Acid/Base-Treated Activated Carbon Catalysts for the Low-Temperature Endothermic Cracking of N-Dodecane with Applications in Hypersonic Vehicle Heat Management Systems


Abstract
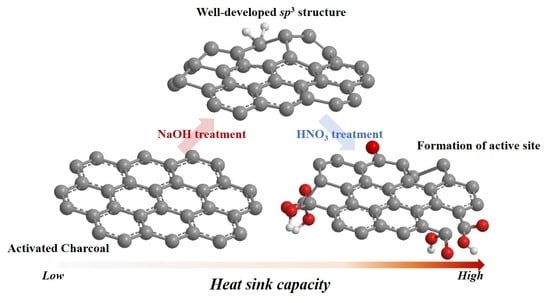
:
1. Introduction
2. Results
2.1. Physical Properties of the AC Catalysts
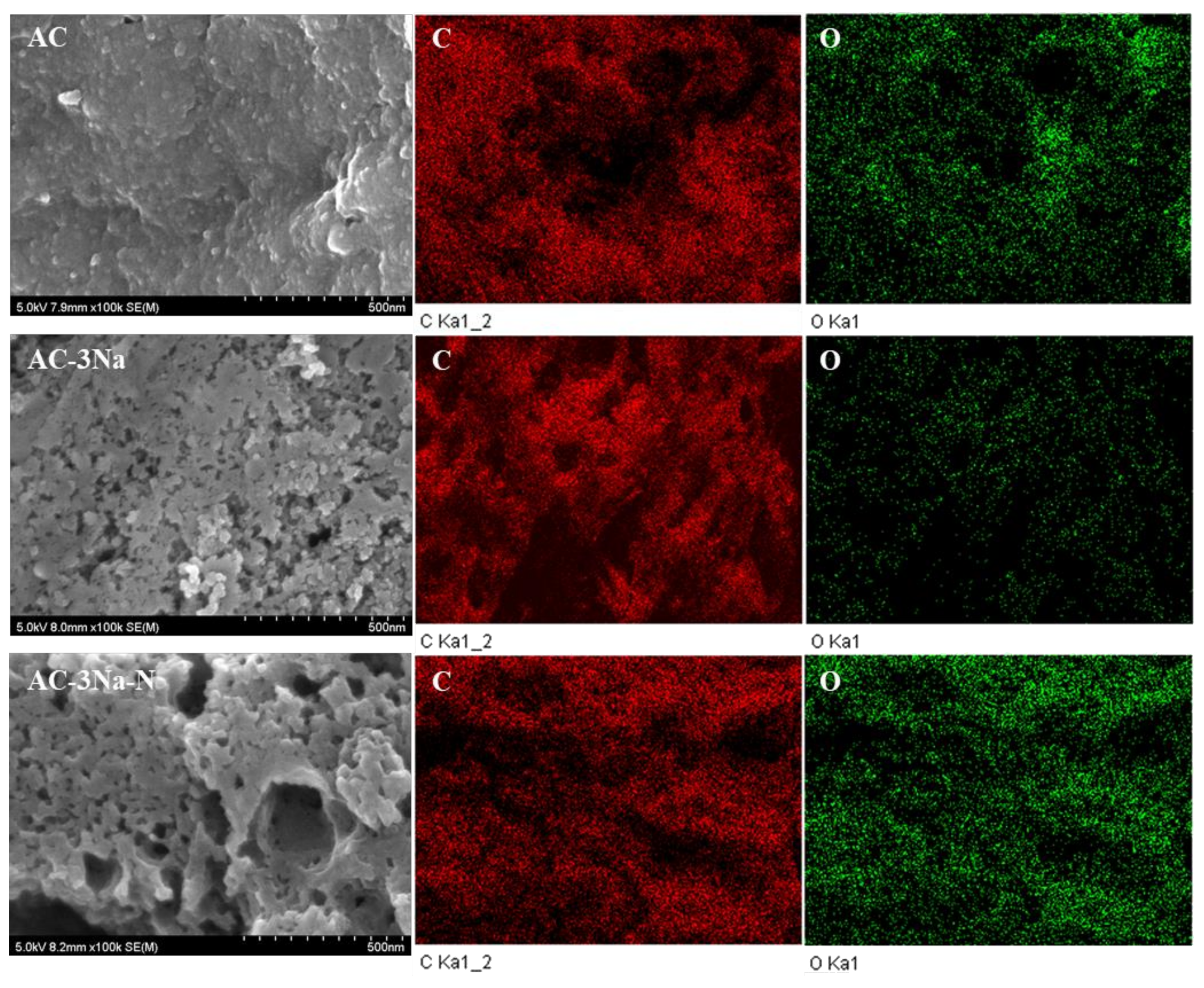
2.2. Surface Texture and EDX Elemental Mapping SEM Images of the AC Catalysts
2.3. XRD Analysis of the ACs and ZSM-5 Zeolite
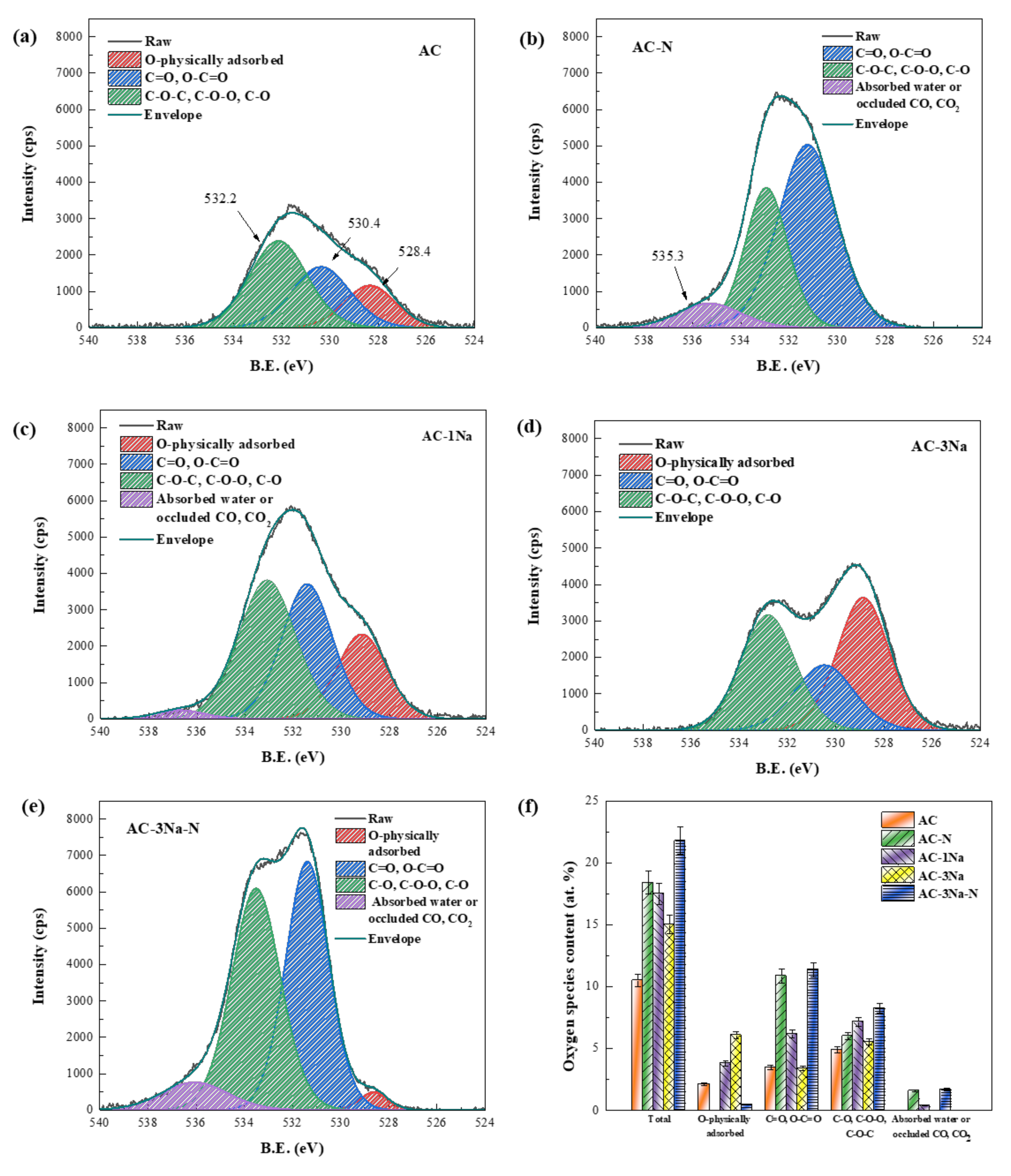
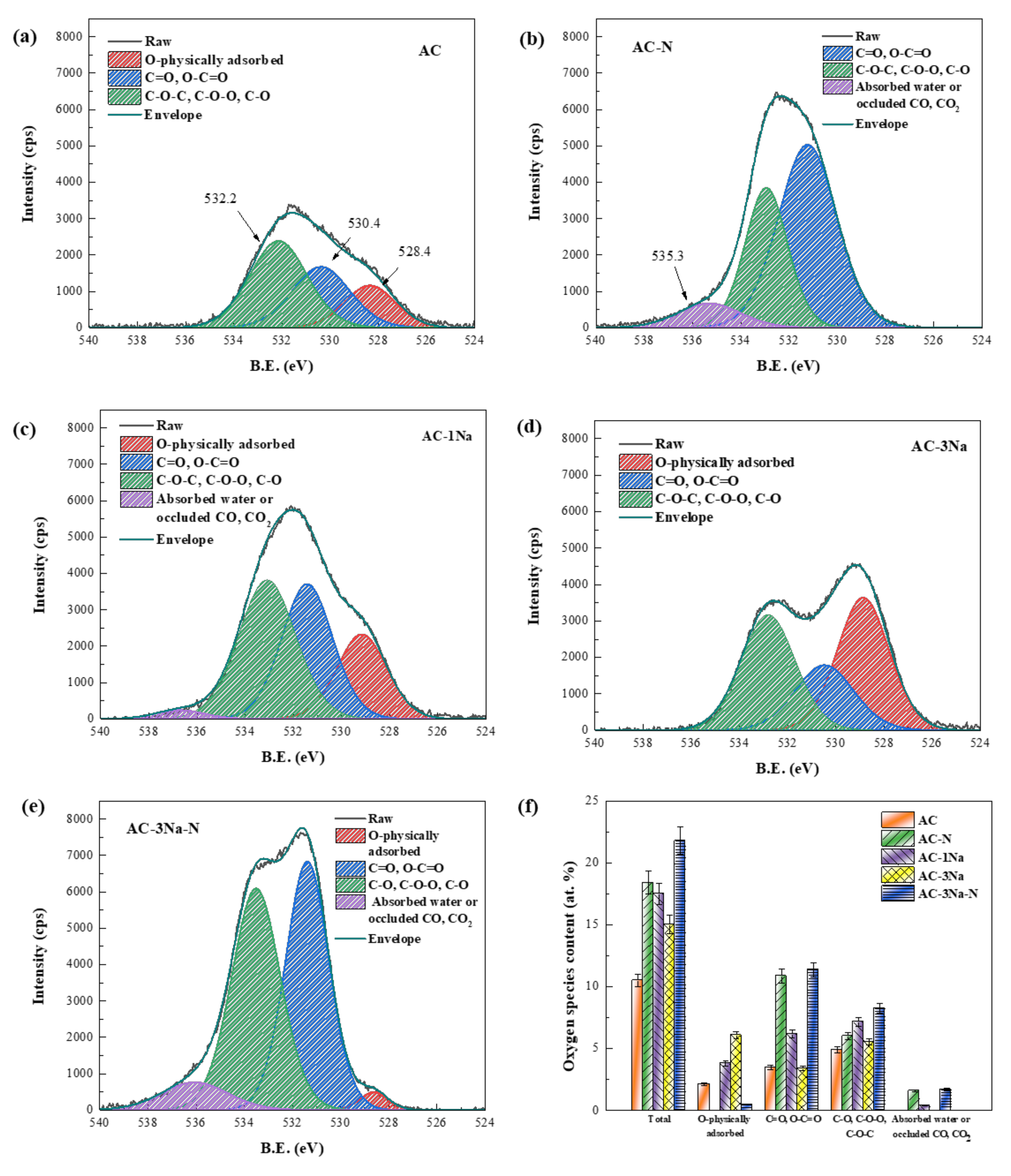
2.4. XPS Analysis of AC Catalysts
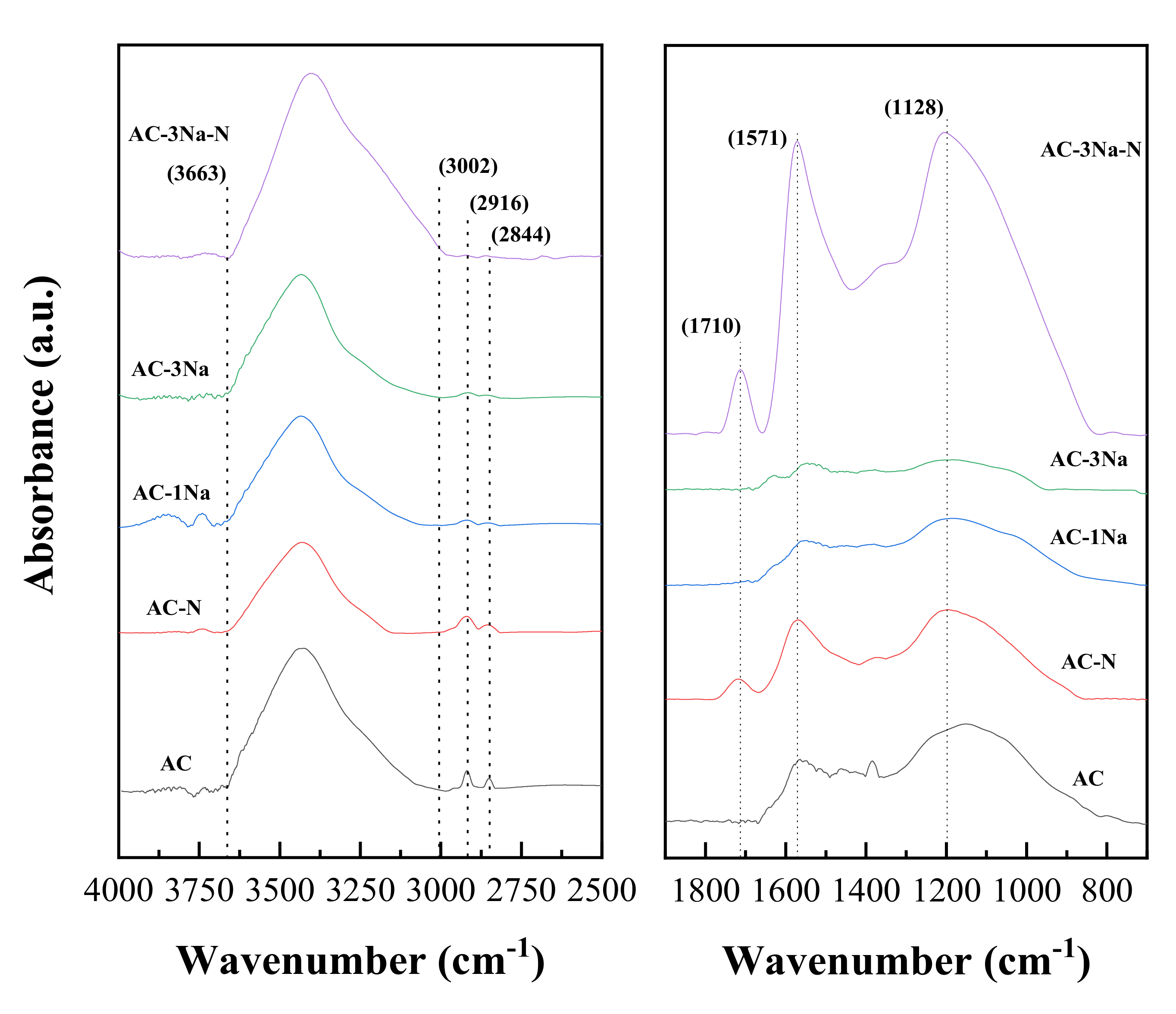
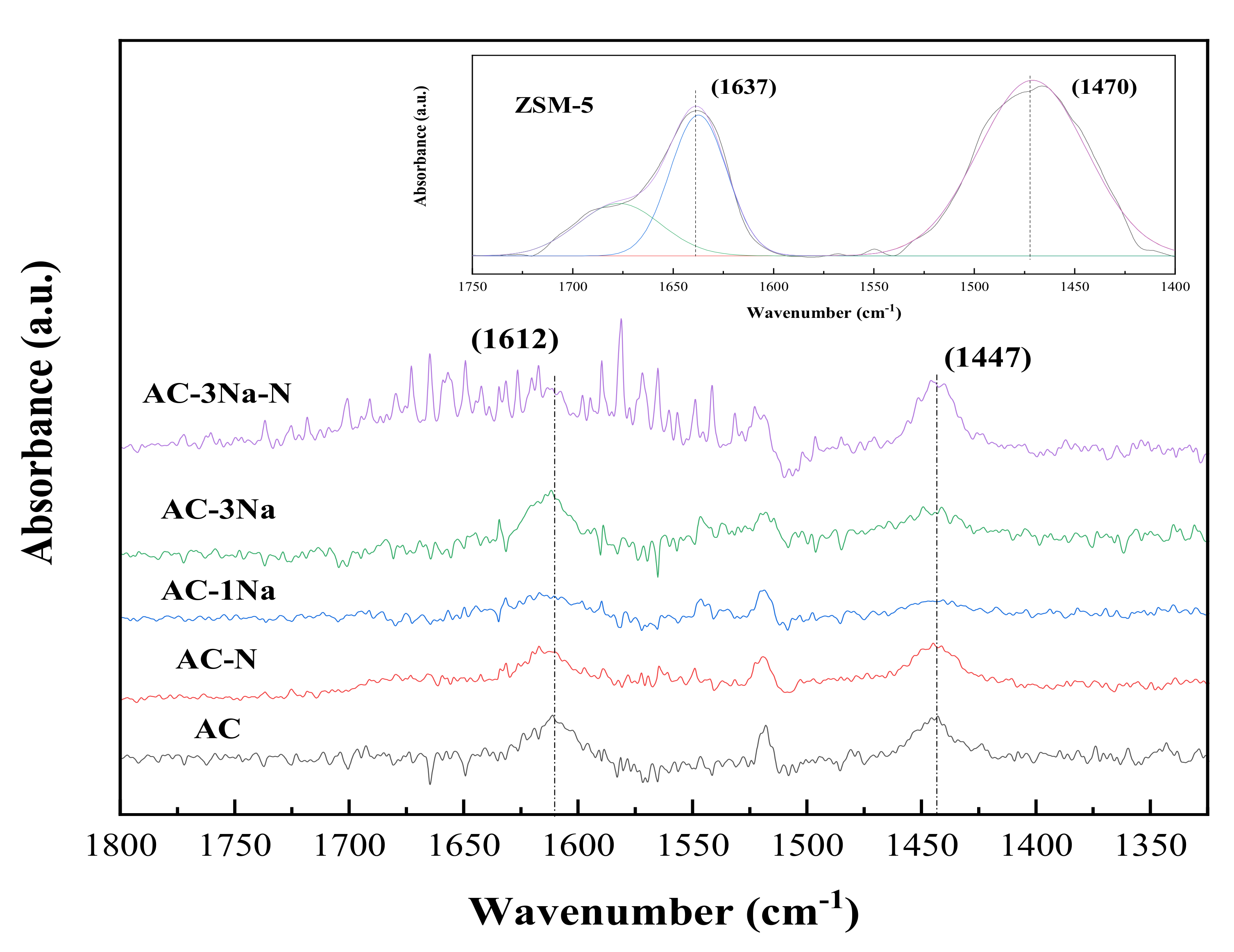
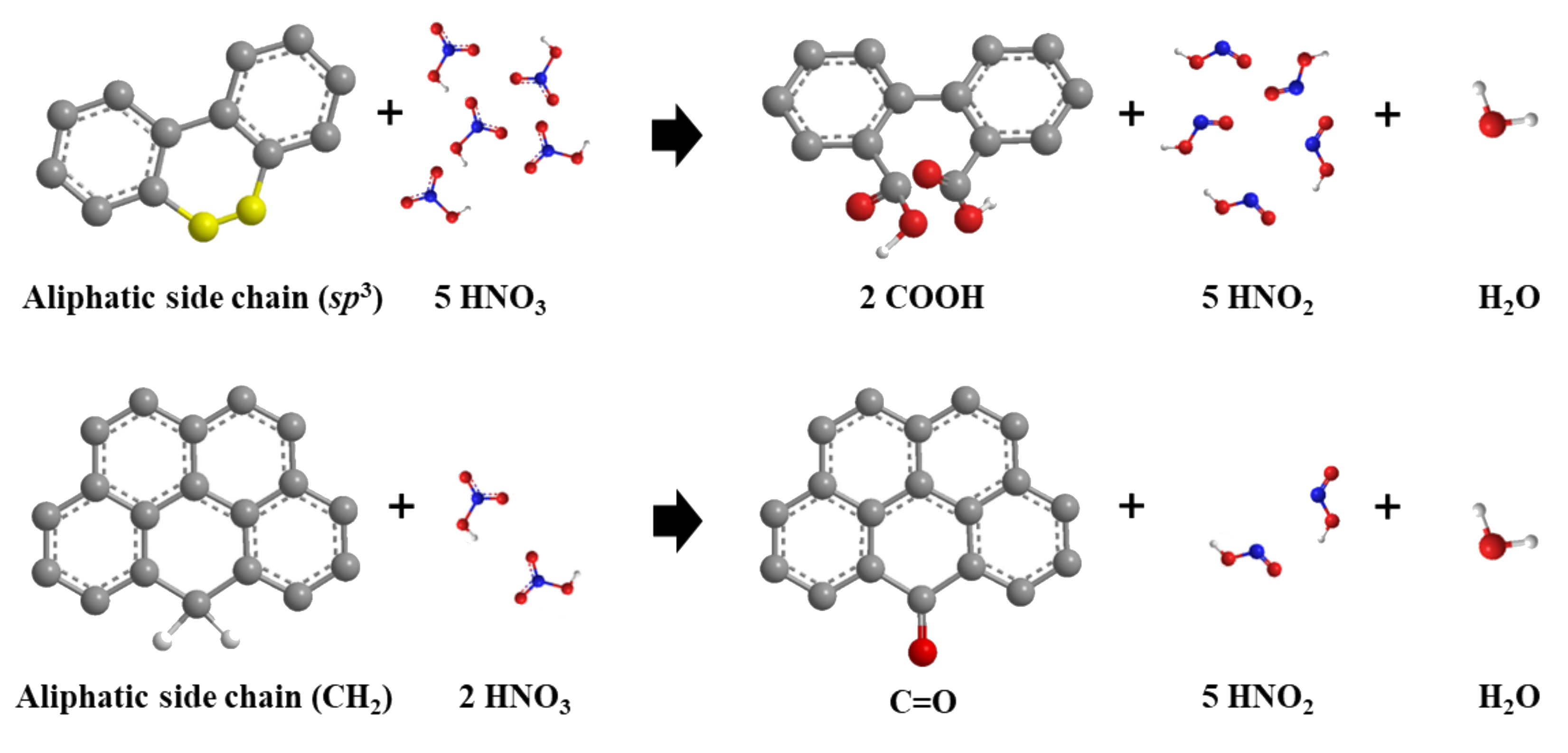
2.5. FT-IR Analysis of AC Catalysts
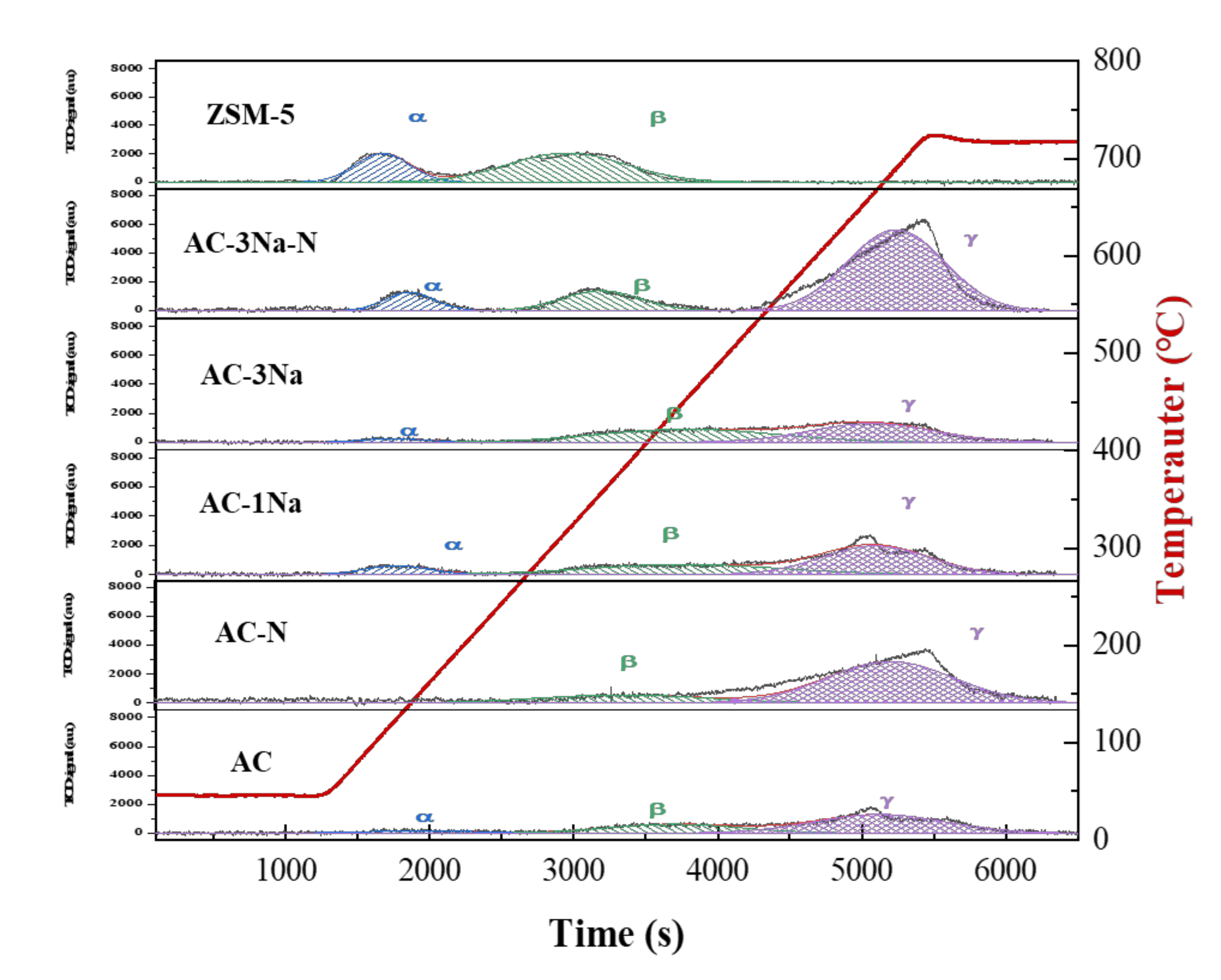
2.6. Acid Properties of AC and ZSM-5 Zeolite Catalysts
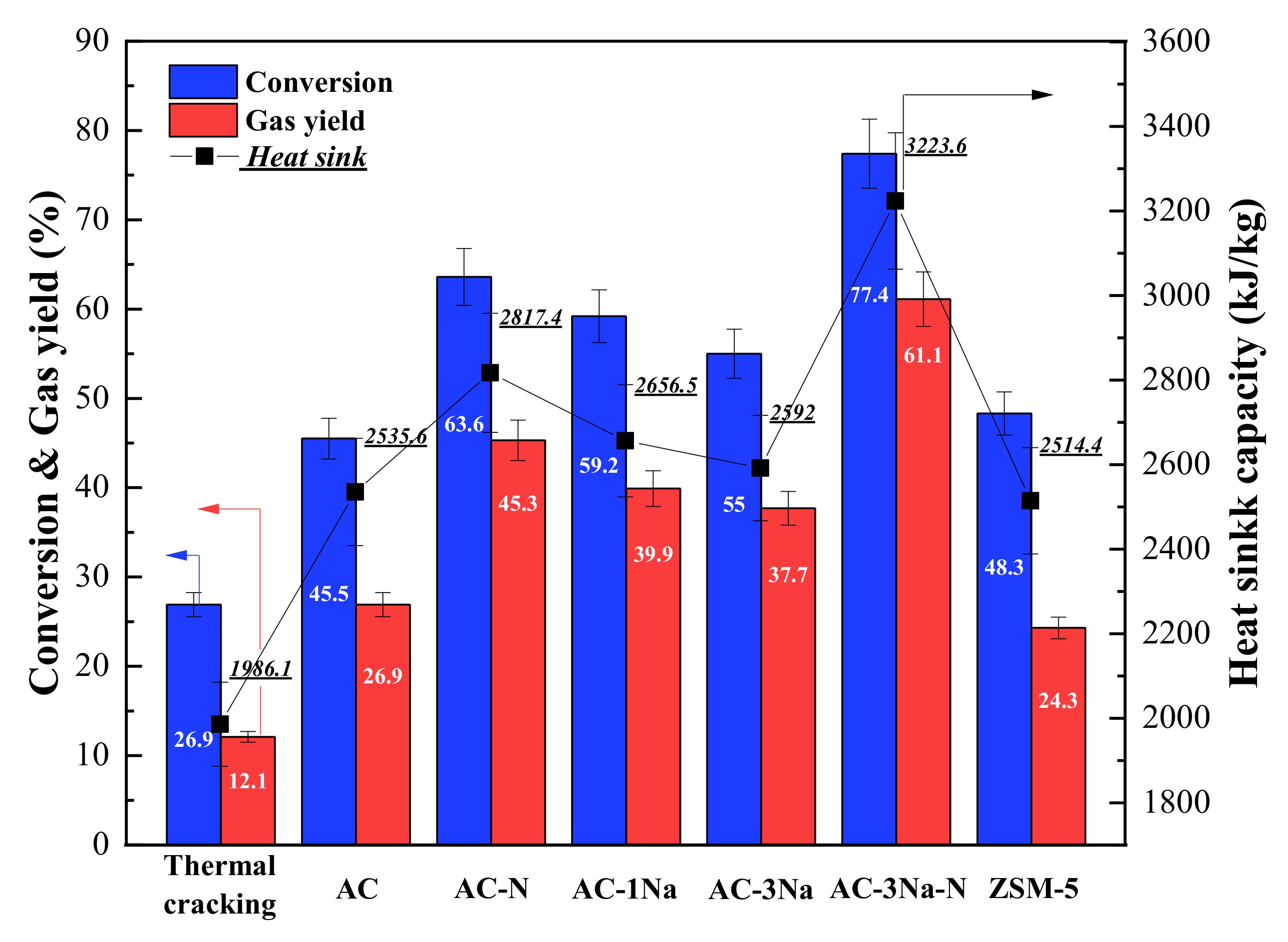
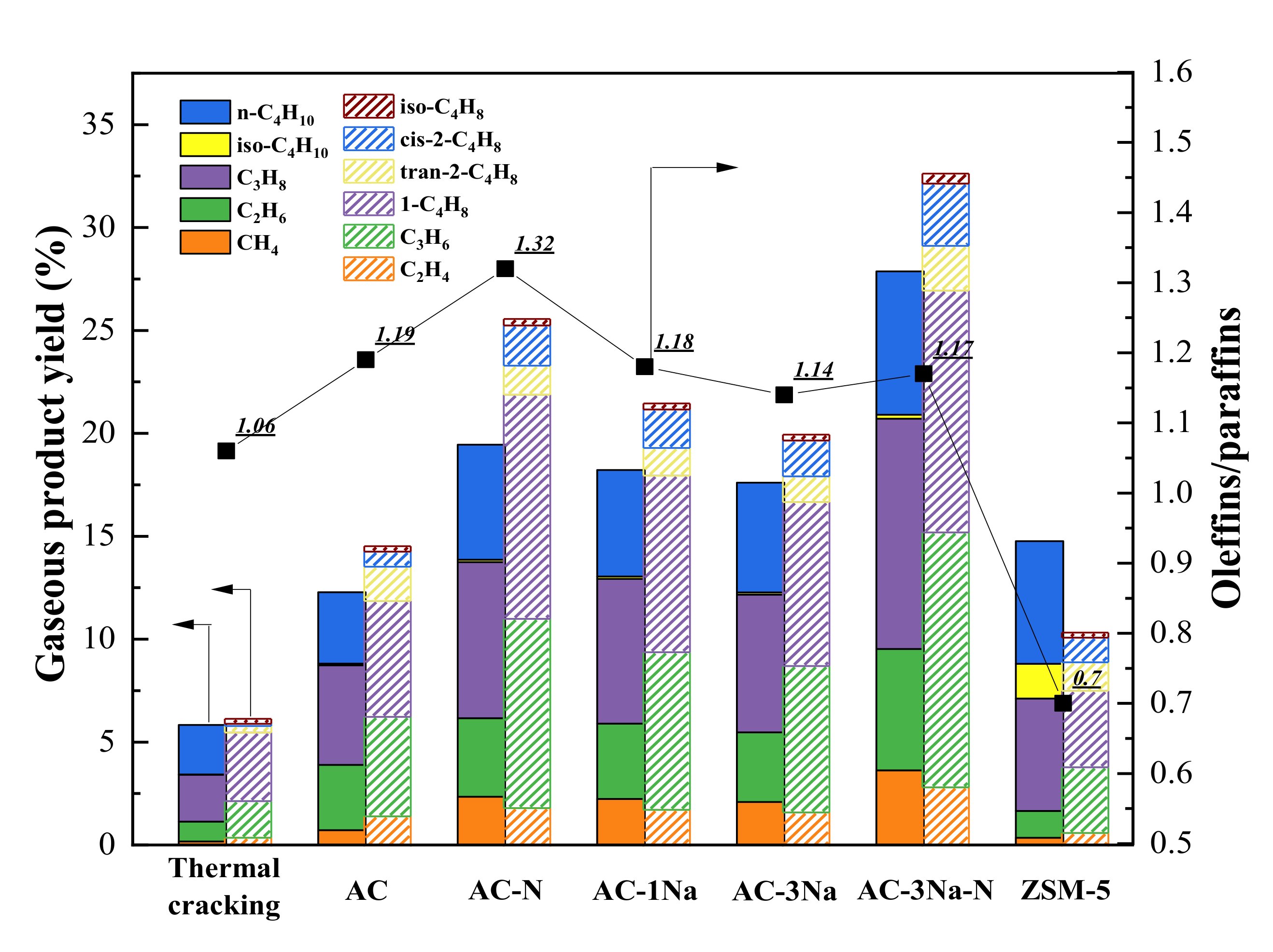
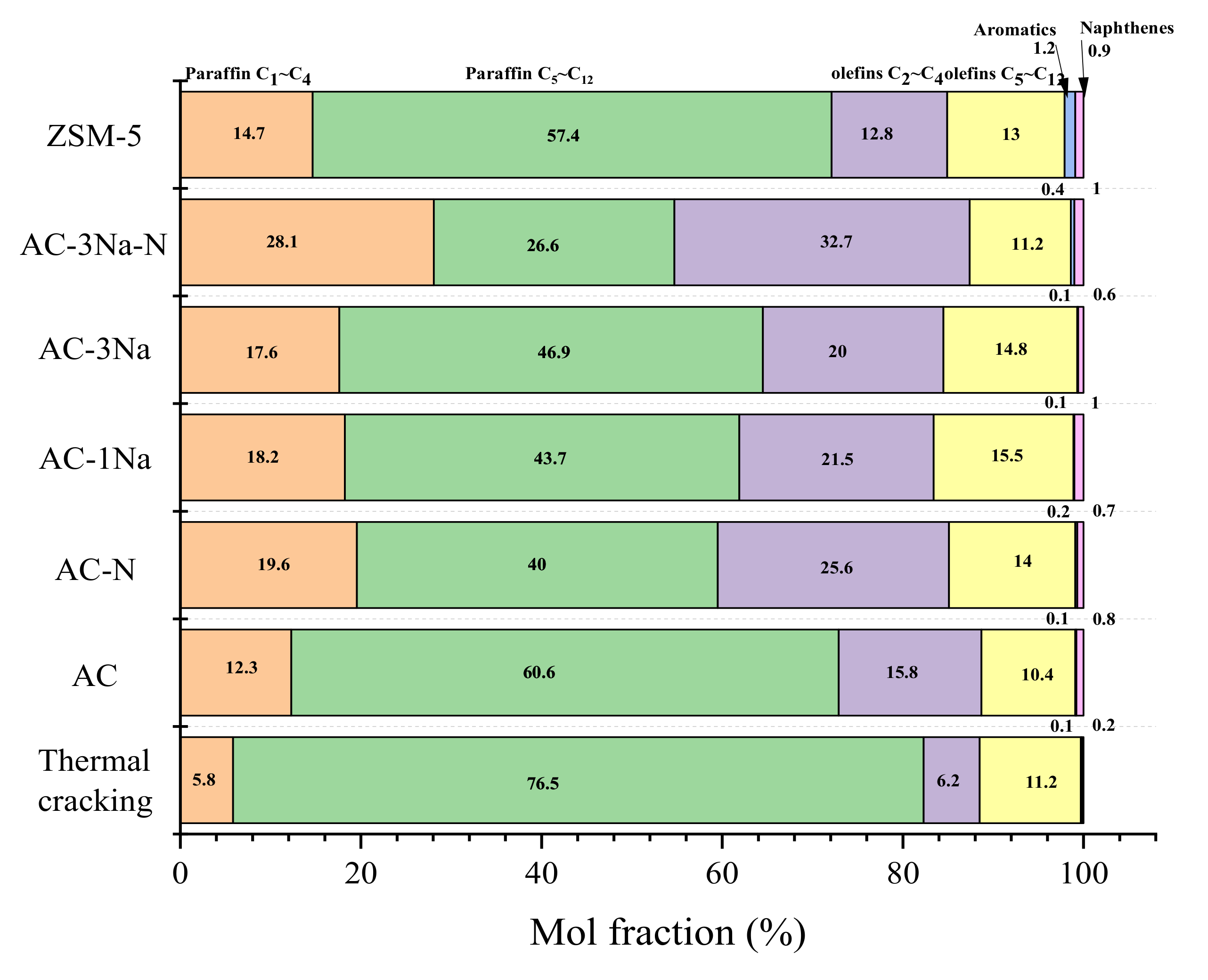
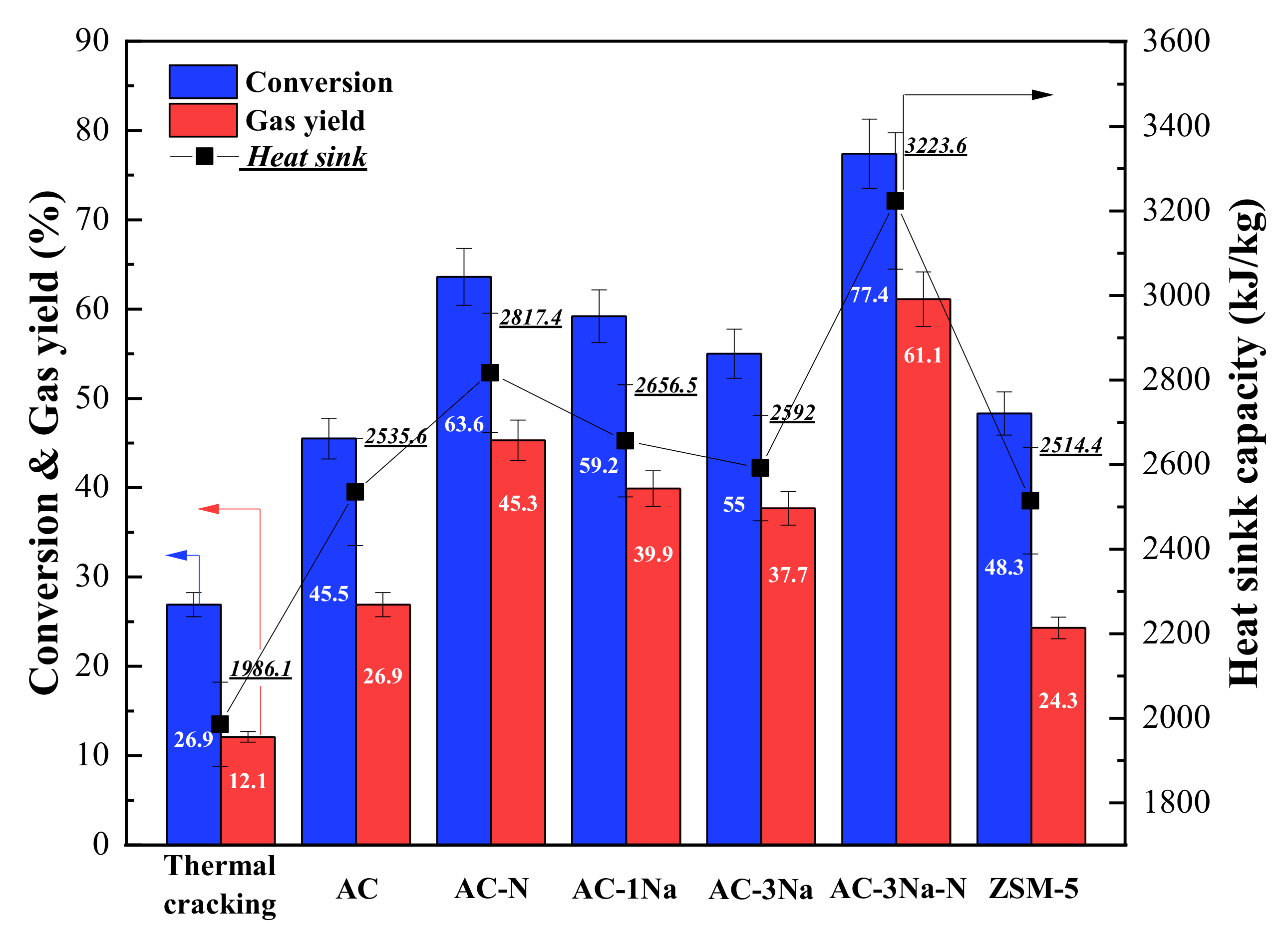
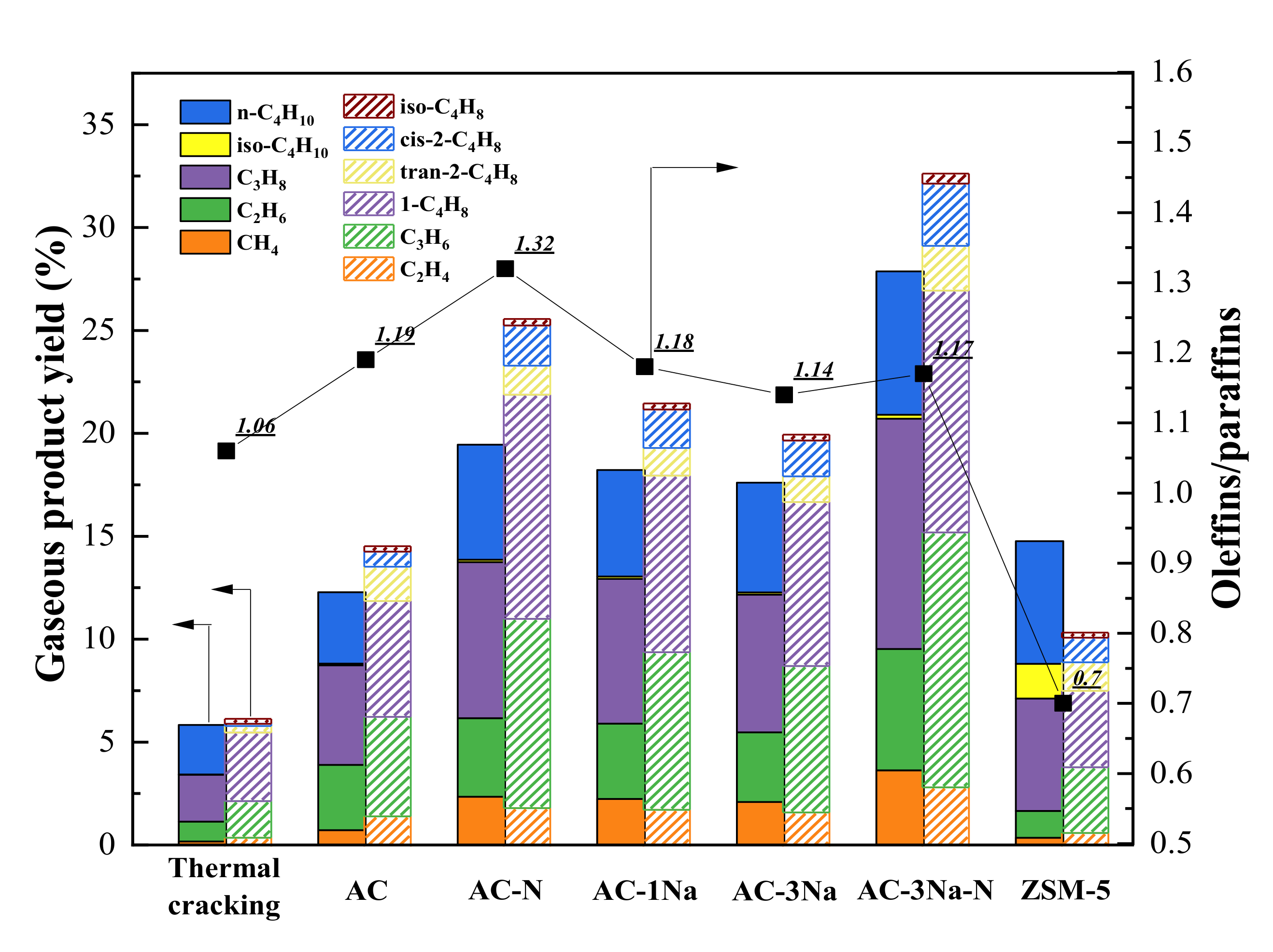
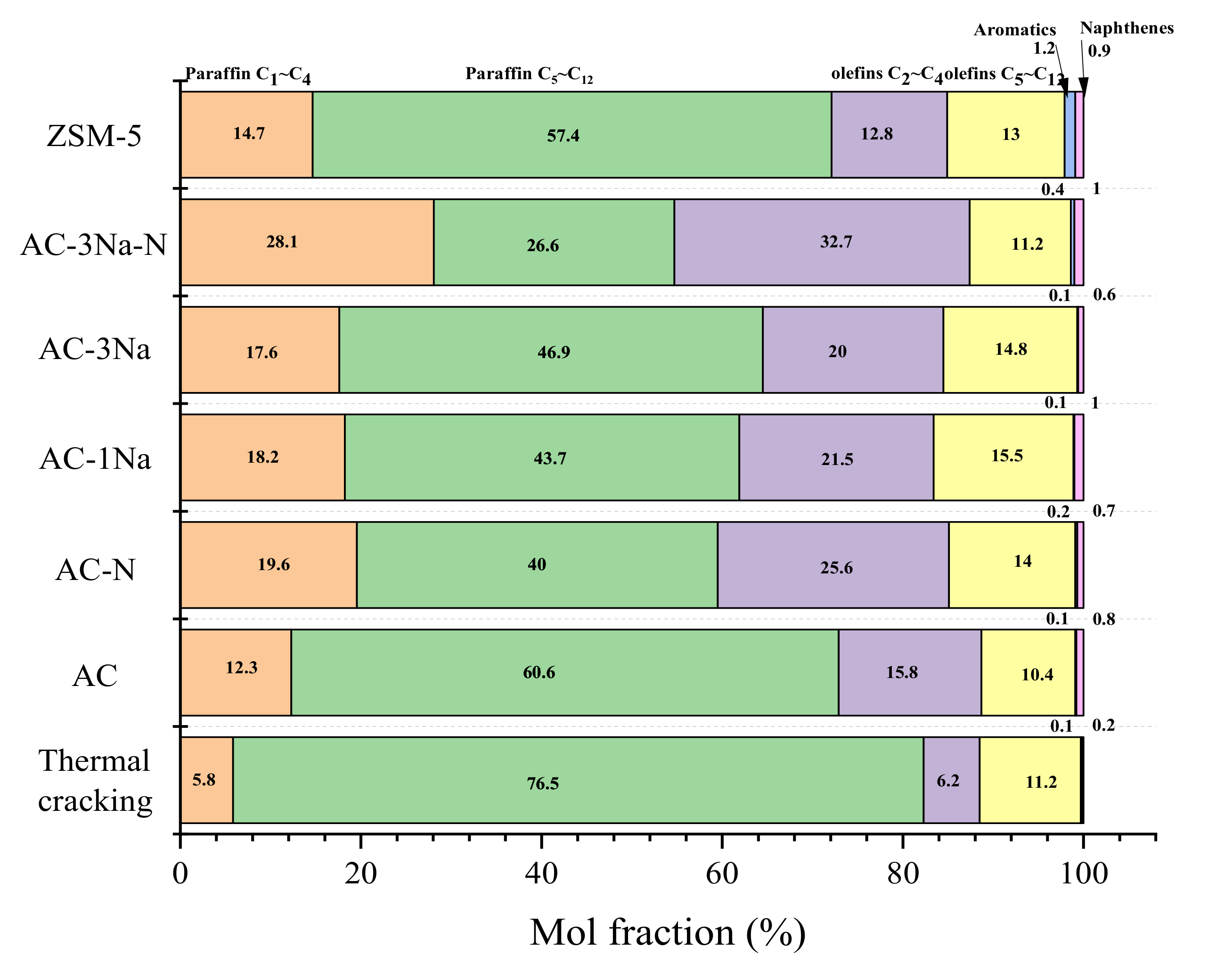
2.7. Cracking Activity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Catalyst Synthesis
4.3. Catalytic Activity Evaluation
4.4. Catalyst Characterization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Edwards, T. Liquid fuels and propellants for aerospace propulsion: 1903–2003. J. Propuls. Power 2003, 19, 1089–1107. [Google Scholar] [CrossRef]
- Kay, I.W.; Peschke, W.T.; Guile, R.N. Hydrocarbon-fueled scramjet combustor investigation. J. Propuls. Power 1992, 8, 507–512. [Google Scholar] [CrossRef]
- Edwards, T. Cracking and deposition behavior of supercritical hydrocarbon aviation fuels. Combust. Sci. Technol. 2006, 178, 307–334. [Google Scholar] [CrossRef]
- Edwards, T. Advancements in gas turbine fuels from 1943 to 2005. J. Eng. Gas Turbines Power 2007, 129, 13–20. [Google Scholar] [CrossRef]
- Puri, P.; Ma, F.; Choi, J.Y.; Yang, V. Ignition characteristics of cracked JP-7 fuel. Combust. Flame 2005, 142, 454–457. [Google Scholar] [CrossRef]
- Ritchie, A.W.; Nixon, A.C. Dehydrogenation of Methylcyclohexane over a Platinum-Alumina Catalyst in Absence of Added Hydrogen. Ind. Eng. Chem. Prod. Res. Dev. 1966, 5, 59–64. [Google Scholar] [CrossRef]
- Jiao, Y.; Liu, A.; Li, C.; Wang, J.; Zhu, Q.; Li, X.; Chena, Y. Catalytic cracking of RP-3 jet fuel over wall-coated Pt/ZrO2-TiO2-Al2O3 catalysts with different Al2O3 ratios. J. Anal. Appl. Pyrolysis 2015, 111, 100–107. [Google Scholar] [CrossRef]
- Xian, X.; Liu, G.; Zhang, X.; Wang, L.; Mi, Z. Catalytic cracking of n-dodecane over HZSM-5 zeolite under supercritical conditions: Experiments and kinetics. Chem. Eng. Sci. 2010, 65, 5588–5604. [Google Scholar] [CrossRef]
- Lee, S.; Lee, S.; Kumbhalkar, M.D.; Wiaderek, K.M.; Dumesic, J.; Winans, R.E. Effect of Particle Size upon Pt/SiO2 Catalytic Cracking of n-Dodecane under Supercritical Conditions: In situ SAXS and XANES Studies. ChemCatChem 2017, 9, 99–102. [Google Scholar] [CrossRef]
- Ji, Y.; Yang, H.; Yan, W. Effect of alkali metal cations modification on the acid/basic properties and catalytic activity of ZSM-5 in cracking of supercritical n-dodecane. Fuel 2019, 243, 155–161. [Google Scholar] [CrossRef]
- Li, S.; Wang, Z.; Zhang, H.; Liu, Z.; Wang, J.; Zhu, Q.; Li, X.; Chen, Y. The effects of MxOy (M = K, Ba, and Sr) promoters on inhibiting carbon deposit during catalytic cracking reactions. J. Anal. Appl. Pyrolysis 2017, 123, 269–277. [Google Scholar] [CrossRef]
- Liu, B.; Wang, Z.; Zhu, Q.; Li, X.; Wang, J. Performance of Pt/ZrO2–TiO2–Al2O3 and coke deposition during methylcyclohexane catalytic cracking. Fuel 2017, 200, 387–394. [Google Scholar] [CrossRef]
- Rownaghi, A.A.; Rezaei, F.; Hedlund, J. Selective formation of light olefin by n-hexane cracking over HZSM-5: Influence of crystal size and acid sites of nano- and micrometer-sized crystals. Chem. Eng. J. 2012, 191, 528–533. [Google Scholar] [CrossRef]
- Viswanadham, N.; Kamble, R.; Singh, M.; Kumar, M.; Murali Dhar, G. Catalytic properties of nano-sized ZSM-5 aggregates. Catal. Today 2009, 141, 182–186. [Google Scholar] [CrossRef]
- Ji, Y.; Yang, H.; Yan, W. Strategies to enhance the catalytic performance of ZSM-5 zeolite in hydrocarbon cracking: A review. Catalysts 2017, 7, 367. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, H.; Liu, B.; Zhu, Q.; Wang, J.; Li, X. Mo-promoted catalysts for supercritical n-decane cracking. Appl. Therm. Eng. 2016, 102, 1238–1240. [Google Scholar] [CrossRef]
- Jentoft, F.C.; Gates, B.C. Solid-acid-catalyzed alkane cracking mechanisms: Evidence from reactions of small probe molecules. Top. Catal. 1997, 4, 1–13. [Google Scholar] [CrossRef]
- Wojciechowski, B.W. The Reaction Mechanism of Catalytic Cracking: Quantifying Activity, Selectivity, and Catalyst Decay. Catal. Rev. 1998, 40, 209–328. [Google Scholar] [CrossRef]
- Gumming, K.A.; Wojciechowski, B.W. Hydrogen transfer, coke formation, and catalyst decay and their role in the chain mechanism of catalytic cracking. Catal. Rev.-Sci. Eng. 1996, 38, 101–157. [Google Scholar] [CrossRef]
- Boronat, M.; Corma, A. Are carbenium and carbonium ions reaction intermediates in zeolite-catalyzed reactions? Appl. Catal. A Gen. 2008, 336, 2–10. [Google Scholar] [CrossRef]
- Kissin, Y.V. Chemical Mechanisms of Catalytic Cracking over Solid Acidic Catalysts: Alkanes and Alkenes. Catal. Rev.-Sci. Eng. 2001, 43, 85–146. [Google Scholar] [CrossRef]
- Liu, C.; Gao, X.; Zhang, Z.; Zhang, H.; Sun, S.; Deng, Y. Surface modification of zeolite Y and mechanism for reducing naphtha olefin formation in catalytic cracking reaction. Appl. Catal. A Gen. 2004, 264, 225–228. [Google Scholar] [CrossRef]
- Abbot, J. Role of Brønsted and Lewis acid sites during cracking reactions of alkanes. Appl. Catal. 1989, 47, 33–44. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, B.J. Ammonia removal of activated carbon fibers produced by oxyfluorination. J. Colloid Interface Sci. 2005, 291, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Park, S.J. Effects of carbonyl group formation on ammonia adsorption of porous carbon surfaces. J. Colloid Interf. Sci. 2007, 311, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, P.R.; Daniel, L.A. A review: Organic matter and ammonia removal by biological activated carbon filtration for water and wastewater treatment. Int. J. Environ. Sci. Technol. 2020, 17, 591–606. [Google Scholar] [CrossRef]
- Bansal, R.C.; Goyal, M. Adsorptive removal of organic from water, activated carbon adsorption. In Activated Carbon Adsorption and Environment; Taylor: Boca Raton, FL, USA, 2005; pp. 297–372. [Google Scholar]
- Park, S.J.; Jin, S.Y. Effect of ozone treatment on ammonia removal of activated carbons. J. Colloid Interface Sci. 2005, 286, 417–419. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, Y.M. Influence of anodic treatment on heavy metal ion removal by activated carbon fibers. J. Colloid Interf. Sci. 2004, 278, 276–281. [Google Scholar] [CrossRef]
- Le Leuch, L.M.; Bandosz, T.J. The role of water and surface acidity on the reactive adsorption of ammonia on modified activated carbons. Carbon N. Y. 2007, 45, 568–578. [Google Scholar] [CrossRef]
- Zawadzki, J.; Wiśniewski, M. In situ characterization of interaction of ammonia with carbon surface in oxygen atmosphere. Carbon N. Y. 2003, 41, 2257–2267. [Google Scholar] [CrossRef]
- Kim, J.H.; Hwang, S.Y.; Park, J.E.; Lee, G.B.; Kim, H.; Kim, S.; Hong, B.U. Impact of the oxygen functional group of nitric acid-treated activated carbon on KOH activation reaction. Carbon Lett. 2019, 29, 281–287. [Google Scholar] [CrossRef]
- Datsyuk, V.; Kalyva, M.; Papagelis, K.; Parthenios, J.; Tasis, D.; Siokou, A.; Kallitsis, I.; Galiotis, C. Chemical oxidation of multiwalled carbon nanotubes. Carbon N. Y. 2008, 46, 833–840. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, Z.; Xu, K.; Zheng, X.; Gao, Q. Surface Modification and Performance of Activated Carbon Electrode Material. Acta Phys.-Chim. Sin. 2008, 24, 1143–1148. [Google Scholar] [CrossRef]
- Chingombe, P.; Saha, B.; Wakeman, R.J. Surface modification and characterisation of a coal-based activated carbon. Carbon N. Y. 2005, 43, 3132–3143. [Google Scholar] [CrossRef]
- Vinke, P.; van der Eijk, M.; Verbree, M.; Voskamp, A.F.; van Bekkum, H. Modification of the surfaces of a gasactivated carbon and a chemically activated carbon with nitric acid, hypochlorite, and ammonia. Carbon N. Y. 1994, 32, 675–686. [Google Scholar] [CrossRef]
- March, J. Advanced Organic Chemistry, 3rd ed.; John Wiley & Sons: New York, NY, USA, 1985; p. 1072. [Google Scholar]
- Ogata, Y. Oxidation in Organic Chemistry; Academic Press: New York, NY, USA, 1978; Volume SC, p. 295. [Google Scholar]
- Raymundo-Piñero, E.; Azaïs, P.; Cacciaguerra, T.; Cazorla-Amorós, D.; Linares-Solano, A.; Béguin, F. KOH and NaOH activation mechanisms of multiwalled carbon nanotubes with different structural organisation. Carbon N. Y. 2005, 43, 786–795. [Google Scholar] [CrossRef]
- Islam, M.A.; Ahmed, M.J.; Khanday, W.A.; Asif, M.; Hameed, B.H. Mesoporous activated carbon prepared from NaOH activation of rattan (Lacosperma secundiflorum) hydrochar for methylene blue removal. Ecotoxicol. Environ. Saf. 2017, 138, 279–285. [Google Scholar] [CrossRef]
- Ramesh, T.; Rajalakshmi, N.; Dhathathreyan, K.S. Synthesis and characterization of activated carbon from jute fibers for hydrogen storage. Renew. Energy Environ. Sustain. 2017, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.W.; Park, S.J.; Ryu, S.K. Effect of modification with HNO3 and NaOH on metal adsorption by pitch-based activated carbon fibers. Carbon N. Y. 2001, 39, 1635–1642. [Google Scholar] [CrossRef]
- Nian, Y.R.; Teng, H. Influence of surface oxides on the impedance behavior of carbon-based electrochemical capacitors. J. Electroanal. Chem. 2003, 540, 119–127. [Google Scholar] [CrossRef]
- Chiang, Y.C.; Lin, W.H.; Chang, Y.C. The influence of treatment duration on multi-walled carbon nanotubes functionalized by H 2 SO 4 /HNO 3 oxidation. Appl. Surf. Sci. 2011, 257, 2401–2410. [Google Scholar] [CrossRef]
- Beamson, G.; Briggs, D. High resolution XPS of organic polymers, the scienta ESCA A300 database, Wiley, Chichester 1992. Adv. Mater. 1993, 5, 778. [Google Scholar]
- De La Puente, G.; Pis, J.J.; Menéndez, J.A.; Grange, P. Thermal stability of oxygenated functions in activated carbons. J. Anal. Appl. Pyrolysis 1997, 43, 125–138. [Google Scholar] [CrossRef]
- Song, K.H.; Jeong, S.K.; Park, K.T.; Lee, K.Y.; Kim, H.J. Supercritical catalytic cracking of n-dodecane over air-oxidized activated charcoal. Fuel 2020, 276, 118010. [Google Scholar] [CrossRef]
- Biniak, S.; Szymański, G.; Siedlewski, J.; Światkoski, A. The characterization of activated carbons with oxygen and nitrogen surface groups. Carbon N. Y. 1997, 35, 1799–1810. [Google Scholar] [CrossRef]
- Lambert, J.B.; Shurvell, F.H.; Lightner, D.A.; Cooks, R.G. Organic Structural Spectroscopy; Prentice Hall: Upper Saddle River, NJ, USA, 1998. [Google Scholar]
- Vasu, A.E. Surface modification of activated carbon for enhancement of nickel(II) adsorption. E-J. Chem. 2008, 5, 814–819. [Google Scholar] [CrossRef] [Green Version]
- Fanning, P.E.; Vannice, M.A. A DRIFTS study of the formation of surface groups on carbon by oxidation. Carbon N. Y. 1993, 31, 721–730. [Google Scholar] [CrossRef]
- Park, S.H.; McClain, S.; Tian, Z.R.; Suib, S.L.; Karwacki, C. Surface and Bulk Measurements of Metals Deposited on Activated Carbon. Chem. Mater. 1997, 9, 176–183. [Google Scholar] [CrossRef]
- Zawadzki, J. Spektroskopia W Podczerwieni Zjawisk Powierzchniowych na Weglach (Infrared Spectroscopy of the Surface Phenomena on Carbons); UMK: Toruń, Poland, 1980. [Google Scholar]
- Socrates, G. Infrared Characteristic Group Frequencies; Wiley: Chichester, UK, 1994. [Google Scholar]
- Solum, M.S.; Pugmire, R.J.; Jagtoyen, M.; Derbyshire, F. Evolution of carbon structure in chemically activated wood. Carbon N. Y. 1995, 33, 1247–1254. [Google Scholar] [CrossRef]
- Barthos, R.; Lónyi, F.; Onyestyák, G.; Valyon, J. IR, FR, and TPD study on the acidity of H-ZSM-5, sulfated zirconia, and sulfated zirconia-titania using ammonia as the probe molecule. J. Phys. Chem. B 2000, 104, 7311–7319. [Google Scholar] [CrossRef]
- Shu, Y.; Sun, H.; Quan, X.; Chen, S. Enhancement of catalytic activity over the iron-modified Ce/TiO2 catalyst for selective catalytic reduction of NOx with ammonia. J. Phys. Chem. C 2012, 116, 25319–25327. [Google Scholar] [CrossRef]
- Gonçalves, M.; Sánchez-García, L.; Oliveira Jardim, E.; De Silvestre-Albero, J.; Rodríguez-Reinoso, F. Ammonia removal using activated carbons: Effect of the surface chemistry in dry and moist conditions. Environ. Sci. Technol. 2011, 45, 10605–10610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serp, P.; Machado, B. Nanostructured Carbon Materials for Catalysis; Royal Society of Chemistry: London, UK, 2015; ISBN 978-1-84973-909-2. [Google Scholar]
- Seo, M.G.; Kim, S.; Lee, D.W.; Jeong, H.E.; Lee, K.Y. Core-shell structured, nano-Pd-embedded SiO2-Al2O3 catalyst (Pd@SiO2-Al2O3) for direct hydrogen peroxide synthesis from hydrogen and oxygen. Appl. Catal. A Gen. 2016, 511, 87–94. [Google Scholar] [CrossRef]
- Guisnet, M.; Magnoux, P. Organic chemistry of coke formation. Appl. Catal. A Gen. 2001, 212, 83–96. [Google Scholar] [CrossRef]
- ASTM D6730-01; ASTM International: West Conshohocken, PA, USA, 2011.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBETa | Micropore S.A b | Mesopore S.A c | Vtotald | Vmicroe | Vmesof | Pg |
|---|---|---|---|---|---|---|---|
| (m2∙g-cat−1) | (cm3∙g-cat−1) | (nm) | |||||
| AC | 565 | 321 | 244 | 0.41 | 0.17 | 0.24 | 4.7 |
| AC-N | 538 | 299 | 239 | 0.39 | 0.16 | 0.23 | 4.6 |
| AC-1Na | 670 | 374 | 296 | 0.47 | 0.20 | 0.27 | 4.4 |
| AC-3Na | 1315 | 673 | 642 | 0.88 | 0.35 | 0.53 | 4.0 |
| AC-3Na-N | 983 | 460 | 523 | 0.63 | 0.24 | 0.39 | 3.5 |
| ZSM-5 | 294 | 248 | 46 | 0.18 | 0.12 | 0.06 | 2.5 |
| Sample | Acid Site Density | B/L ASD Ratio b | Acid Site Density | ||||
|---|---|---|---|---|---|---|---|
| Weak (α) | Medium (β) | Strong (γ) | Total a | Brønsted c | Lewis c | ||
| (mmol-NH3/g-cat) | (mmol-NH3/g-cat) | ||||||
| AC | 0.061 (7.1%) | 0.258 (30.0%) | 0.54 (62.9%) | 0.859 | 0.99 | 0.43 | 0.43 |
| AC-N | 0 (0%) | 0.376 (20.3%) | 1.473 (79.7%) | 1.849 | 1.30 | 1.04 | 0.81 |
| AC-1Na | 0.138 (10.3%) | 0.468 (35.0%) | 0.733 (54.7%) | 1.339 | 0.52 | 0.46 | 0.88 |
| AC-3Na | 0.057 (4.7%) | 0.574 (47.8%) | 0.571 (47.5%) | 1.202 | 0.74 | 0.51 | 0.69 |
| AC-3Na-N | 0.239 (8.8%) | 0.418 (15.3%) | 2.071 (75.9%) | 2.728 | 3.16 | 2.07 | 0.66 |
| ZSM-5 | 0.511 (52.6%) | 0.461 (47.4%) | 0 (0%) | 0.972 | 2.36 | 0.58 | 0.39 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, K.H.; Jeong, S.K.; Jeong, B.H.; Lee, K.-Y.; Kim, H.J. Acid/Base-Treated Activated Carbon Catalysts for the Low-Temperature Endothermic Cracking of N-Dodecane with Applications in Hypersonic Vehicle Heat Management Systems. Catalysts 2020, 10, 1149. https://doi.org/10.3390/catal10101149
Song KH, Jeong SK, Jeong BH, Lee K-Y, Kim HJ. Acid/Base-Treated Activated Carbon Catalysts for the Low-Temperature Endothermic Cracking of N-Dodecane with Applications in Hypersonic Vehicle Heat Management Systems. Catalysts. 2020; 10(10):1149. https://doi.org/10.3390/catal10101149
Chicago/Turabian StyleSong, Kyoung Ho, Soon Kwan Jeong, Byung Hun Jeong, Kwan-Young Lee, and Hak Joo Kim. 2020. "Acid/Base-Treated Activated Carbon Catalysts for the Low-Temperature Endothermic Cracking of N-Dodecane with Applications in Hypersonic Vehicle Heat Management Systems" Catalysts 10, no. 10: 1149. https://doi.org/10.3390/catal10101149