
KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer

 ,
, 
Abstract
:1. Introduction
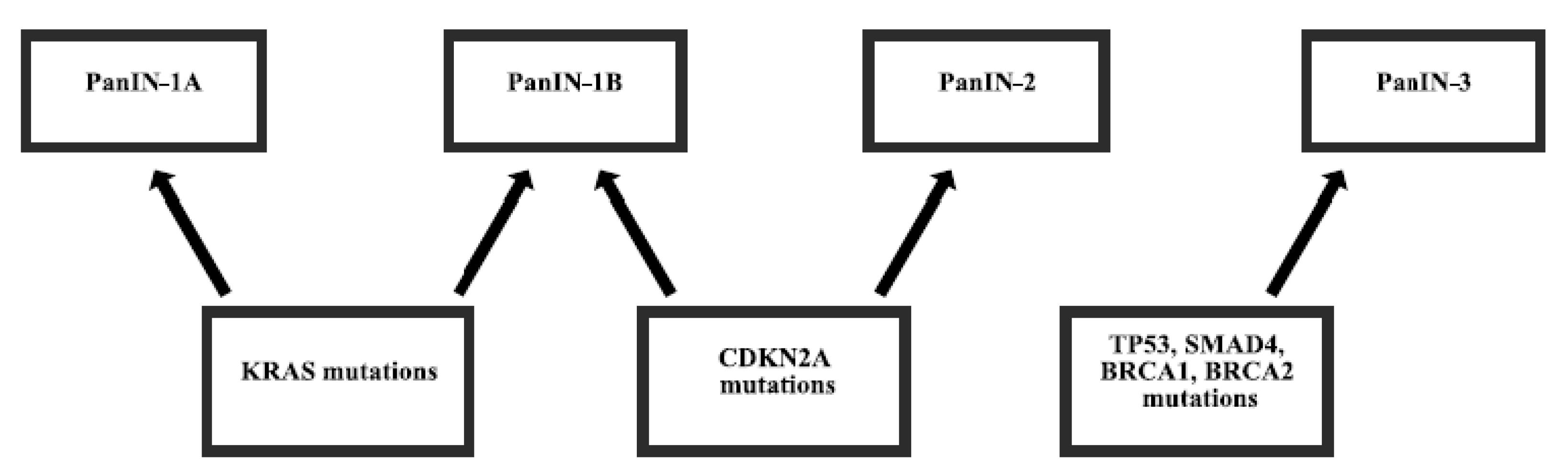
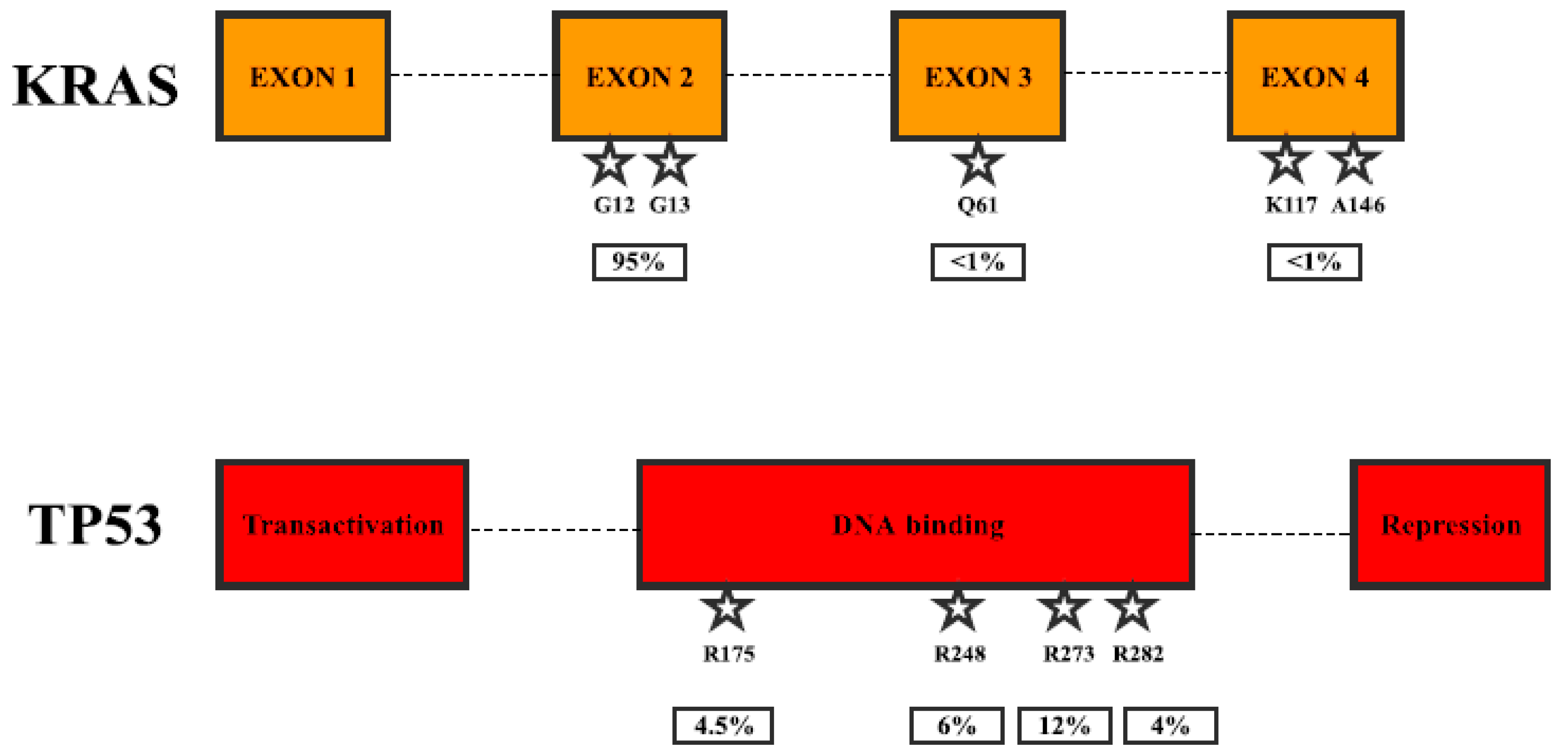
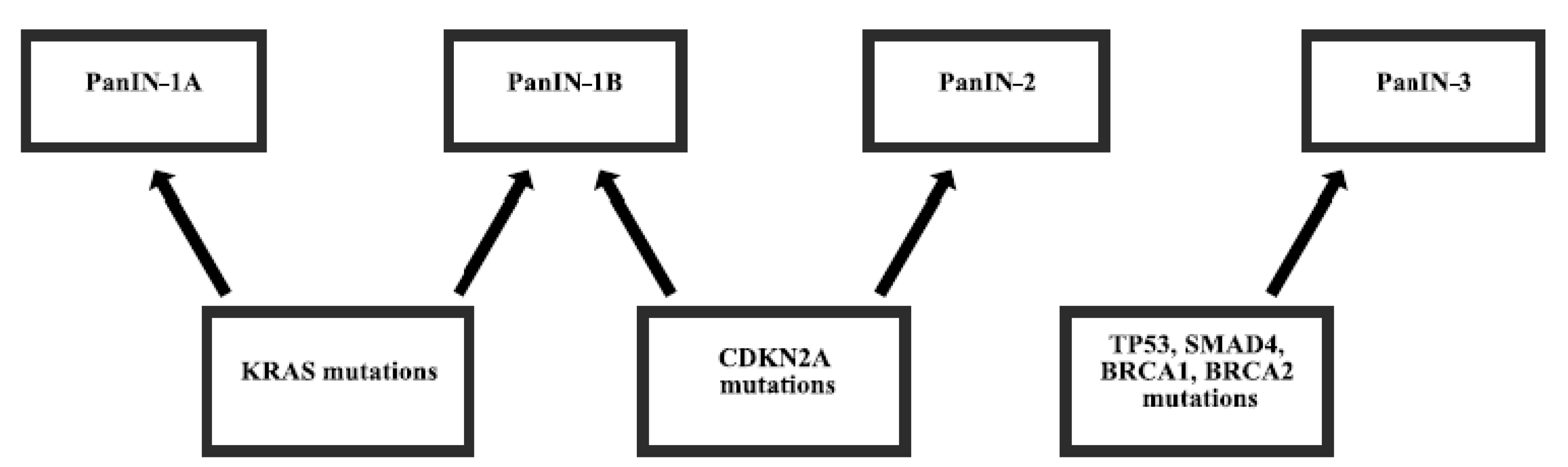
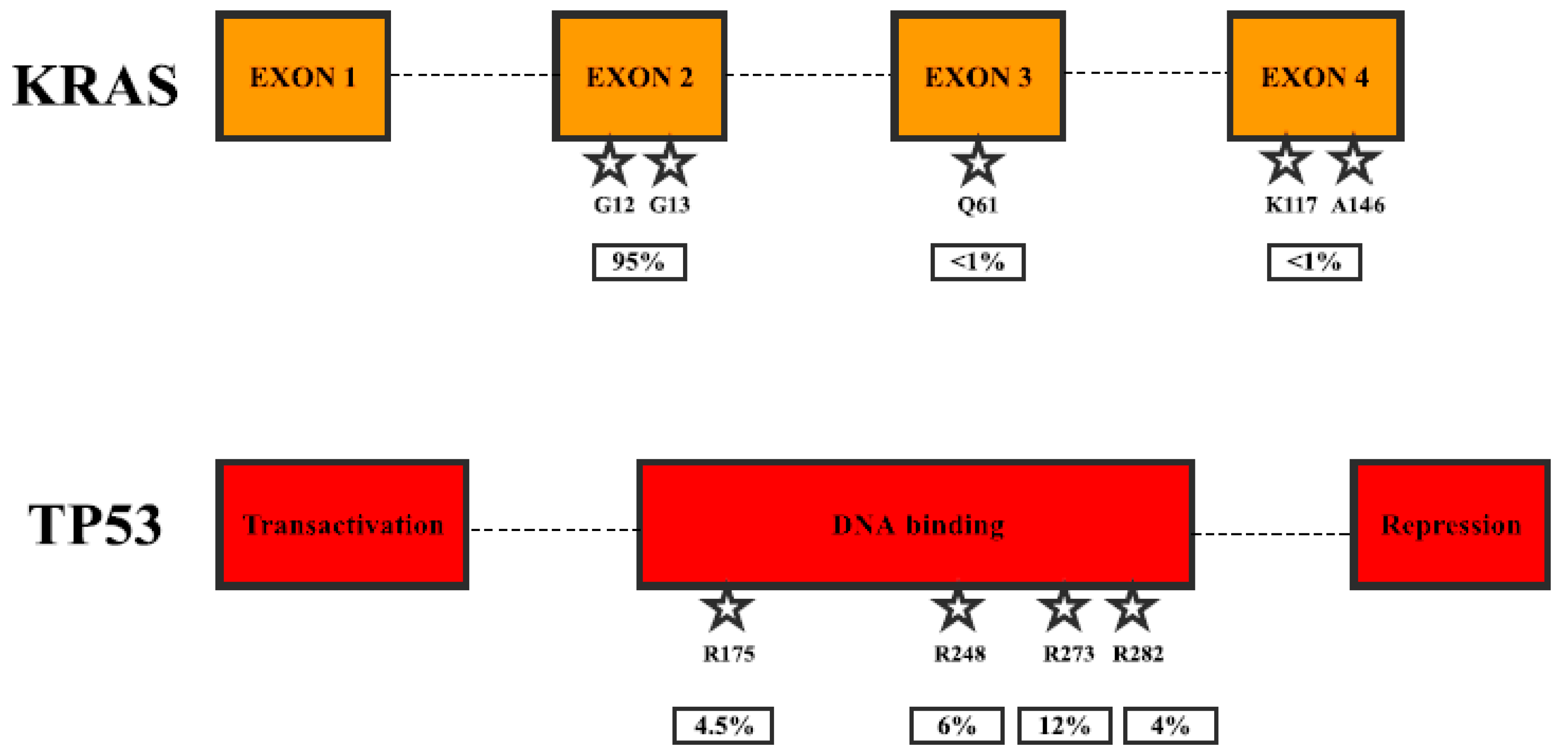
2. Gene Mutations in Pancreatic Cancer
3. Conclusions
Author Contributions
Conflicts of Interest
References
- American Cancer Society. Key Statistics for Pancreatic Cancer. How Common is Pancreatic Cancer? Available online: https://www.cancer.org/cancer/pancreatic-cancer/about/key-statistics.html (accessed on 16 February 2017).
- United European Gastroenterology. Pancreatic Cancer Set to Become Third Biggest Cancer Killer in EU Next Year. Available online: https://www.ueg.eu/press/releases/ueg-press-release/article/pancreatic-cancer-set-to-become-third-biggest-cancer-killer-in-eu-next-year/ (accessed on 16 February 2017).
- Carrato, A.; Falcone, A.; Ducreux, M.; Valle, J.W.; Parnaby, A.; Djazouli, K.; Alnwick-Allu, K.; Hutchings, A.; Palaska, C.; Parthenaki, I. A systematic review of the burden of pancreatic cancer in Europe: Real-world impact on survival, quality of life and costs. J. Gastrointest. Cancer 2015, 46, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Smit, V.T.; Boot, A.J.; Smits, A.M.; Fleuren, G.J.; Cornelisse, C.J.; Bos, J.L. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988, 16, 7773–7782. [Google Scholar] [CrossRef] [PubMed]
- Caldas, C.; Hahn, S.A.; da Costa, L.T.; Redston, M.S.; Schutte, M.; Seymour, A.B.; Weinstein, C.L.; Hruban, R.H.; Yeo, C.J.; Kern, S.E. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat. Genet. 1994, 8, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Redston, M.S.; Caldas, C.; Seymour, A.B.; Hruban, R.H.; da Costa, L.; Yeo, C.J.; Kern, S.E. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994, 54, 3025–3033. [Google Scholar] [PubMed]
- Maurice, D.; Pierreux, C.E.; Howell, M.; Wilentz, R.E.; Owen, M.J.; Hill, C.S. Loss of SMAD4 function in pancreatic tumors: C-terminal truncation leads to decreased stability. J. Boil. Chem. 2001, 276, 43175–43181. [Google Scholar] [CrossRef] [PubMed]
- Goggins, M.; Schutte, M.; Lu, J.; Moskaluk, C.A.; Weinstein, C.L.; Petersen, G.M.; Yeo, C.J.; Jackson, C.E.; Lynch, H.T.; Hruban, R.H.; et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996, 56, 5360–5364. [Google Scholar] [PubMed]
- Carnevale, J.; Ashworth, A. Assessing the significance of BRCA1 and BRCA2 mutations in pancreatic cancer. J. Clin. Oncol. 2015, 33, 3080–3081. [Google Scholar] [CrossRef] [PubMed]
- Marais, R.; Light, Y.; Paterson, H.F.; Marshall, C.J. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995, 14, 3136–3145. [Google Scholar] [PubMed]
- Gallo, A.; Cuozzo, C.; Esposito, I.; Maggiolini, M.; Bonofiglio, D.; Vivacqua, A.; Garramone, M.; Weiss, C.; Bohmann, D.; Musti, A.M. Menin uncouples Elk-1, JunD and c-Jun phosphorylation from MAP kinase activation. Oncogene 2002, 21, 6434–6445. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Tamosaitis, L.; Kvederaviciute, K.; Tarvydas, R.; Staniute, G.; Kalyan, K.; Meskinyte-Kausiliene, E.; Stankevicius, V.; Valius, M. KRAS, NRAS and BRAF mutations in colorectal cancer and melanoma. Med. Oncol. 2017, 34, 26. [Google Scholar] [CrossRef] [PubMed]
- Al-Kzayer, L.F.; Sakashita, K.; Al-Jadiry, M.F.; Al-Hadad, S.A.; Ghali, H.H.; Uyen, L.T.N.; Liu, T.; Matsuda, K.; Abdulkadhim, J.M.; Al-Shujairi, T.A.; et al. Analysis of KRAS and NRAS Gene mutations in Arab Asian Children with acute leukemia: High frequency of RAS mutations in acute lymphoblastic leukemia. Pediatr. Blood Cancer 2015, 62, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The cosmic (catalogue of somatic mutations in cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lim, D.H.; Jang, K.T.; Lim, T.; Lee, J.; Choi, Y.L.; Jang, H.L.; Yi, J.H.; Baek, K.K.; Park, S.H.; et al. Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol. Cancer Ther. 2011, 10, 1993–1999. [Google Scholar] [CrossRef] [PubMed]
- Boeck, S.; Jung, A.; Laubender, R.P.; Neumann, J.; Egg, R.; Goritschan, C.; Ormanns, S.; Haas, M.; Modest, D.P.; Kirchner, T.; et al. KRAS mutation status is not predictive for objective response to anti-EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J. Gastroenterol. 2013, 48, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. Rnai therapy targeting kras in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Kohno, T.; Ueno, H.; Hiraoka, N.; Kondo, S.; Saito, M.; Shimada, Y.; Ichikawa, H.; Kato, M.; Shibata, T.; et al. Utility of assessing the number of mutated KRAS, CDKN2A, Tp53, and SMAD4 genes using a targeted deep sequencing assay as a prognostic biomarker for pancreatic cancer. Pancreas 2017, 46, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, E.S.; Jones, J.B.; Ashfaq, R.; Adsay, V.; Baker, S.J.; Valentine, V.; Hempen, P.M.; Hilgers, W.; Yeo, C.J.; Hruban, R.H.; et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: Potential therapeutic targets. Am. J. Pathol. 2003, 163, 1255–1260. [Google Scholar] [CrossRef]
- Levy, N.; Yonish-Rouach, E.; Oren, M.; Kimchi, A. Complementation by wild-type p53 of interleukin-6 effects on m1 cells: Induction of cell cycle exit and cooperativity with c-Myc suppression. Mol. Cell. Boil. 1993, 13, 7942–7952. [Google Scholar] [CrossRef]
- Bates, S.; Ryan, K.M.; Phillips, A.C.; Vousden, K.H. Cell cycle arrest and DNA endoreduplication following p21WAF1/CIP1 expression. Oncogene 1998, 17, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Gordon, E.M.; Anderson, W.F.; Parekh, D. Gene therapy for primary and metastatic pancreatic cancer with intraperitoneal retroviral vector bearing the wild-type p53 gene. Surgery 1998, 124, 143–150. [Google Scholar] [CrossRef]
- Kern, S.E.; Pietenpol, J.A.; Thiagalingam, S.; Seymour, A.; Kinzler, K.W.; Vogelstein, B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science 1992, 256, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Grochola, L.F.; Taubert, H.; Greither, T.; Bhanot, U.; Udelnow, A.; Wurl, P. Elevated transcript levels from the MDM2 p1 promoter and low p53 transcript levels are associated with poor prognosis in human pancreatic ductal adenocarcinoma. Pancreas 2011, 40, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.F.; Wang, W.Q.; Liu, L.; Xu, H.X.; Wu, C.T.; Yang, J.X.; Qi, Z.H.; Wang, Y.Q.; Xu, J.; Liu, C.; et al. Mutant p53 determines pancreatic cancer poor prognosis to pancreatectomy through upregulation of cavin-1 in patients with preoperative serum CA19-9 ≥ 1000 u/mL. Sci. Rep. 2016, 6, 19222. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.F.; Wang, W.Q.; Liu, L.; Xu, H.X.; Wu, C.T.; Yang, J.X.; Qi, Z.H.; Wang, Y.Q.; Xu, J.; Liu, C.; et al. Retraction: Mutant p53 determines pancreatic cancer poor prognosis to pancreatectomy through upregulation of cavin-1 in patients with preoperative serum CA19-9 ≥ 1000 u/mL. Sci. Rep. 2016, 6, 25115. [Google Scholar] [CrossRef] [PubMed]
- Ormanns, S.; Siveke, J.T.; Heinemann, V.; Haas, M.; Sipos, B.; Schlitter, A.M.; Esposito, I.; Jung, A.; Laubender, R.P.; Kruger, S.; et al. Perk, PAKT and p53 as tissue biomarkers in erlotinib-treated patients with advanced pancreatic cancer: A translational subgroup analysis from AIO-PK0104. BMC Cancer 2014, 14, 624. [Google Scholar] [CrossRef] [PubMed]
- Hussussian, C.J.; Struewing, J.P.; Goldstein, A.M.; Higgins, P.A.; Ally, D.S.; Sheahan, M.D.; Clark, W.H., Jr.; Tucker, M.A.; Dracopoli, N.C. Germline p16 mutations in familial melanoma. Nat. Genet. 1994, 8, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.; Gruis, N.A.; Frants, R.R.; van Der Velden, P.A.; Hille, E.T.; Bergman, W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-leiden). Int. J. Cancer 2000, 87, 809–811. [Google Scholar] [CrossRef]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the RB/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar] [PubMed]
- Chen, J.; Li, D.; Killary, A.M.; Sen, S.; Amos, C.I.; Evans, D.B.; Abbruzzese, J.L.; Frazier, M.L. Polymorphisms of p16, p27, p73, and MDM2 modulate response and survival of pancreatic cancer patients treated with preoperative chemoradiation. Ann. Surg. Oncol. 2009, 16, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Ghiorzo, P.; Gargiulo, S.; Nasti, S.; Pastorino, L.; Battistuzzi, L.; Bruno, W.; Bonelli, L.; Taveggia, P.; Pugliese, V.; Borgonovo, G.; et al. Predicting the risk of pancreatic cancer: On CDKN2A mutations in the melanoma-pancreatic cancer syndrome in Italy. J. Clin. Oncol. 2007, 25, 5336–5337. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Tian, L.; Feng, Y.; Yi, M.; Chen, X.; Huang, Q. The predictive role of p16 deletion, p53 deletion, and polysomy 9 and 17 in pancreatic ductal adenocarcinoma. Pathol. Oncol. Res. 2013, 19, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.L.; Turnacioglu, K.K.; Schutte, M.; Sugar, A.Y.; Kern, S.E. DPC4 transcriptional activation and dysfunction in cancer cells. Cancer Res. 1998, 58, 4592–4597. [Google Scholar] [PubMed]
- Cao, D.; Ashfaq, R.; Goggins, M.G.; Hruban, R.H.; Kern, S.E.; Iacobuzio-Donahue, C.A. Differential expression of multiple genes in association with MADH4/DPC4/SMAD4 inactivation in pancreatic cancer. Int. J. Clin. Exp. Pathol. 2008, 1, 510–517. [Google Scholar] [PubMed]
- De Bosscher, K.; Hill, C.S.; Nicolas, F.J. Molecular and functional consequences of SMAD4 c-terminal missense mutations in colorectal tumour cells. Biochem. J. 2004, 379, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.L.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Eshleman, J.R.; et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin. Cancer Res. 2009, 15, 4674–4679. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Srinivasan, R.; Wig, J.D. SMAD4 genetic alterations predict a worse prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas 2012, 41, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Boulton, S.J. Brca1-mediated ubiquitylation. Cell Cycle 2006, 5, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.X.; Wang, R.H. Roles of BRCA1 in DNA damage repair: A link between development and cancer. Hum. Mol. Genet. 2003, R113–R123. [Google Scholar] [CrossRef]
- Xu, C.F.; Solomon, E. Mutations of the BRCA1 gene in human cancer. Semin. Cancer Boil. 1996, 7, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Stadler, Z.K.; Salo-Mullen, E.; Patil, S.M.; Pietanza, M.C.; Vijai, J.; Saloustros, E.; Hansen, N.A.; Kauff, N.D.; Kurtz, R.C.; Kelsen, D.P.; et al. Prevalence of brca1 and BRCA2 mutations in ashkenazi jewish families with breast and pancreatic cancer. Cancer 2012, 118, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Lohse, I.; Borgida, A.; Cao, P.; Cheung, M.; Pintilie, M.; Bianco, T.; Holter, S.; Ibrahimov, E.; Kumareswaran, R.; Bristow, R.G.; et al. BRCA1 and BRCA2 mutations sensitize to chemotherapy in patient-derived pancreatic cancer xenografts. Br. J. Cancer 2015, 113, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Rudkin, T.M.; Foulkes, W.D. BRCA2: Breaks, mistakes and failed separations. Trends Mol. Med. 2005, 11, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Friedenson, B. BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian. MedGenMed 2005, 7, 60. [Google Scholar] [PubMed]
- Welcsh, P.L.; King, M.C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, C.; Yu, D.; Wang, C.; Che, X.; Miao, X.; Zhai, K.; Chang, J.; Jiang, G.; Yang, X.; et al. Identification of common variants in BRCA2 and MAP2K4 for susceptibility to sporadic pancreatic cancer. Carcinogenesis 2013, 34, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Kadouri, L.; Appelbaum, L.; Peretz, T.; Sagi, M.; Goldberg, Y.; Hubert, A. Complete remission, in BRCA2 mutation carrier with metastatic pancreatic adenocarcinoma, treated with cisplatin based therapy. Cancer Boil. Ther. 2011, 12, 165–168. [Google Scholar] [CrossRef]
- James, E.; Waldron-Lynch, M.G.; Saif, M.W. Prolonged survival in a patient with BRCA2 associated metastatic pancreatic cancer after exposure to camptothecin: A case report and review of literature. Anticancer Drugs 2009, 20, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, P.; Kurtin, S.; Dragovich, T. Response to a third-line mitomycin C (MMC)-based chemotherapy in a patient with metastatic pancreatic adenocarcinoma carrying germline BRCA2 mutation. JOP 2008, 9, 305–308. [Google Scholar] [PubMed]
- Meldrum, C.; Doyle, M.A.; Tothill, R.W. Next-generation sequencing for cancer diagnostics: A practical perspective. Clin. Biochem. Rev. 2011, 32, 177–195. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. https://doi.org/10.3390/cancers9050042
Cicenas J, Kvederaviciute K, Meskinyte I, Meskinyte-Kausiliene E, Skeberdyte A, Cicenas J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers. 2017; 9(5):42. https://doi.org/10.3390/cancers9050042
Chicago/Turabian StyleCicenas, Jonas, Kotryna Kvederaviciute, Ingrida Meskinyte, Edita Meskinyte-Kausiliene, Aiste Skeberdyte, and Jonas Cicenas. 2017. "KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer" Cancers 9, no. 5: 42. https://doi.org/10.3390/cancers9050042
APA StyleCicenas, J., Kvederaviciute, K., Meskinyte, I., Meskinyte-Kausiliene, E., Skeberdyte, A., & Cicenas, J. (2017). KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers, 9(5), 42. https://doi.org/10.3390/cancers9050042