
Targeting the ATR-CHK1 Axis in Cancer Therapy

Abstract
:1. Introduction
2. The Role of ATR and CHK1 in the DDR
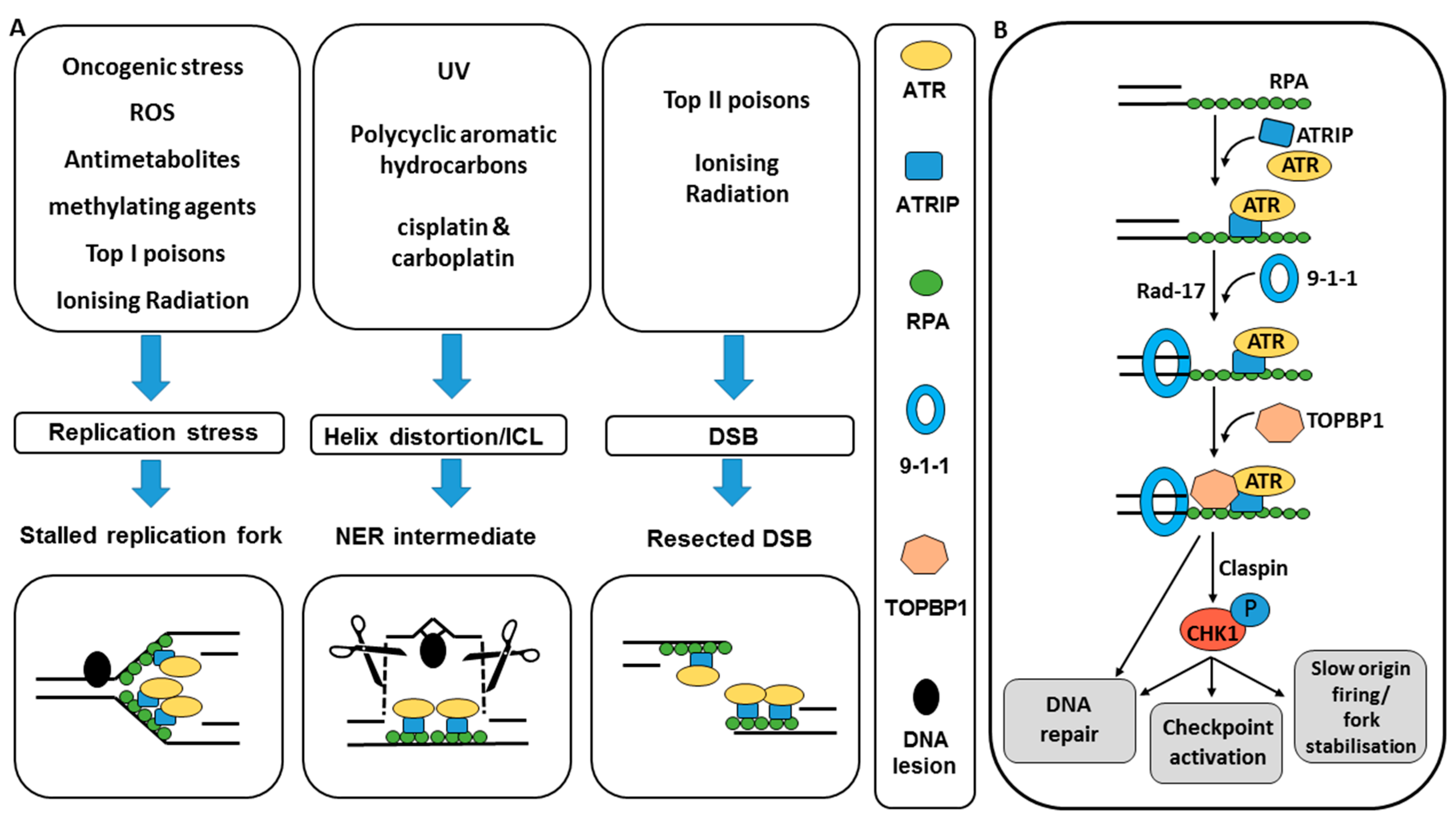
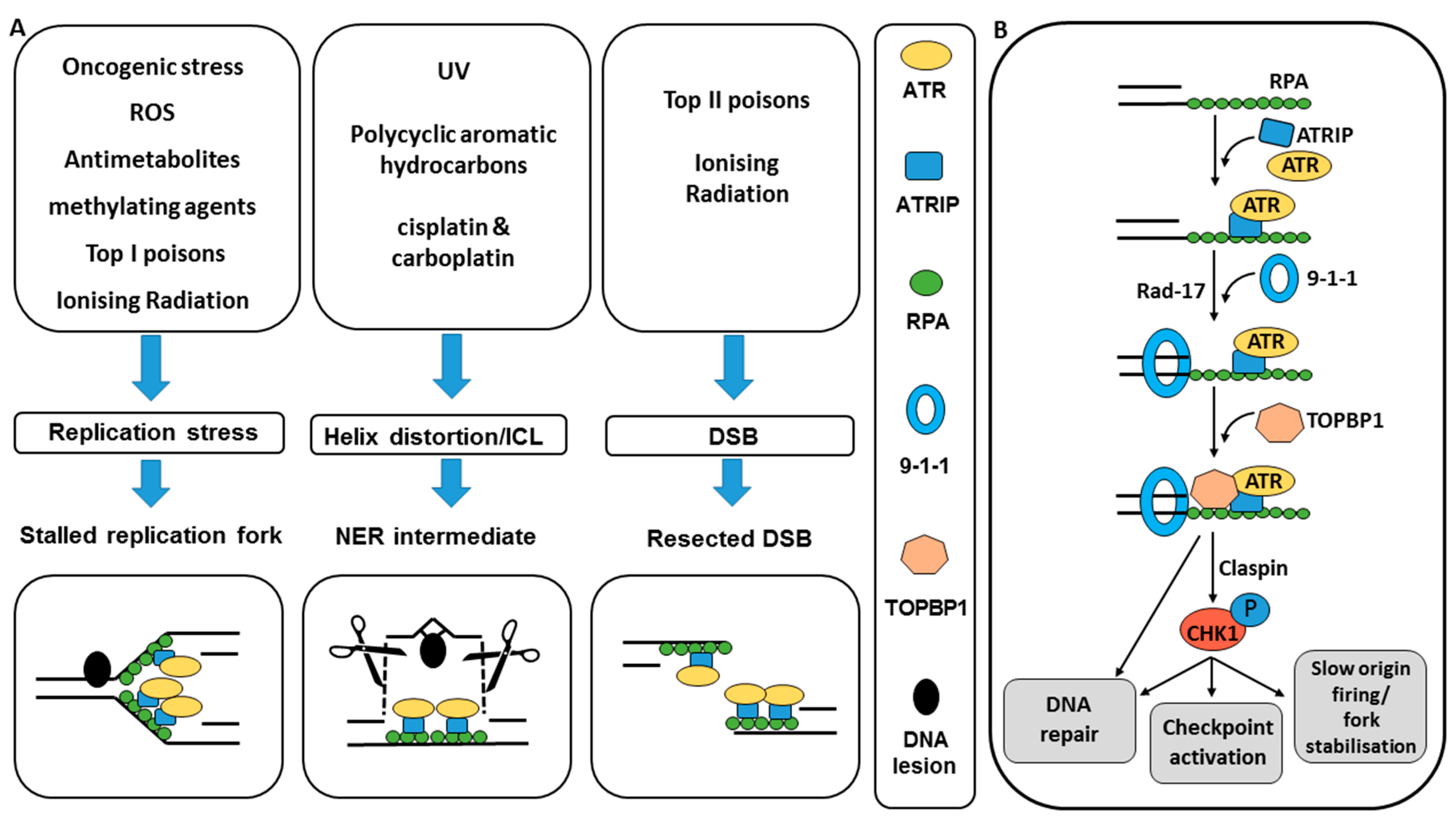
2.1. ATR and CHK1 Activation by DNA Damage
2.2. ATR and CHK1 Signal to DNA Damage Checkpoints
2.3. ATR and CHK1 Signal to Reduce Replication Stress
2.4. ATR and CHK1 Signal to DNA Repair
3. Validation of Target and Rationale for Cancer Specificity
4. Development of ATR and CHK1 Inhibitors
4.1. CHK1 Inhibitors
4.2. ATR Inhibitors
5. Pre-Clinical Data: Chemo- and Radio-Sensitisation In Vitro and In Vivo
5.1. Combinations with Chemotherapy Agents
5.1.1. ATR and CHK1 Inhibitors in Combination with Antimetabolite Drugs In Vitro
5.1.2. ATR and CHK1 Inhibitors in Combination with Topoisomerase Poisons In Vitro
5.1.3. ATR Inhibitors in Combination with Platinum-Based Chemotherapy Agents In Vitro
5.1.4. CHK1 Inhibitors in Combination with Platinum Agents In Vitro
5.1.5. CHK1 Inhibitors in Combination with Taxanes In Vitro
5.1.6. ATR and CHK1 Inhibitor- Cytotoxic Drug Combinations In Vivo
5.2. Combinations with Ionising Radiation (IR)
5.2.1. Radiopotentiation and Chemo-Radiopotentiation by CHK1 Inhibitors In Vitro
5.2.2. Radiopotentiation and Chemo-Radiopotentiation by CHK1 Inhibitors In Vivo
5.2.3. Radiopotentiation and Chemo-Radiopotentiation by ATR Inhibitors In Vitro and In Vivo
6. Combinations with Other Molecular Targeted Agents
7. Single Agent Activity and Determinants of Sensitivity
7.1. Replication Stress
7.2. DNA Damage Response
8. ATR and CHK1 Inhibitors in Clinical Trials
9. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan Coyne, G.; Chen, A.P.; Meehan, R.; Doroshow, J.H. PARP Inhibitors in Reproductive System Cancers: Current Use and Developments. Drugs 2017, 77, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. G1 cell-cycle control and cancer. Nature 2004, 432, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Takai, H.; Tominaga, K.; Motoyama, N.; Minamishima, Y.A.; Nagahama, H.; Tsukiyama, T.; Ikeda, K.; Nakayama, K.; Nakanishi, M.; Nakayama, K. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev. 2000, 14, 1439–1447. [Google Scholar] [PubMed]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar] [PubMed]
- Cliby, W.A.; Roberts, C.J.; Cimprich, K.A.; Stringer, C.M.; Lamb, J.R.; Schreiber, S.L.; Friend, S.H. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998, 17, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- MacDougall, C.A.; Byun, T.S.; Van, C.; Yee, M.; Cimprich, K.A. The structural determinants of checkpoint activation. Genes Dev. 2007, 21, 898–903. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Lindsey-Boltz, L.A.; Kemp, M.; Mason, A.C.; Wold, M.S.; Sancar, A. Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 13660–13665. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.L.; Ehrhardt, M.R.; Mordes, D.A.; Glick, G.G.; Chazin, W.J.; Cortez, D. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol. Cell. Biol. 2007, 27, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, B.; Zou, L. ATR signaling at a glance. J. Cell Sci. 2009, 122, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Walworth, N.C.; Bernards, R. Rad-dependent response of the CHK1-encoded protein kinase at the DNA damage checkpoint. Science 1996, 271, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Walworth, N.C.; Capasso, H.; Dunaway, S.; Liu, H.Y.; Rao, H.; Wan, S.H. DNA damage checkpoint signaling through the protein kinase Chk1. FASEB J. 2000, 14, A1582. [Google Scholar]
- Liu, Q.; Guntuku, S.; Cui, X.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [PubMed]
- Liu, S.; Bekker-Jensen, S.; Mailand, N.; Lukas, C.; Bartek, J.; Lukas, J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol. Cell. Biol. 2006, 26, 6056–6064. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Dunphy, W.G. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell 2000, 6, 839–849. [Google Scholar] [CrossRef]
- Wang, X.; Zou, L.; Lu, T.; Bao, S.; Hurov, K.E.; Hittelman, W.N.; Elledge, S.J.; Li, L. Rad17 phosphorylation is required for claspin recruitment and Chk1 activation in response to replication stress. Mol. Cell 2006, 23, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Smits, V.A.; Gillespie, D.A. DNA damage control: Regulation and functions of checkpoint kinase 1. FEBS J. 2015, 282, 3681–3692. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.S.; Syljuasen, R.G. Safeguarding genome integrity: The checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012, 40, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Ryan, C.E.; Piwnica-Worms, H. Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14–3-3 binding. Mol. Cell. Biol. 2003, 23, 7488–7497. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Grant, S. New Insights into Checkpoint Kinase 1 in the DNA Damage Response Signaling Network. Clin. Cancer Res. 2010, 16, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kumagai, A.; Dunphy, W.G. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol. Biol. Cell 2001, 12, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.A.; Cortez, D. AIR signalling: More than meeting at the fork. Biochem. J. 2011, 436, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.S.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef]
- Paulsen, R.D.; Cimprich, K.A. The ATR pathway: Fine-tuning the fork. DNA Repair 2007, 6, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Shigechi, T.; Tomida, J.; Sato, K.; Kobayashi, M.; Eykelenboom, J.K.; Pessina, F.; Zhang, Y.; Uchida, E.; Ishiai, M.; Lowndes, N.F.; et al. ATR-ATRIP Kinase Complex Triggers Activation of the Fanconi Anemia DNA Repair Pathway. Cancer Res. 2012, 72, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Shell, S.M.; Liu, Y.; Zou, Y. ATR-dependent checkpoint modulates XPA nuclear import in response to UV irradiation. Oncogene 2007, 26, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, R.S.; Cortez, D.; Brumbaugh, K.M.; Scully, R.; Livingston, D.; Elledge, S.J.; Abraham, R.T. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000, 14, 2989–3002. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuåsen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Peasland, A.; Wang, L.Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Parsels, L.A.; Morgan, M.A.; Tanska, D.M.; Parsels, J.D.; Palmer, B.D.; Booth, R.J.; Denny, W.A.; Canman, C.E.; Kraker, A.J.; Lawrence, T.S.; et al. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol. Cancer Ther. 2009, 8, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [PubMed]
- Haaf, T.; Golub, E.I.; Reddy, G.; Radding, C.M.; Ward, D.C. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. USA 1995, 92, 2298–2302. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Niraj, J.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol. Cell 2017, 65, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hartwell, L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell 1992, 71, 543–546. [Google Scholar] [CrossRef]
- Sherr, C.J. Cancer Cell Cycles. Science 1996, 274, 1672. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Oncogenes and Cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Ruiz-Perez, V.L.; Woods, C.G.; Jeggo, P.A.; Goodship, J.A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 2003, 33, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Cliby, W.A.; Lewis, K.A.; Lilly, K.K.; Kaufmann, S.H. S Phase and G2 Arrests Induced by Topoisomerase I Poisons Are Dependent on ATR Kinase Function. J. Biol. Chem. 2002, 277, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.; Park, P.K.; Kim, Y.; Vaziri, C.; Schreiber, S.L. ATR inhibition selectively sensitizes G(1) checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. USA 2001, 98, 9092–9097. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Broggini, M.; Erba, E.; Damia, G. Chk1, but not Chk2, is Involved in the Cellular Response to DNA Damaging Agents: Differential Activity in Cells Expressing, or not, p53. Cell Cycle 2004, 3, 1175–1179. [Google Scholar] [CrossRef]
- Flatten, K.; Dai, N.T.; Vroman, B.T.; Loegering, D.; Erlichman, C.; Karnitz, L.M.; Kaufmann, S.H. The Role of Checkpoint Kinase 1 in Sensitivity to Topoisomerase I Poisons. J. Biol. Chem. 2005, 280, 14349–14355. [Google Scholar] [CrossRef] [PubMed]
- Ganzinelli, M.; Carrassa, L.; Crippa, F.; Tavecchio, M.; Broggini, M.; Damia, G. Checkpoint Kinase 1 Down-Regulation by an Inducible Small Interfering RNA Expression System Sensitized in vivo Tumors to Treatment with 5-Fluorouracil. Clin. Cancer Res. 2008, 14, 5131. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Ren, K.H.; He, H.W.; Shao, R.G. Knockdown of Chk1 sensitizes human colon carcinoma HCT116 cells in a p53-dependent manner to lidamycin through abrogation of a G2/M checkpoint and induction of apoptosis. Cancer Biol. Ther. 2009, 8, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Azorsa, D.O.; Gonzales, I.M.; Basu, G.D.; Choudhary, A.; Arora, S.; Bisanz, K.M.; Kiefer, J.A.; Henderson, M.C.; Trent, J.M.; Von Hoff, D.D.; et al. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J. Transl. Med. 2009, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Karnitz, L.M. Cisplatin-Induced DNA Damage Activates Replication Checkpoint Signaling Components that Differentially Affect Tumor Cell Survival. Mol. Pharmacol. 2009, 76, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Zenvirt, S.; Kravchenko-Balasha, N.; Levitzki, A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene 2010, 29, 6149–6159. [Google Scholar] [CrossRef] [PubMed]
- Senderowicz, A.M. Small molecule modulators of cyclin-dependent kinases for cancer therapy. Oncogene 2000, 19, 6600–6606. [Google Scholar] [CrossRef] [PubMed]
- Oza, V.; Ashwell, S.; Almeida, L.; Brassil, P.; Breed, J.; Deng, C.; Gero, T.; Grondine, M.; Horn, C.; Ioannidis, S.; et al. Discovery of checkpoint kinase inhibitor (S)-5-(3-fluorophenyl)-N-(piperidin-3-yl)-3-ureidothiophene-2-carboxamide (AZD7762) by structure-based design and optimization of thiophenecarboxamide ureas. J. Med. Chem. 2012, 55, 5130–5142. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J.; Stokes, S.; Browne, H.; Foloppe, N.; Fiumana, A.; Scrace, S.; Fallowfield, M.; Bedford, S.; Webb, P.; Baker, L.; et al. Identification of novel, in vivo active Chk1 inhibitors utilizing structure guided drug design. Oncotarget 2015, 6, 35797–35812. [Google Scholar] [PubMed]
- Blasina, A.; Hallin, J.; Chen, E.; Arango, M.E.; Kraynov, E.; Register, J.; Grant, S.; Ninkovic, S.; Chen, P.; Nichols, T.; et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 2008, 7, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Guzi, T.J.; Paruch, K.; Dwyer, M.P.; Labroli, M.; Shanahan, F.; Davis, N.; Taricani, L.; Wiswell, D.; Seghezzi, W.; Penaflor, E.; et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol. Cancer Ther. 2011, 10, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Matthews, T.P.; Jones, A.M.; Collins, I. Structure-based design, discovery and development of checkpoint kinase inhibitors as potential anticancer therapies. Expert Opin. Drug. Discov. 2013, 8, 621–640. [Google Scholar] [CrossRef] [PubMed]
- Walton, M.I.; Eve, P.D.; Hayes, A.; Valenti, M.R.; De Haven Brandon, A.K.; Box, G.; Hallsworth, A.; Smith, E.L.; Boxall, K.J.; Lainchbury, M.; et al. CCT244747 is a novel potent and selective CHK1 inhibitor with oral efficacy alone and in combination with genotoxic anticancer drugs. Clin. Cancer Res. 2012, 18, 5650–5661. [Google Scholar] [CrossRef] [PubMed]
- Walton, M.I.; Eve, P.D.; Hayes, A.; Henley, A.T.; Valenti, M.R.; De Haven Brandon, A.K.; Box, G.; Boxall, K.J.; Tall, M.; Swales, K.; et al. The clinical development candidate CCT245737 is an orally active CHK1 inhibitor with preclinical activity in RAS mutant NSCLC and Emicro-MYC driven B-cell lymphoma. Oncotarget 2016, 7, 2329–2342. [Google Scholar] [PubMed]
- King, C.; Diaz, H.; Barnard, D.; Barda, D.; Clawson, D.; Blosser, W.; Cox, K.; Guo, S.; Marshall, M. Characterization and preclinical development of LY2603618: A selective and potent Chk1 inhibitor. Investig. New Drugs 2014, 32, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar] [PubMed]
- Nishida, H.; Tatewaki, N.; Nakajima, Y.; Magara, T.; Ko, K.M.; Hamamori, Y.; Konishi, T. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 2009, 37, 5678–5689. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Charrier, J.D.; Durrant, S.J.; Golec, J.M.; Kay, D.P.; Knegtel, R.M.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Lau, A.; Nissink, J.W. Drugging ATR: Progress in the development of specific inhibitors for the treatment of cancer. Future Med. Chem. 2015, 7, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-y l}-1H-indole (AZ20): A potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Stephens, P.A.; Middleton, F.K.; Curtin, N.J. Targeting the S and G2 checkpoint to treat cancer. Drug Discov. Today 2012, 17, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Zabludoff, S.D.; Deng, C.; Grondine, M.R.; Sheehy, A.M.; Ashwell, S.; Caleb, B.L.; Green, S.; Haye, H.R.; Horn, C.L.; Janetka, J.W.; et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 2008, 7, 2955–2966. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Svensson, S.; Romania, P.; Dattilo, R.; Patrizii, M.; Signore, M.; Navarra, S.; Lotti, F.; Biffoni, M.; Pilozzi, E.; et al. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Differ. 2012, 19, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheung, I.Y.; Wei, X.X.; Tran, H.; Gao, X.; Cheung, N.K. Checkpoint kinase inhibitor synergizes with DNA-damaging agents in G1 checkpoint-defective neuroblastoma. Int. J. Cancer 2011, 129, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Montano, R.; Chung, I.; Garner, K.M.; Parry, D.; Eastman, A. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol. Cancer Ther. 2012, 11, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Engelke, C.G.; Parsels, L.A.; Qian, Y.; Zhang, Q.; Karnak, D.; Robertson, J.R.; Tanska, D.M.; Wei, D.; Davis, M.A.; Parsels, J.D.; et al. Sensitization of pancreatic cancer to chemoradiation by the Chk1 inhibitor MK8776. Clin. Cancer Res. 2013, 19, 4412–4421. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.; Rawlinson, R.; Massey, A.J. Chk1 inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer 2014, 14, 570. [Google Scholar] [CrossRef] [PubMed]
- Schenk, E.L.; Koh, B.D.; Flatten, K.S.; Peterson, K.L.; Parry, D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Karnitz, L.M.; Kaufmann, S.H. Effects of selective checkpoint kinase 1 inhibition on cytarabine cytotoxicity in acute myelogenous leukemia cells in vitro. Clin. Cancer Res. 2012, 18, 5364–5373. [Google Scholar] [CrossRef] [PubMed]
- Didier, C.; Demur, C.; Grimal, F.; Jullien, D.; Manenti, S.; Ducommun, B. Evaluation of checkpoint kinase targeting therapy in acute myeloid leukemia with complex karyotype. Cancer Biol. Ther. 2012, 13, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Milton, S.; Murphy, C.E.; et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; James, J.; Annunziata, C.M. Topotecan synergizes with CHEK1 (CHK1) inhibitor to induce apoptosis in ovarian cancer cells. BMC Cancer 2015, 15, 196. [Google Scholar] [CrossRef] [PubMed]
- Josse, R.; Martin, S.E.; Guha, R.; Ormanoglu, P.; Pfister, T.D.; Reaper, P.M.; Barnes, C.S.; Jones, J.; Charlton, P.; Pollard, J.R.; et al. ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 2014, 74, 6968–6979. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Activation of programmed cell death by anticancer agents: Cisplatin as a model system. Cancer Cells 1990, 2, 275–280. [Google Scholar] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Demarcq, C.; Bunch, R.T.; Creswell, D.; Eastman, A. The role of cell cycle progression in cisplatin-induced apoptosis in Chinese hamster ovary cells. Cell Growth Differ. 1994, 5, 983–993. [Google Scholar] [PubMed]
- Raaphorst, G.P.; Leblanc, M.; Li, L.F. A comparison of response to cisplatin, radiation and combined treatment for cells deficient in recombination repair pathways. Anticancer Res. 2005, 25, 53–58. [Google Scholar] [PubMed]
- Damia, G.; Filiberti, L.; Vikhanskaya, F.; Carrassa, L.; Taya, Y.; D'Incalci, M.; Broggini, M. Cisplatinum and taxol induce different patterns of p53 phosphorylation. Neoplasia 2001, 3, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.J.; Kotsaki-Kovatsi, V.P. Potentiation of sulphur mustard or cisplatin-induced toxicity by caffeine in Chinese hamster cells correlates with formation of DNA double-strand breaks during replication on a damaged template. Mutat. Res. 1986, 165, 207–220. [Google Scholar] [CrossRef]
- Yazlovitskaya, E.M.; Persons, D.L. Inhibition of cisplatin-induced ATR activity and enhanced sensitivity to cisplatin. Anticancer Res. 2003, 23, 2275–2279. [Google Scholar] [PubMed]
- Vendetti, F.P.; Lau, A.; Schamus, S.; Conrads, T.P.; O’Connor, M.J.; Bakkenist, C.J. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget 2015, 6, 44289–44305. [Google Scholar] [PubMed]
- Teng, P.N.; Bateman, N.W.; Darcy, K.M.; Hamilton, C.A.; Maxwell, G.L.; Bakkenist, C.J.; Conrads, T.P. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol. Oncol. 2015, 136, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Karnitz, L.M. Cisplatin-induced DNA damage activates replication checkpoint signaling components that differentially affect tumor cell survival. Mol. Pharmacol. 2009, 76, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Itamochi, H.; Nishimura, M.; Oumi, N.; Kato, M.; Oishi, T.; Shimada, M.; Sato, S.; Naniwa, J.; Sato, S.; Kudoh, A.; et al. Checkpoint kinase inhibitor AZD7762 overcomes cisplatin resistance in clear cell carcinoma of the ovary. Int. J. Gynecol. Cancer 2014, 24, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Gadhikar, M.A.; Sciuto, M.R.; Alves, M.V.; Pickering, C.R.; Osman, A.A.; Neskey, D.M.; Zhao, M.; Fitzgerald, A.L.; Myers, J.N.; Frederick, M.J. CHK1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol. Cancer Ther. 2013, 12, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yan, Z.; Painter, C.L.; Zhang, Q.; Chen, E.; Arango, M.E.; Kuszpit, K.; Zasadny, K.; Hallin, M.; Hallin, J.; et al. PF-00477736 mediates checkpoint kinase 1 signaling pathway and potentiates docetaxel-induced efficacy in xenografts. Clin. Cancer Res. 2009, 15, 4630–4640. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Patel, R.; McLaughlin, M.; Schick, U.; Zaidi, S.; Nutting, C.M.; Newbold, K.L.; Bhide, S.; Harrington, K.J. CHK1 Inhibition Radiosensitizes Head and Neck Cancers to Paclitaxel-Based Chemoradiotherapy. Mol. Cancer Ther. 2016, 15, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yoon, S.J.; Jin, J.; Choi, S.H.; Seol, H.J.; Lee, J.I.; Nam, D.H.; Yoo, H.Y. Inhibition of checkpoint kinase 1 sensitizes lung cancer brain metastases to radiotherapy. Biochem. Biophys. Res. Commun. 2011, 406, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.; Choudhuri, R.; Fabre, K.; Sowers, A.L.; Citrin, D.; Zabludoff, S.D.; Cook, J.A. In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin. Cancer Res. 2010, 16, 2076–2084. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.T.; Barker, H.E.; Pedersen, M.; Hafsi, H.; Bhide, S.A.; Newbold, K.L.; Nutting, C.M.; McLaughlin, M.; Harrington, K.J. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol. Cancer Ther. 2017, 16, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lai, J.; Du, Z.; Gao, J.; Yang, S.; Gorityala, S.; Xiong, X.; Deng, O.; Ma, Z.; Yan, C.; et al. Targeting radioresistant breast cancer cells by single agent CHK1 inhibitor via enhancing replication stress. Oncotarget 2016, 7, 34688–34702. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, L.B.; Krebs, A.M.; Kim, S.Y.; Hong, T.S.; Haigis, K.M. Comparative analysis of radiosensitizers for K-RAS mutant rectal cancers. PLoS ONE 2013, 8, e82982. [Google Scholar] [CrossRef] [PubMed]
- Pires, I.M.; Olcina, M.M.; Anbalagan, S.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; McKenna, W.G.; Hammond, E.M. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer 2012, 107, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [PubMed]
- Graeber, T.G.; Osmanian, C.; Jacks, T.; Housman, D.E. Koch Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996, 379, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Huntoon, C.J.; Flatten, K.S.; Wahner Hendrickson, A.E.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.; Park, M.; Eulitt, P.; Yang, C.; Yacoub, A.; Dent, P. Poly(ADP-Ribose) Polymerase 1 Modulates the Lethality of CHK1 Inhibitors in Carcinoma Cells. Mol. Pharmacol. 2010, 78, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Hamed, H.A.; Poklepovic, A.; Dai, Y.; Grant, S.; Dent, P. Poly(ADP-ribose) Polymerase 1 Modulates the Lethality of CHK1 Inhibitors in Mammary Tumors. Mol. Pharmacol. 2012, 82, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Cruickshanks, N.; Ridder, T.; Dai, Y.; Grant, S.; Dent, P. PARP and CHK inhibitors interact to cause DNA damage and cell death in mammary carcinoma cells. Cancer Biol. Ther. 2013, 14, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Vance, S.M.; Liu, E.; Zhao, L.; Parsels, J.D.; Parsels, L.A.; Brown, J.L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle 2011, 10, 4321–4329. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Brown, E.; Thomason, A.; Odedra, R.; Sheridan, V.; Cadogan, E.; Xu, S.; Cui, A.; Gavine, P.R.; Connor, M. Abstract C60: Pre-clinical efficacy of the ATR inhibitor AZD6738 in combination with the PARP inhibitor olaparib. Mol. Cancer Ther. 2016, 14 (Suppl. 2), C60. [Google Scholar] [CrossRef]
- Sanjiv, K.; Hagenkort, A.; Calderon-Montano, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R.V.; Schultz, N.; Scobie, M.; et al. Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep. 2016, 14, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.D.; Cable, P.L.; Garrus, J.E.; Sullivan, F.X.; von Carlowitz, I.; Huerou, Y.L.; Wallace, E.; Woessner, R.D.; Gross, S. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol. Ther. 2011, 12, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Chilà, R.; Lupi, M.; Ricci, F.; Celenza, C.; Mazzoletti, M.; Broggini, M.; Damia, G. Combined inhibition of Chk1 and Wee1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 2012, 11, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; Artista, L.D.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar]
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010, 70, 9693–9702. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Fernandez-Capetillo, O. Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs. Mol. Oncol. 2011, 5, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Palacin, I.; Day, A.; Murga, M.; Lafarga, V.; Anton, M.E.; Tubbs, A.; Chen, H.-T.; Ergen, A.V.; Anderson, R.; Bhandoola, A.; et al. Targeting the kinase activities of ATR and ATM exhibits antitumoral activity in mouse models of MLL-rearranged AML. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef] [PubMed]
- Hocke, S.; Guo, Y.; Job, A.; Orth, M.; Ziesch, A.; Lauber, K.; De Toni, E.N.; Gress, T.M.; Herbst, A.; Göke, B.; et al. A synthetic lethal screen identifies ATR-inhibition as a novel therapeutic approach for POLD1-deficient cancers. Oncotarget 2016, 7, 7080–7095. [Google Scholar] [PubMed]
- Taricani, L.; Shanahan, F.; Parry, D. Replication stress activates DNA polymerase alpha-associated Chk1. Cell Cycle 2009, 8, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Abdel-Fatah, T.; Perry, C.; Moseley, P.; Albarakti, N.; Mohan, V.; Seedhouse, C.; Chan, S.; Madhusudan, S. Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Protein Kinase Inhibition Is Synthetically Lethal in XRCC1 Deficient Ovarian Cancer Cells. PLoS ONE 2013, 8, e57098. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.K.; Patterson, M.J.; Elstob, C.J.; Fordham, S.; Herriott, A.; Wade, M.A.; McCormick, A.; Edmondson, R.; May, F.E.B.; Allan, J.M.; et al. Common cancer-associated imbalances in the DNA damage response confer sensitivity to single agent ATR inhibition. Oncotarget 2015, 6, 32396–32409. [Google Scholar] [PubMed]
- Mohni, K.N.; Kavanaugh, G.M.; Cortez, D. ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 2014, 74, 2835–2845. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base Excision Repair and Cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J.; Stephens, P.; Rawlinson, R.; McGurk, L.; Plummer, R.; Curtin, N.J. mTORC1 and DNA-PKcs as novel molecular determinants of sensitivity to Chk1 inhibition. Mol. Oncol. 2016, 10, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Kennedy, R.D.; Sidi, S.; Look, A.T.; D’Andrea, A. CHK1 inhibition as a strategy for targeting fanconi anemia (FA) DNA repair pathway deficient tumors. Mol. Cancer 2009, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Andrea, A.D. Molecular pathogenesis of Fanconi anemia: Recent progress. Blood 2006, 107, 4223. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM- defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Fehrmann, R.S.N.; Schoonen, P.M.; Labib, S.; de Vries, E.G.E.; Franke, L.; van Vugt, M.A.T.M. ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene 2015, 34, 3474–3481. [Google Scholar] [CrossRef] [PubMed]
- Mohni, K.N.; Thompson, P.S.; Luzwick, J.W.; Glick, G.G.; Pendleton, C.S.; Lehmann, B.D.; Pietenpol, J.A.; Cortez, D. A Synthetic Lethal Screen Identifies DNA Repair Pathways that Sensitize Cancer Cells to Combined ATR Inhibition and Cisplatin Treatments. PLoS ONE 2015, 10, e0125482. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Resources for Information on Approved Drugs. 2017. Available online: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs (accessed on 20 April 2017).
- European Medicines Agency. 2017. Available online: http://www.ema.europa.eu (accessed on 20 April 2017).
- O’Carrigan, B.; de Miguel Luken, M. J.; Papadatos-Pastos, D.; Brown, J.; Tunariu, N.; Lopez, R. P. Phase I trial of a first-in-class ATR inhibitor VX-970 as monotherapy (mono) or in combination (combo) with carboplatin (CP) incorporating pharmacodynamics (PD) studies. J. Clin. Oncol. 2016, 34, 2504. [Google Scholar]
- Plummer, E.R.; Dean, E.J.; Evans, T.R.J.; Greystoke, A.; Herbschleb, K.; Ranson, M.; Brown, J.; Zhang, Y.; Karan, S.; Pollard, J.; et al. Phase I trial of first-in-class ATR inhibitor VX-970 in combination with gemcitabine (Gem) in advanced solid tumors (NCT02157792). J. Clin. Oncol. 2016, 34, 2513. [Google Scholar]
- Shapiro, G.; Wesolowski, R.; Middleton, M.; Devoe, C.; Constantinidou, A.; Papadatos-Pastos, D.; Fricano, M.; Zhang, Y.; Karan, S.; Pollard, J.; et al. Phase 1 trial of first-in-class ATR inhibitor VX-970 in combination with cisplatin (Cis) in patients (pts) with advanced solid tumors (NCT02157792). Cancer Res. 2016. [Google Scholar] [CrossRef]
- Dillon, M.; Ellis, S.; Grove, L.; McLellan, L.; Clack, G.; Smith, S.; Laude, J.; Viney, Z.; Adeleke, S.; Lazaridis, G.; et al. PATRIOT: A phase I study to assess the tolerability, safety and biological effects of a specific ataxia telangiectasia and Rad3-related (ATR) inhibitor (AZD6738) as a single agent and in combination with palliative radiation therapy in patients with solid tumours. J. Clin. Oncol. 2016, 34, TPS2603. [Google Scholar]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial Watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol. Cell. Oncol. 2015, 2, e1012976. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Ashworth, M.T.; Strosberg, J.; Goldman, J.W.; Mendelson, D.; Springett, G.; Venook, A.P.; Loechner, S.; Rosen, L.S.; Shanahan, F.; et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Karp, J.E.; Thomas, B.M.; Greer, J.M.; Sorge, C.; Gore, S.D.; Pratz, K.W.; Smith, B.D.; Flatten, K.S.; Peterson, K.; Schneider, P.; et al. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin. Cancer Res. 2012, 18, 6723–6731. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Yoshino, T.; Shitara, K.; Matsubara, N.; Fuse, N.; Naito, Y.; Uenaka, K.; Nakamura, T.; Hynes, S.M.; Lin, A.B. Phase I study of LY2603618, a CHK1 inhibitor, in combination with gemcitabine in Japanese patients with solid tumors. Anticancer Drugs 2015, 26, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Braiteh, F.; Von Hoff, D.; McWilliams, R.; Becerra, C.; Galsky, M.D.; Jameson, G.; Lin, J.; McKane, S.; Wickremsinhe, E.R.; et al. Phase I Study of CHK1 Inhibitor LY2603618 in Combination with Gemcitabine in Patients with Solid Tumors. Oncology 2016, 91, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.; Kang, J.H.; Smith, D.; Rosenberg, R.; Park, K.; Kim, S.W.; Su, W.C.; Boyd, T.E.; Richards, D.A.; Novello, S.; et al. Phase II evaluation of LY2603618, a first-generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non-small cell lung cancer. Investig. New Drugs 2016, 34, 625–635. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| CHK1 Inhibitors | |||
| Name | Structure | IC50/Ki | Specificity |
| AZD7762 | 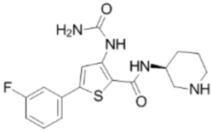 | CHK1 IC50 = 5 nM | Equally potent: CHK1/CHK2 |
| V158411 | 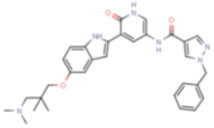 | CHK1 IC50 = 4.4 nM | Equally potent: CHK1/CHK2; 20-fold CHK1 vs. CHK2 in cells (Cellular CHK1 IC50 = 48 nM vs. CHK2 IC50 = 904 nM) |
| PF477736 |  | CHK1 Ki = 4.9 nM | 100-fold CHK1 vs. CHK2 |
| MK8776/SCH900776 | 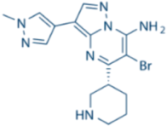 | CHK1 IC50 = 3 nM | 500-fold CHK1 vs. CHK2 |
| CCT244747 | 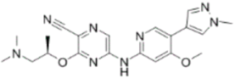 | CHK1 IC50 = 8 nM | >1000-fold CHK1 vs. CHK2 |
| CCT245737 | 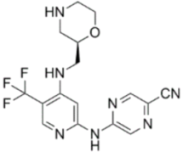 | CHK1 IC50 = 1.3 nM | >1500-fold CHK1 vs. CHK2 |
| LY2603618 | 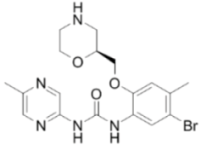 | CHK1 IC50 = 7 nM | >1500-fold CHK1 vs. CHK2 |
| ATR Inhibitors | |||
| Name | Structure | IC50/Ki | Specificity |
| NU6027 | 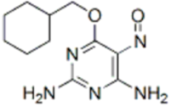 | ATR IC50 = 1 nM | ATR, CDK1 (Ki = 2.5 µM), CDK2 (Ki = 1.3 µM) |
| ETP-46464 | 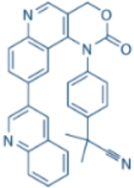 | ATR IC50 = 25 nM | ATR |
| VE-821 | 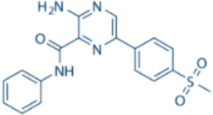 | ATR IC50 = 26 nM | >100-fold ATR vs. ATM/DNA-PK |
| VE-822/VX-970 | 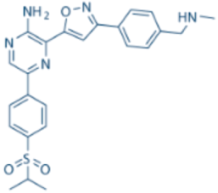 | ATR IC50 = 0.2 nM | >100-fold ATR vs. ATM/DNA-PK |
| AZ20 | 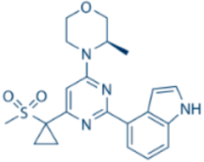 | ATR IC50 = 5 nM | >600-fold ATR vs. ATM/DNA-PK/PI-3K |
| AZD6738 | 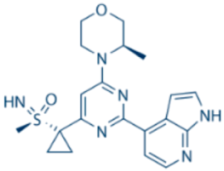 | ATR IC50 = 1 nM | ATR |
| Target, Agent | Phase | Combination | Indication | NCT No. |
|---|---|---|---|---|
| ATR-VX-970, intravenous, Vertex pharmaceuticals (recently licenced to Merck KGaA, Germany) | 1 | irinotecan | Advanced solid tumours | NCT02595931 |
| 1 | Veliparib + Cisplatin | Advanced solid tumours | NCT02723864 | |
| 1/2 | Topotecan | Advanced small cell lung, cervical, endometrial, ovarian cancers | NCT02487095 | |
| 2 | Gemcitabine | Advanced ovarian/fallopian tube/primary peritoneal cancer(OC/FT/PP) | NCT02595892 | |
| 2 | Carboplatin/Gemcitabine | Advanced OC/FT/PP | NCT02627443 | |
| 2 | Cisplatin/Gemcitabine | Advanced urothelial cancers | NCT02567409 | |
| 1 | Cisplatin/Radiotherapy | Locally dvanced HPV negative SCC head and neck cancers | NCT02567422 | |
| 1 | Whole brain radiotherapy | Non- small cell lung cancers with brain mets | NCT02589522 | |
| 1 | Gemcitabine, Cisplatin, Etoposide, Carboplatin | Multiple parts including p53mut NSCLC, triple negative breast cancers | NCT02157792 | |
| ATR—AZD6738, oral, Astrazeneca | 1 | Single agent and in combination with radiotherapy | Advanced solid tumours | NCT62223923 |
| 1 | Paclitaxel | Advanced solid tumours | NCT02630199 | |
| 1 | Carboplatin, Olaparib, MEDI4736 | Advanced solid tumours | NCT02264678 | |
| CHK1—LY2606368, (Prexasertib), intravenous, Eli Lilly | 1 | Cytarabine and Fludarabine | Relapsed/Refractory Acute Myelogenous Leukemia (AML) and High-Risk Myelodysplastic Syndrome | NCT02649764 |
| 1 | 14C radiolabelled LY2606368 | Advanced solid tumours | NCT02778126 | |
| 1 | Single agent | Japanese patients with Advanced solid tumours | NCT02514603 | |
| 2 | Single agent | Advanced small cell lung cancer | NCT02735980 | |
| 1 | Ralimetinib (p38 MAPK inhibitor) | Advanced solid tumours | NCT02860780 | |
| 1 | Cisplatin, Cetuximab, Intensity Modulated Radiation Therapy | Advanced solid tumours, Head and Neck | NCT02555644 | |
| 1 | Cisplatin, Cetuximab, Pemetrexed, Fluorouraci | Advanced solid tumours | NCT02124148 | |
| 2 | Single agent | BRCA1/2 Mutation Associated Breast or Ovarian Cancer, Triple Negative Breast Cancer, High Grade Serous OC, and Metastatic CRPC | NCT02203513 | |
| 2 | Single agent | Advanced Solid Tumours Exhibiting Replicative Stress or Homologous Recombination Repair Deficiency | NCT02873975 | |
| CHK1—SRA 737 previously known CCT245737, Sierra Oncology Inc. | 1 | Gemcitabine + Cisplatin or Gemcitabine Alone | Advanced solid tumours | NCT02797977 |
| 1 | Single agent | Advanced solid tumours | NCT02797964 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rundle, S.; Bradbury, A.; Drew, Y.; Curtin, N.J. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers 2017, 9, 41. https://doi.org/10.3390/cancers9050041
Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers. 2017; 9(5):41. https://doi.org/10.3390/cancers9050041
Chicago/Turabian StyleRundle, Stuart, Alice Bradbury, Yvette Drew, and Nicola J. Curtin. 2017. "Targeting the ATR-CHK1 Axis in Cancer Therapy" Cancers 9, no. 5: 41. https://doi.org/10.3390/cancers9050041
APA StyleRundle, S., Bradbury, A., Drew, Y., & Curtin, N. J. (2017). Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers, 9(5), 41. https://doi.org/10.3390/cancers9050041