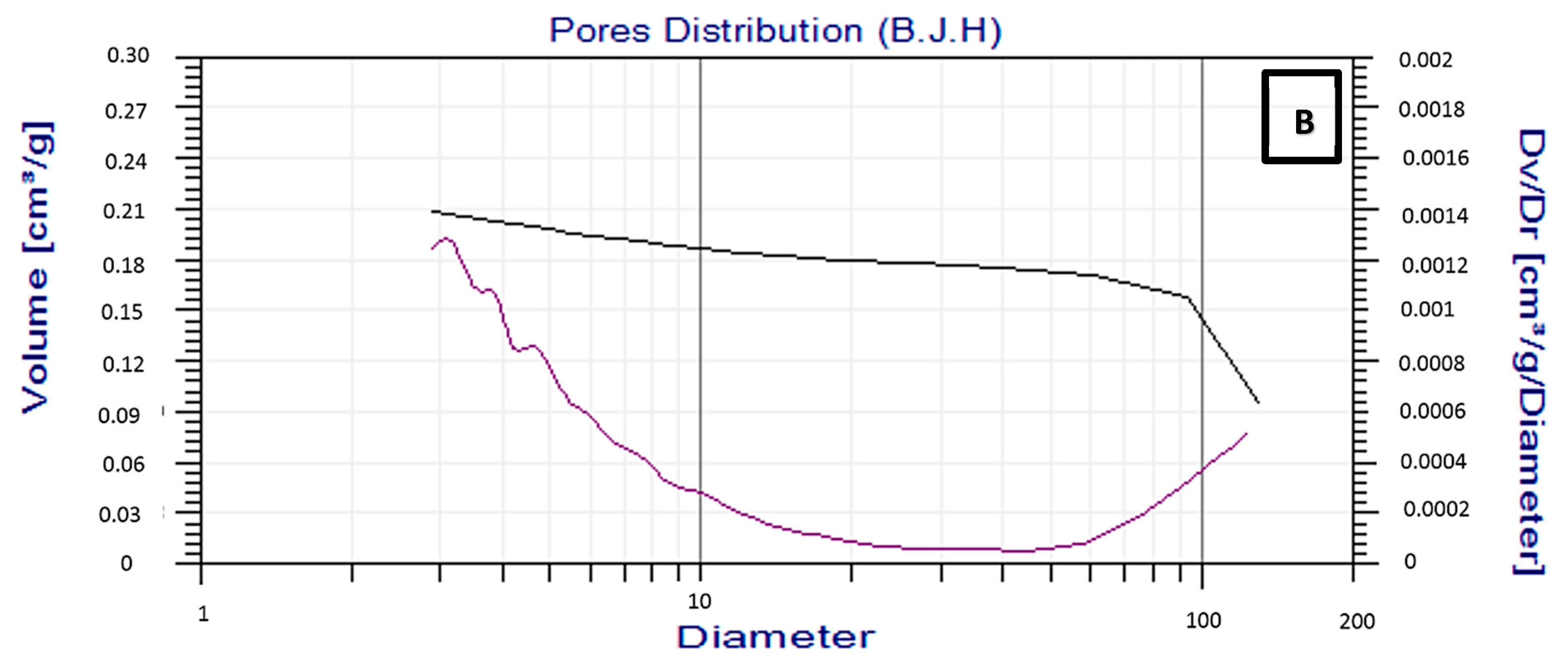
2.1.4. Surface are of Brunauer–Emmett–Teller (BET)
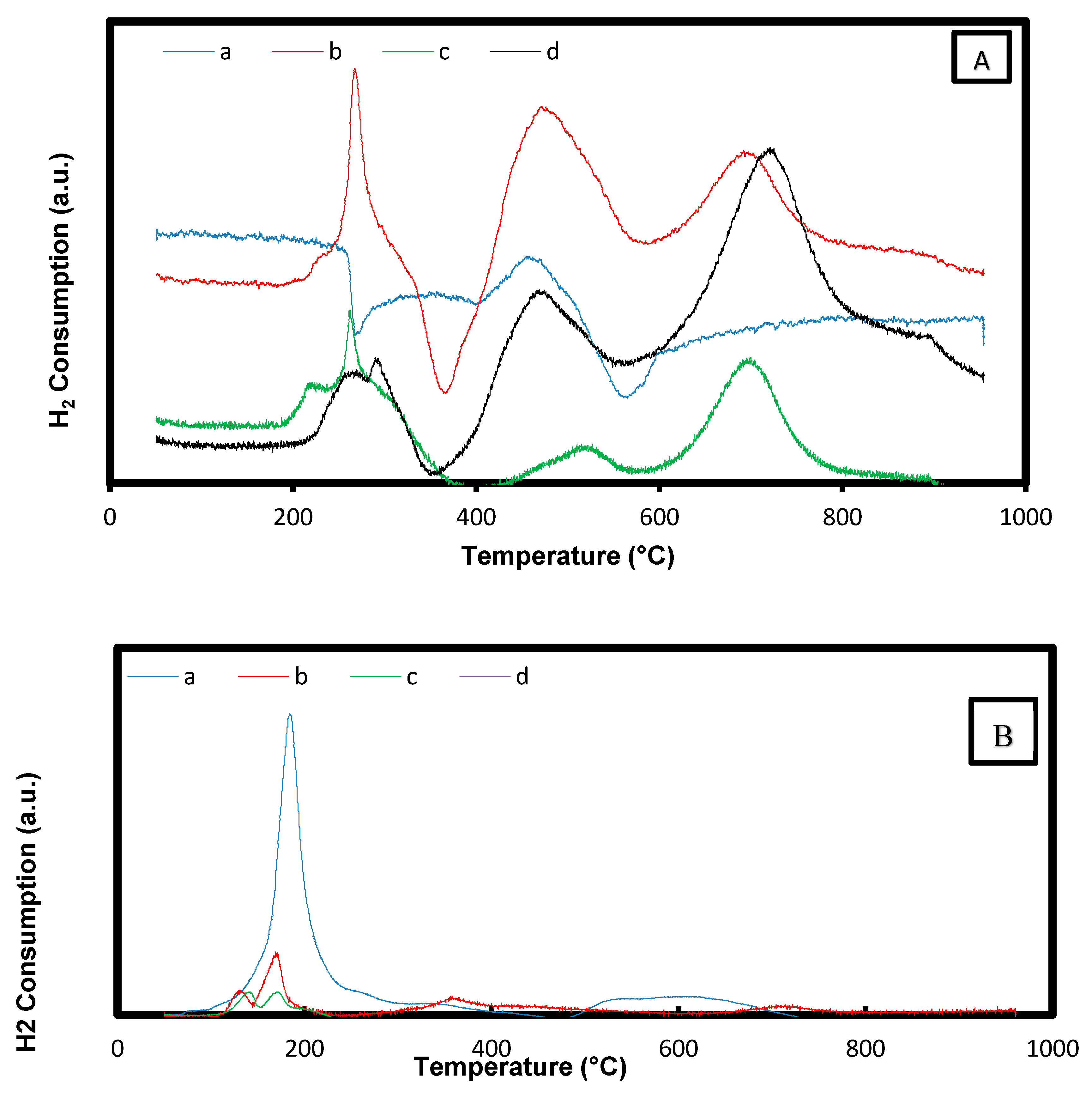
2.1.5. Temperature Programmed Reduction (H2-TPR)
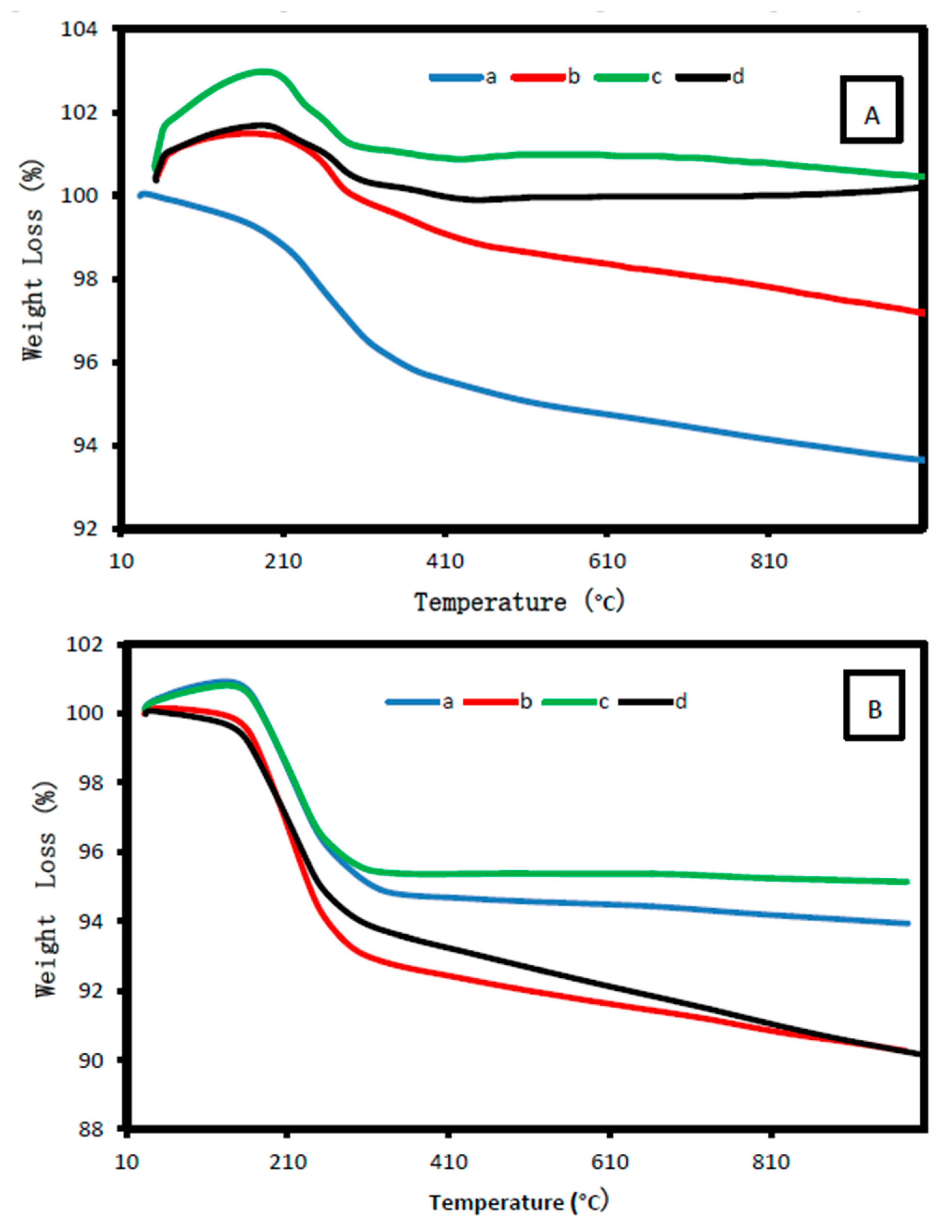
2.1.6. Thermal Analysis
2.2. The Performance of Catalysts in DRM Reaction
2.2.1. Effects of the Concentration of Reactants on the Conversion
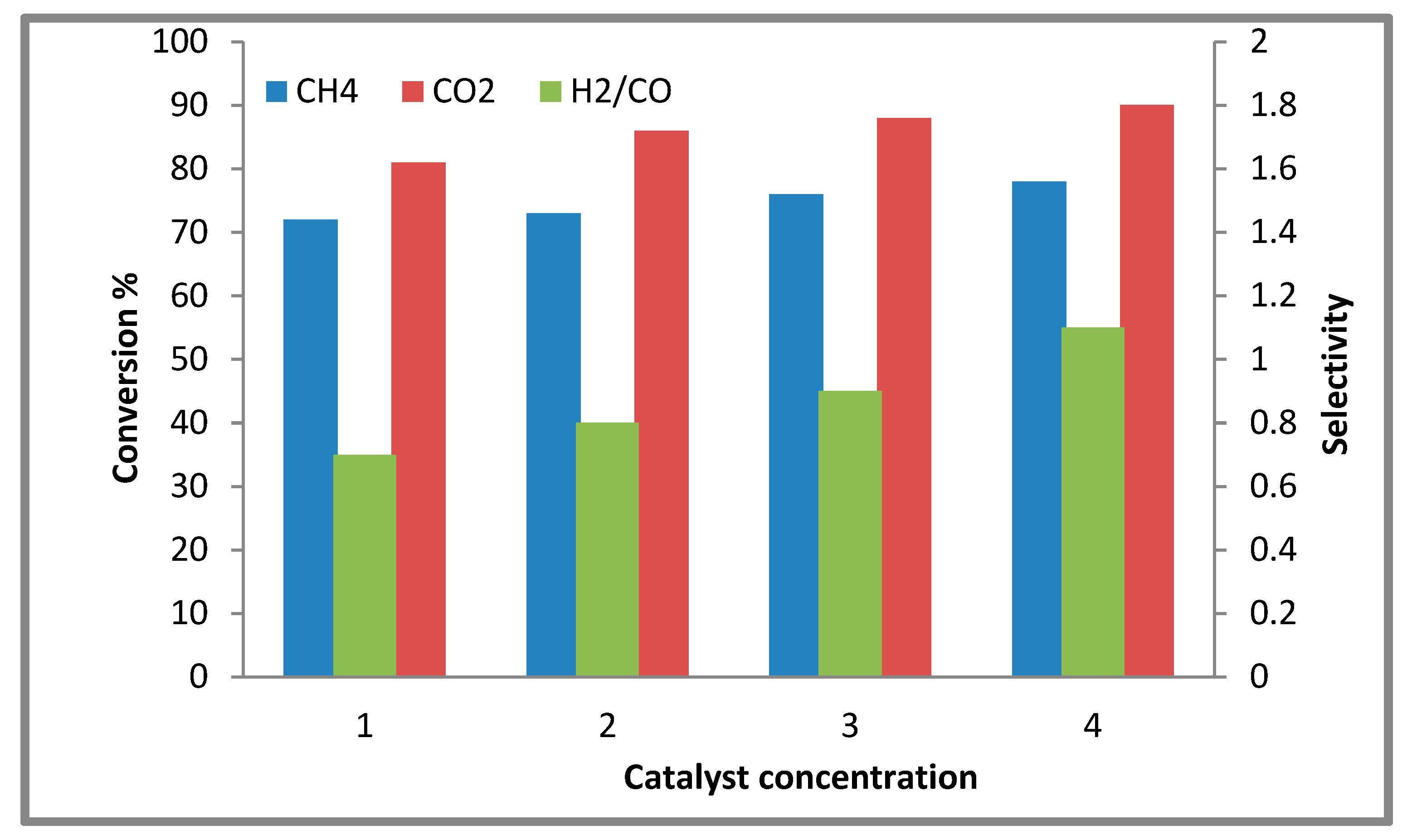
2.2.2. Effect of the Catalyst Concentration on the Conversion
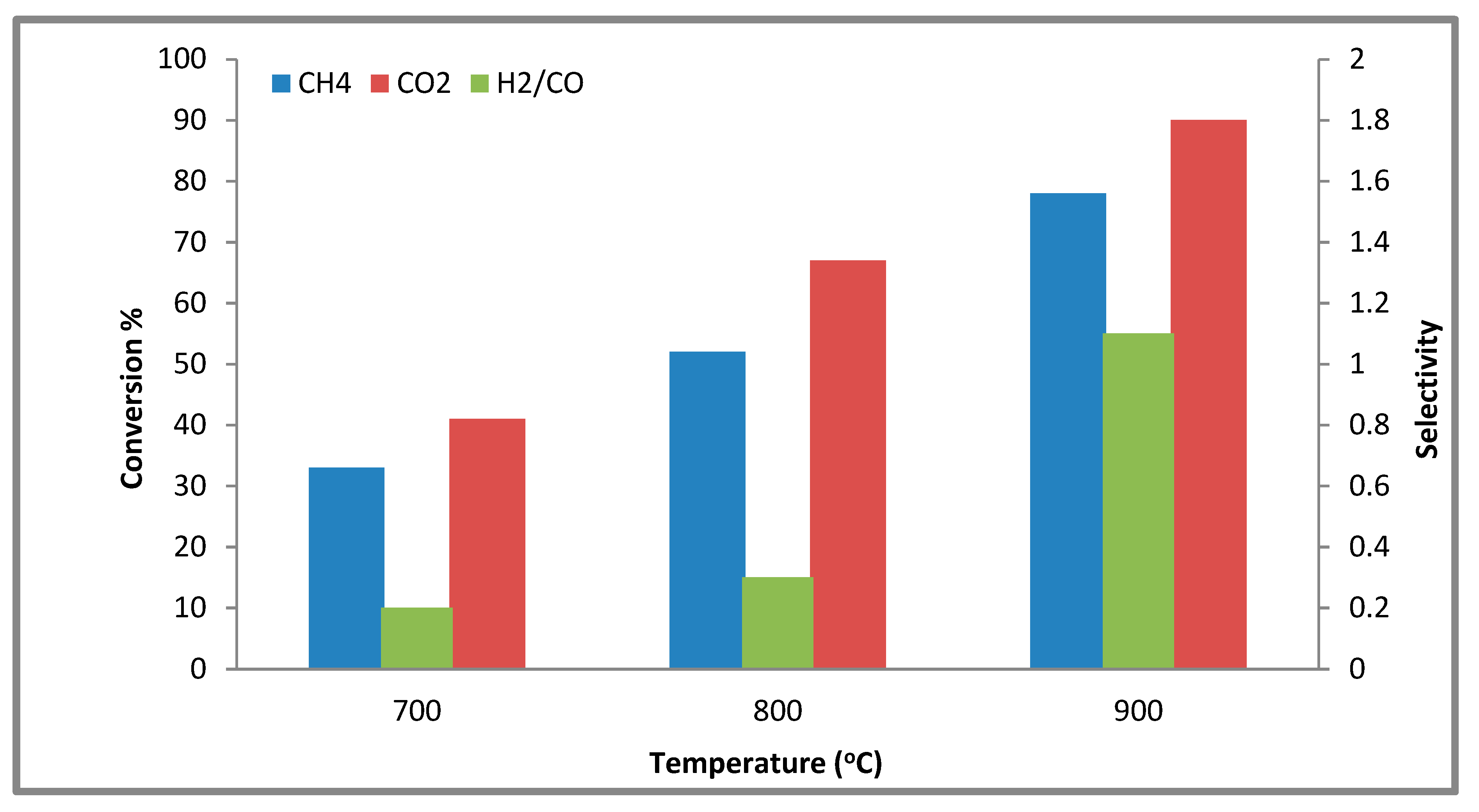
2.2.3. The Effect of Various Temperatures on the Conversion
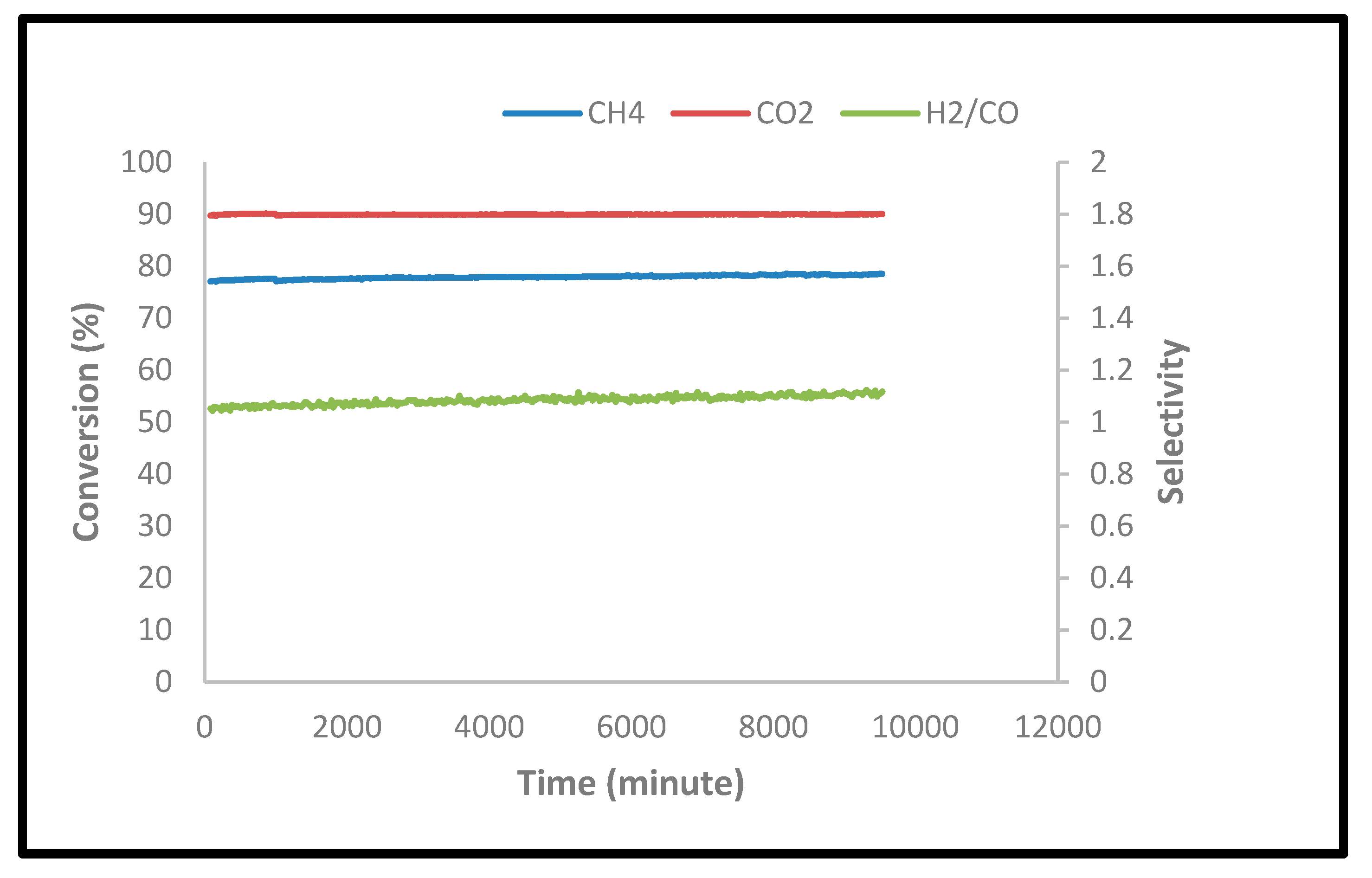
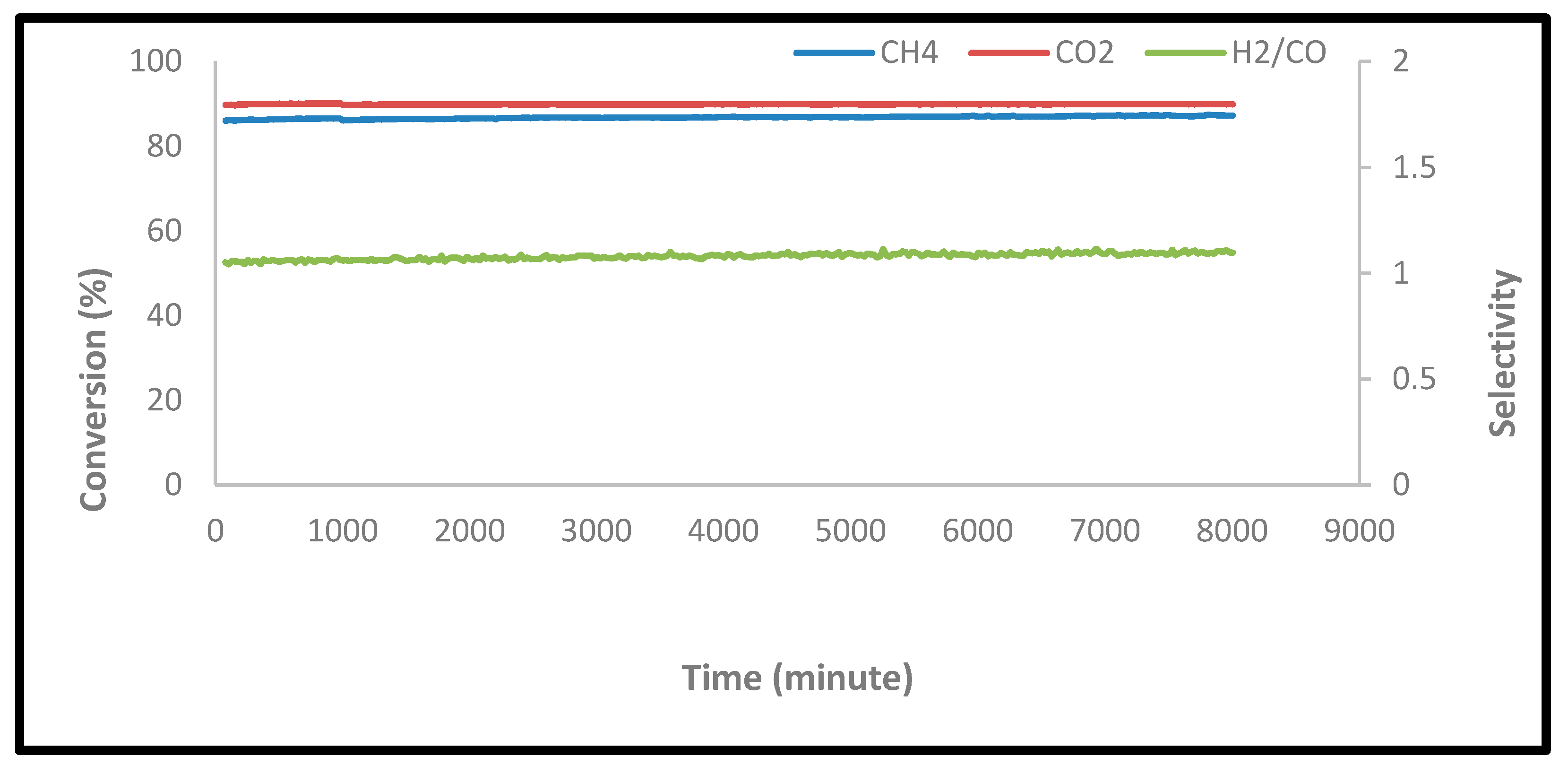
2.2.4. Stability Tests
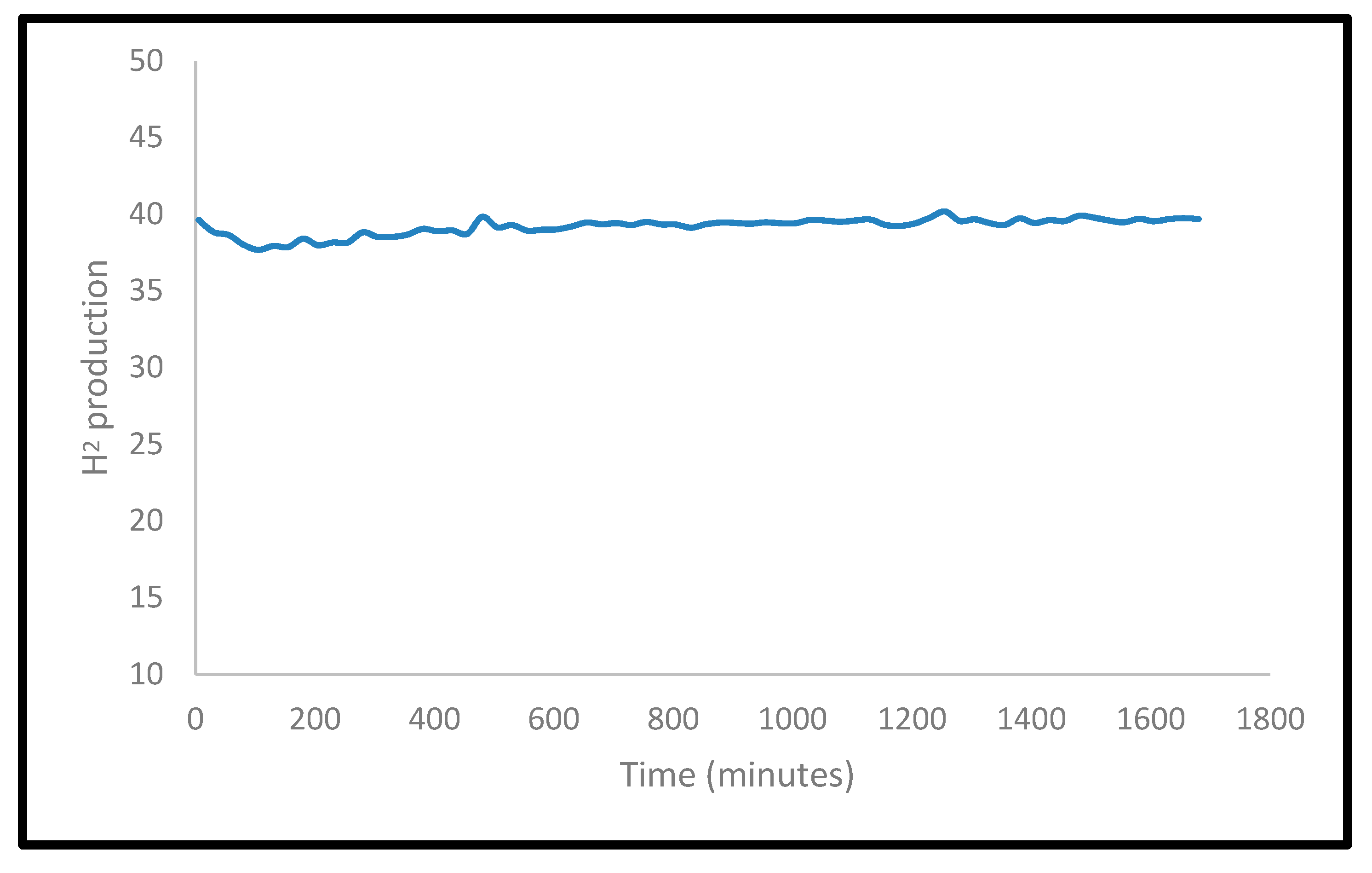
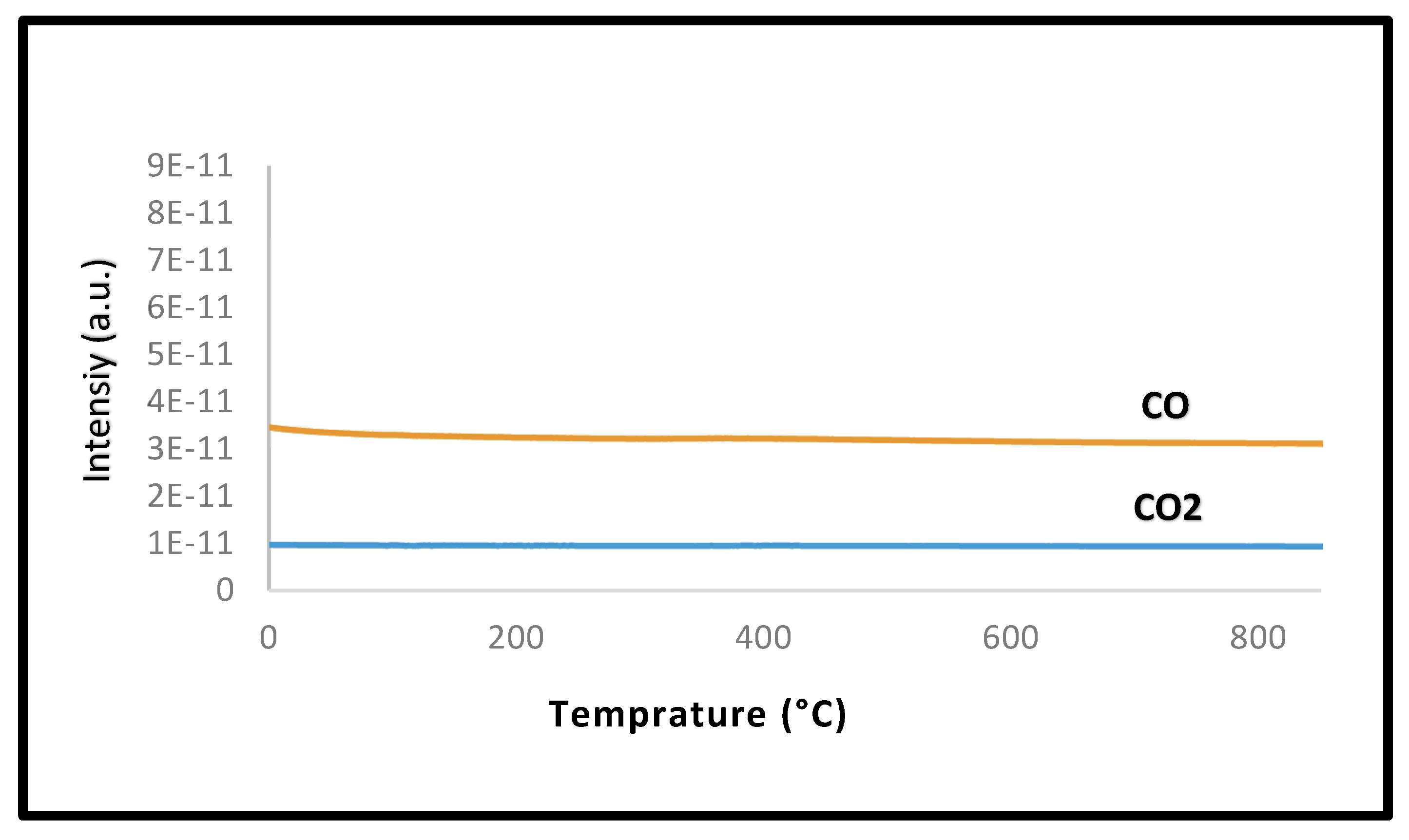
2.2.5. Post-Reaction Characterization
2.2.6. Improvement in the Catalytic Stability and Selectivity
3. Experimental Section
3.1. Materials
3.2. Catalysts Preparation
3.3. Catalysts Characterization
3.4. Catalytic Evaluations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Al-Doghachi, F.A.; Taufiq-Yap, Y.H. Syngas production from the CO2 reforming of methane over Co/Mg1−xNixO catalysts. J. Chem. Sci. 2017, 129, 1781–1786. [Google Scholar] [CrossRef]
- Al-Doghachi, F.A.J.; Rashid, U.; Taufiq-Yap, Y.H. Investigation of Ce(iii) promoter effects on the tri-metallic Pt, Pd, Ni/MgO catalyst in dry-reforming of methane. RSC Adv. 2016, 6, 10372–10384. [Google Scholar] [CrossRef]
- Al-Doghachi, F.A.J.; Taufiq-Yap, Y. CO2 Reforming of Methane over Ni/MgO Catalysts Promoted with Zr and La Oxides. ChemistrySelect 2018, 3, 816–827. [Google Scholar] [CrossRef]
- Akbari, E.; Alavi, S.M.; Rezaei, M. CeO2 Promoted Ni-MgO-Al2O3 nanocatalysts for carbon dioxide reforming of methane. J. CO2 Util. 2018, 24, 128–138. [Google Scholar] [CrossRef]
- Jin, B.; Shang, Z.; Li, S.; Jiang, Y.-B.; Gu, X.; Liang, X. Reforming of methane with carbon dioxide over cerium oxide promoted nickel nanoparticles deposited on 4-channel hollow fibers by atomic layer deposition. Catal. Sci. Technol. 2020, 10, 3212–3222. [Google Scholar] [CrossRef]
- Luisetto, I.; Tuti, G.S.; Di Bartolomeo, E. Co and Ni supported on CeO2 as selective bimetallic catalyst for dry reforming of methane. Int. J. Hydrogen Energy 2012, 37, 15992–15999. [Google Scholar] [CrossRef]
- Meshkani, F.; Rezaei, M. Nano crystalline MgO supported nickel-based bimetallic catalysts for carbon dioxide reforming of methane. Int. J. Hydrogen Energy 2010, 35, 10295–10301. [Google Scholar] [CrossRef]
- Al-Mubaddel, F.; Kasim, S.; Ibrahim, A.A.; Al-Awadi, A.S.; Fakeeha, A.H.; Al-Fatesh, A.S. H2 Production from Catalytic Methane Decomposition Using Fe/x-ZrO2 and Fe-Ni/(x-ZrO2) (x = 0, La2O3, WO3) Catalysts. Catalysts 2020, 10, 793. [Google Scholar] [CrossRef]
- Wu, H.; Pantaleo, G.; La Parola, V.; Venezia, A.M.; Collard, X.; Aprile, C.; Liotta, L.F. Bi-and tri-metallic Ni catalysts over Al2O3 and Al2O3-MOx (M = Ce or Mg) oxides for methane dry reforming: Au and Pt additive effects. Appl. Catal. B Environ. 2014, 156, 350–361. [Google Scholar] [CrossRef]
- Al-Doghachi, F.A.J.; Rashid, U.; Zainal, Z.; Saiman, M.I.; Taufiq-Yap, Y.H. Influence of Ce2O3 and CeO2 promoters on Pd/MgO catalysts in the dry-reforming of methane. RSC Adv. 2015, 5, 81739–81752. [Google Scholar] [CrossRef]
- Yu, Q.; Wu, X.; Tang, C.; Qi, L.; Liu, B.; Gao, B.; Sun, K.; Dong, L.; Chen, Y. Textural, structural, and morphological characterizations and catalytic activity of nanosized CeO2–MOx (M = Mg2+, Al3+, Si4+) mixed oxides for CO oxidation. J. Colloid Interface Sci. 2011, 354, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Al-Doghachi, F.A.J.; Islam, A.; Zainal, Z.; Saiman, M.I.; Embong, Z.; Taufiq-Yap, Y. High Coke-Resistance Pt/Mg1-xNixO Catalyst for Dry Reforming of Methane. PLoS ONE 2016, 11, e0145862. [Google Scholar] [CrossRef] [PubMed]
- Al-Doghachi, F.J.A. Effects of Platinum and Palladium Metals on Ni/Mg1-xZrxO Catalysts in the CO2 Reforming of Methane. Bull. Chem. React. Eng. Catal. 2018, 13, 295–310. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, J.; Tian, S.; Li, K. Biogas dry reforming for syngas production: Catalytic performance of nickel supported on waste-derived SiO2. Catal. Sci. Technol. 2015, 5, 860–868. [Google Scholar] [CrossRef]
- Hidalgo-Carrillo, J.; Sebti, J.; Marinas, A.; Marinas, J.M.; Sebti, S.; Urbano, F.J. XPS evidence for structure-performance relationship in selective hydrogenation of croton aldehyde to crotyl alcohol on platinum systems supported on natural phosphates. J. Colloid Interface Sci. 2012, 382, 67–73. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, J.; Yan, F.; Tian, S.; Li, K. A novel low temperature vapor phase hydrolysis method for the production of nano-structured silica materials using silicon tetrachloride. RSC Adv. 2014, 4, 8703–8710. [Google Scholar] [CrossRef]
- Usman, M.; Daud, W.W.; Abbas, H.F. Dry reforming of methane: Influence of process parameters—A review. Renew. Sustain. Energy Rev. 2015, 45, 710–744. [Google Scholar] [CrossRef]
- Saha, B.; Khan, A.; Ibrahim, H.; Idem, R. Evaluating the performance of non-precious metal based catalysts for sulfur-tolerance during the dry reforming of biogas. Fuel 2014, 120, 202–217. [Google Scholar] [CrossRef]
- Taufiq-Yap, Y.; Sudarno; Rashid, U.; Zainal, Z. CeO2–SiO2 supported nickel catalysts for dry reforming of methane toward syngas production. Appl. Catal. A Gen. 2013, 468, 359–369. [Google Scholar] [CrossRef]
- Li, G.; Hu, L.; Hill, J.M. Comparison of reducibility and stability of alumina-supported Ni catalysts prepared by impregnation and co-precipitation. Appl. Catal. A Gen. 2006, 301, 16–24. [Google Scholar] [CrossRef]
- Fernández-Garcıa, M.; Martınez-Arias, A.; Iglesias-Juez, A.; Belver, C.; Hungrıa, A.B.; Conesa, J.C.; Soria, J. Structural characteristics and redox behavior of CeO2-ZrO2/Al2O3 supports. J. Catal. 2000, 194, 385–392. [Google Scholar]
- Tada, S.; Shimizu, T.; Kameyama, H.; Haneda, T.; Kikuchi, R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int. J. Hydrogen Energy 2012, 37, 5527–5531. [Google Scholar] [CrossRef]
- Mahoney, E.G.; Pusel, J.M.; Stagg-Williams, S.M.; Faraji, S. The effects of Pt addition to supported Ni catalysts on dry (CO2) reforming of methane to syngas. J. CO2 Util. 2014, 6, 40–44. [Google Scholar] [CrossRef]
- Bao, Z.; Lu, Y.; Han, J.; Li, Y.; Yu, F. Highly active and stable Ni-based bimodal pore catalyst for dry reforming of methane. Appl. Catal. A Gen. 2015, 491, 116–126. [Google Scholar] [CrossRef]
- Rotaru, C.; Postole, G.; Florea, M.; Matei-Rutkovska, F.; Parvulescu, V.; Gelin, P. Dry reforming of methane on ceria prepared by modified precipitation route. Appl. Catal. A Gen. 2015, 494, 29–40. [Google Scholar] [CrossRef]
- Gonzalez-Delacruz, V.M.; Ternero, F.; Pereñíguez, R.; Caballero, A.; Holgado, J.P. Study of nanostructured Ni/CeO2 catalysts prepared by combustion synthesis in dry reforming of methane. Appl. Catal. A Gen. 2010, 384, 1–9. [Google Scholar] [CrossRef]
- Gaddalla, A.M.; Sommer, M.E. Carbon dioxide reforming of methane on nickel catalysts. Chem. Eng. Sci. 1989, 44, 2825–2829. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Assabumrungrat, S. Catalytic dry reforming of methane over high surface area ceria. Appl. Catal. B Environ. 2005, 60, 107–116. [Google Scholar] [CrossRef]
- Guo, J.; Lou, H.; Zhao, H.; Chai, D.; Zheng, X. Dry reforming of methane over nickel catalysts supported on magnesium aluminate spinels. Appl. Catal. A Gen. 2004, 273, 75–82. [Google Scholar] [CrossRef]
- Aramouni, N.A.; Zeaiter, J.; Kwapinski, W.; Leahy, J.J.; Ahmad, M.N. Eclectic tri-metallic Ni–Co–Ru catalyst for the dry reforming of methane. Int. J. Hydrogen Energy 2020, 45, 17153–17163. [Google Scholar]
- Kehres, J.; Jakobsen, J.G.; Andreasen, J.W.; Wagner, J.B.; Liu, H.; Molenbroek, A.; Sehested, J.; Chorkendorff, I.; Vegge, T. Dynamical Properties of a Ru/MgAl2O4 Catalyst during Reduction and Dry Methane Reforming. J. Phys. Chem. C 2012, 116, 21407–21415. [Google Scholar] [CrossRef]
- Topalidis, A.; Petrakis, D.; Ladavos, A.; Loukatzikou, L.; Pomonis, P. A kinetic study of methane and carbon dioxide interconversion over 0.5%Pt/SrTiO3 catalysts. Catal. Today 2007, 127, 238–245. [Google Scholar] [CrossRef]
- Zhu, J.; Peng, X.; Yao, L.; Shen, J.; Tong, D.; Hu, C. The promoting effect of La, Mg, Co and Zn on the activity and stability of Ni/SiO2 catalyst for CO2 reforming of methane. Int. J. Hydrogen Energy 2011, 36, 7094–7104. [Google Scholar] [CrossRef]
- Al-Najar, A.M.A.; Al-Doghachi, F.A.J.; Al-Riyahee, A.; Taufiq-Yap, Y.H. Effect of La2O3 as a Promoter on the Pt,Pd,Ni/MgO Catalyst in Dry Reforming of Methane Reaction. Catalysts 2020, 10, 750. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | TEM (nm) | a Crystal Size (D) Debye-Sherrer’s Equation (nm) | XRF | |||
|---|---|---|---|---|---|---|
| Ni% | Pd% | Pt% | Mg & Ce% | |||
| Ni/MgO | 44 | 39 | 0.91 | 98.7 | ||
| Ni/Mg0.97Ce4+0.03O | 56 | 52 | 0.95 | 98.3 | ||
| Ni/Mg0.93Ce4+0.07O | 58 | 52 | 1.03 | 98.6 | ||
| Ni/Mg0.85Ce4+0.15O | 59 | 53 | 0.93 | 98.5 | ||
| Ni,Pd,Pt/MgO | 53 | 42 | 0.96 | 1.09 | 1.15 | 96.2 |
| Ni,Pd,Pt/Mg0.97Ce4+0.03O | 54 | 51 | 1.03 | 1.24 | 1.03 | 96.0 |
| Ni,Pd,Pt/Mg0.93Ce4+0.07O | 55 | 52 | 0.93 | 1.04 | 1.15 | 96.4 |
| Ni,Pd,Pt/Mg0.85Ce4+0.15O | 57 | 52 | 0.96 | 1.22 | 0.98 | 95.8 |
| Sample | Specific Surface | Pore Volume | Pore Radius |
|---|---|---|---|
| Area (m2/g) | (cm3/g) | (Å) | |
| Ni/MgO | 10.7 | 0.089 | 28.7 |
| Ni/Mg0.97Ce4+0.03O | 12.1 | 0.116 | 54.8 |
| Ni/Mg0.93Ce4+0.07O | 14.4 | 0.134 | 63.3 |
| Ni/Mg0.85Ce4+0.15O | 17.2 | 0.185 | 68.5 |
| Ni,Pd,Pt/MgO | 10.9 | 0.122 | 44.4 |
| Ni,Pd,Pt/Mg0.97Ce4+0.03O | 13.5 | 0.132 | 60.9 |
| Ni,Pd,Pt/Mg0.93Ce4+0.07O | 17.1 | 0.182 | 73.3 |
| Ni,Pd,Pt/Mg0.85Ce4+0.15O | 19.8 | 0.213 | 86.3 |
| Catalysts | Temp. | Temp. | Temp. | Temp. | Temp. | Amount H2-Consumed |
|---|---|---|---|---|---|---|
| °C | °C | °C | °C | °C | (µmol/g) | |
| CeO2 | 459 | 871 | 72.25 | |||
| Ni/MgO | 475 | 314.8 | ||||
| Ni/Mg0.97Ce4+0.03O | 279 | 480 | 695 | 415.0 | ||
| Ni/Mg0.93Ce4+0.07O | 283 | 488 | 701 | 623.7 | ||
| Ni/Mg0.85Ce4+0.15O | 310 | 529 | 710 | 706.5 | ||
| Ni,Pd,Pt/MgO | 130 | 184 | 520 | 488.6 | ||
| Ni,Pd,Pt/Mg0.97Ce4+0.03O | 122 | 164 | 485 | 516 | 719 | 524.3 |
| Ni,Pd,Pt/Mg0.93Ce4+0.07O | 134 | 166 | 483 | 522 | 734 | 667.0 |
| Ni,Pd,Pt/Mg0.85Ce4+0.15O | 139 | 178 | 475 | 527 | 783 | 782.3 |
| Sample Name | CH4 Conversion % | CO2 Conversion % | H2/CO Ratio |
|---|---|---|---|
| MgO | 21 | 32 | 0.1 |
| Mg0.85Ce4+0.15O | 32 | 41 | 0.3 |
| Ni/MgO | 45 | 66 | 0.4 |
| Ni/Mg0.97Ce4+0.03O | 40 | 67 | 0.5 |
| Ni/Mg0.93Ce4+0.07O | 48 | 66 | 0.5 |
| Ni/Mg0.85Ce4+0.15O | 56 | 74 | 0.6 |
| Ni,Pd,Pt/MgO | 72 | 81 | 0.7 |
| Ni,Pd,Pt/Mg0.97Ce4+0.03O | 73 | 86 | 0.8 |
| Ni,Pd,Pt/Mg0.93Ce4+0.07O | 76 | 88 | 0.9 |
| Ni,Pd,Pt/Mg0.85Ce4+0.15O | 78 | 90 | 1.1 |
| Catalysts | Support (MgO) Mg (NO3)2.6H2O (g) | Promoter (CeO2) (NH4)2Ce (NO3)6.6H2O (g) | Total Weight of MgO and CeO2 after Calcination (g) | Impregnation of the Main Catalyst (1% Pt) (1% Pd) (1% Ni) (g) | ||
|---|---|---|---|---|---|---|
| Pt (acac)2.H2O | Pd (acac)2 | Ni (acac)2 | ||||
| Ni,Pd,Pt/MgO | 25.0 | 0.0 | 1 | 0.02 | 0.029 | 0.044 |
| Ni,Pd,Pt/Mg0.97Ce4+0.03O | 24.9 | 1.3 | 1 | 0.02 | 0.029 | 0.044 |
| Ni,Pd,Pt/Mg0.93 Ce4+0.07O | 23.8 | 3.0 | 1 | 0.02 | 0.029 | 0.044 |
| Ni,Pd,Pt/Mg0.85 Ce4+0.15O | 21.8 | 6.5 | 1 | 0.02 | 0.029 | 0.044 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Doghachi, F.A.J.; Jassim, A.F.A.; Taufiq-Yap, Y.H. Enhancement of CO2 Reforming of CH4 Reaction Using Ni,Pd,Pt/Mg1−xCex4+O and Ni/Mg1−xCex4+O Catalysts. Catalysts 2020, 10, 1240. https://doi.org/10.3390/catal10111240
Al-Doghachi FAJ, Jassim AFA, Taufiq-Yap YH. Enhancement of CO2 Reforming of CH4 Reaction Using Ni,Pd,Pt/Mg1−xCex4+O and Ni/Mg1−xCex4+O Catalysts. Catalysts. 2020; 10(11):1240. https://doi.org/10.3390/catal10111240
Chicago/Turabian StyleAl-Doghachi, Faris A. J., Ali F. A. Jassim, and Yun Hin Taufiq-Yap. 2020. "Enhancement of CO2 Reforming of CH4 Reaction Using Ni,Pd,Pt/Mg1−xCex4+O and Ni/Mg1−xCex4+O Catalysts" Catalysts 10, no. 11: 1240. https://doi.org/10.3390/catal10111240
APA StyleAl-Doghachi, F. A. J., Jassim, A. F. A., & Taufiq-Yap, Y. H. (2020). Enhancement of CO2 Reforming of CH4 Reaction Using Ni,Pd,Pt/Mg1−xCex4+O and Ni/Mg1−xCex4+O Catalysts. Catalysts, 10(11), 1240. https://doi.org/10.3390/catal10111240