
Cancer-Associated Fibroblasts: Immunosuppressive Crosstalk with Tumor-Infiltrating Immune Cells and Implications for Therapeutic Resistance

 ,
,  ,
,  ,
,  , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Biology of CAF
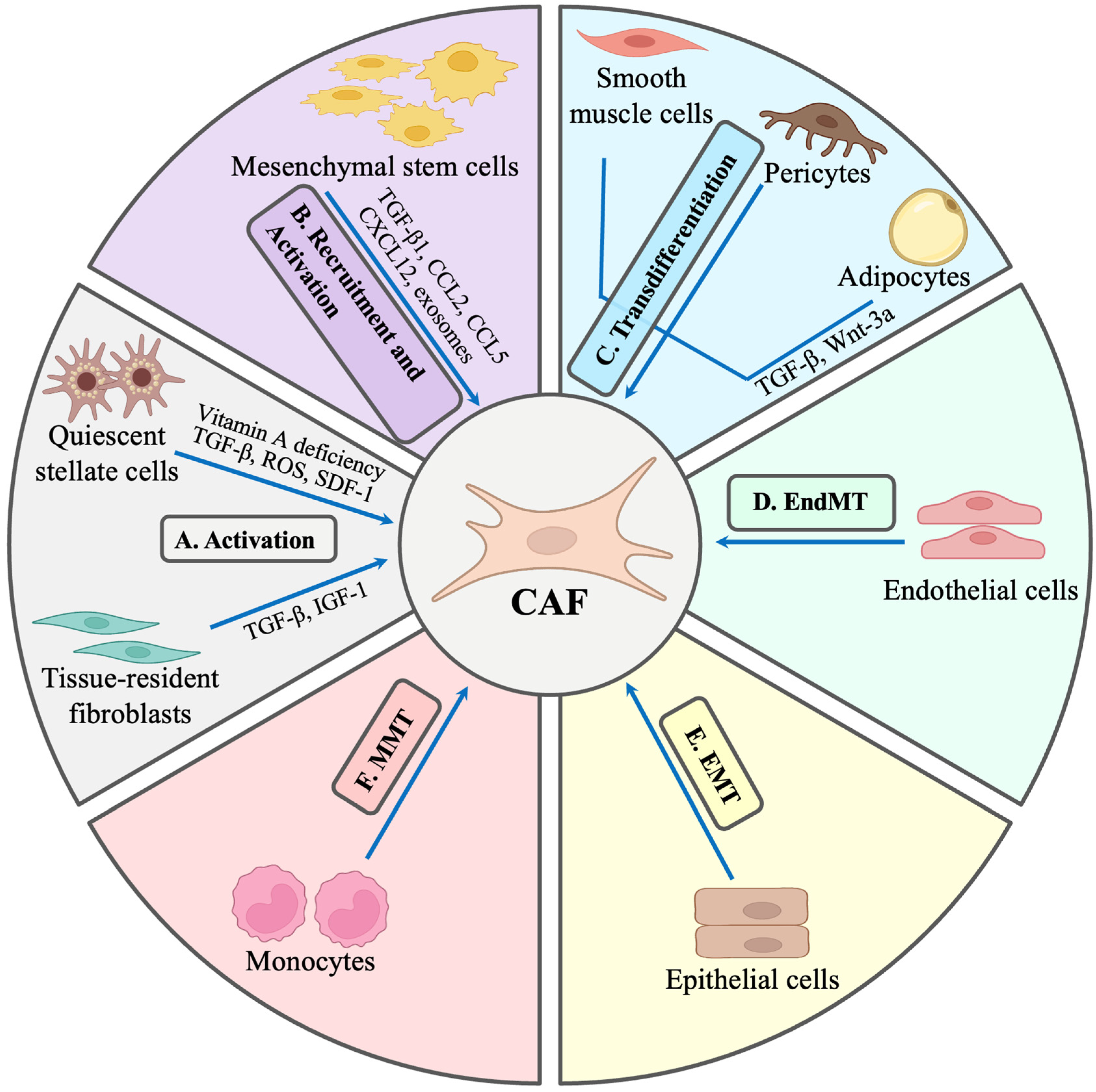
2.1. Origin of CAFs
2.2. Activators of CAFs
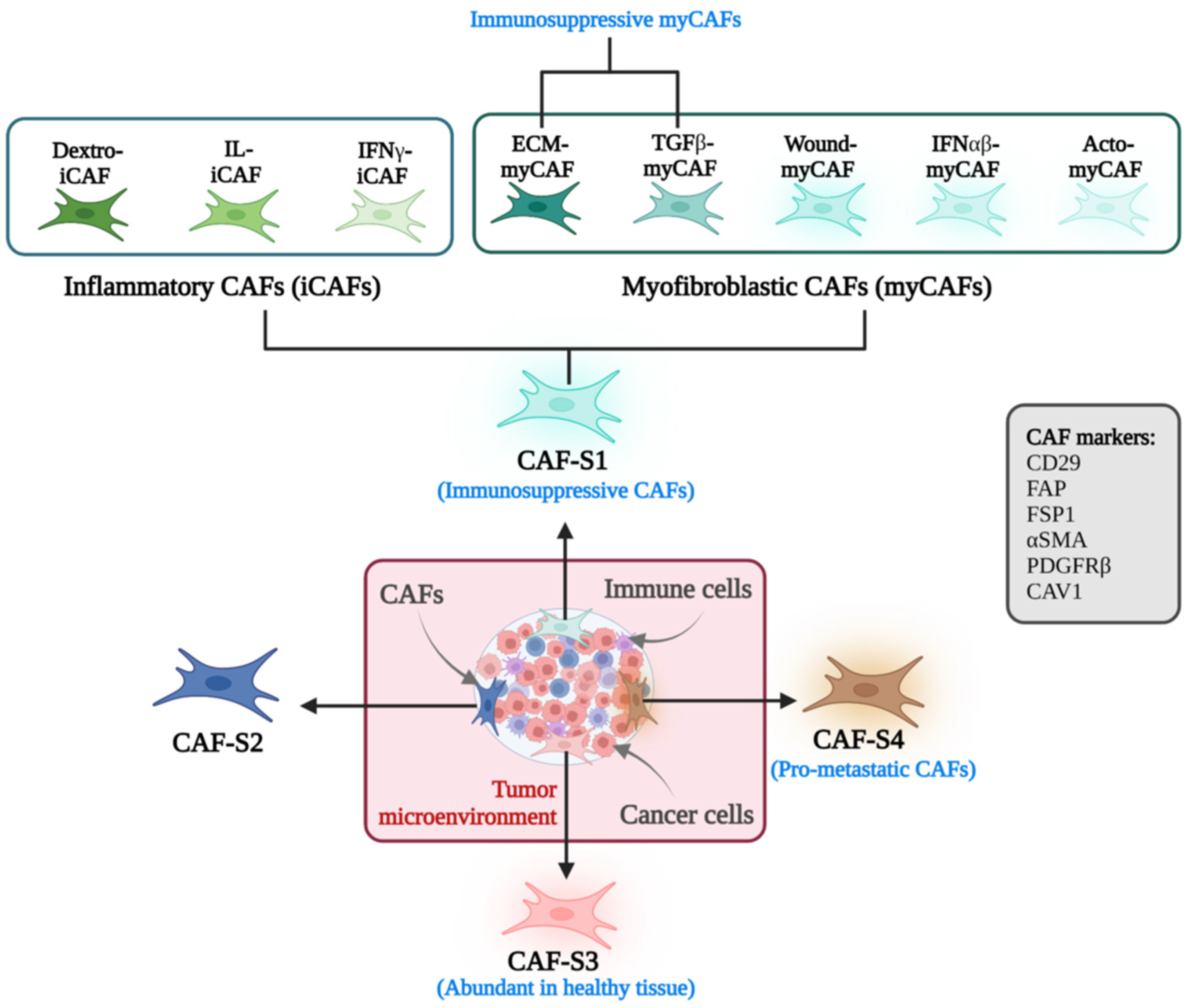
2.3. Markers and Functional Phenotypes of CAFs
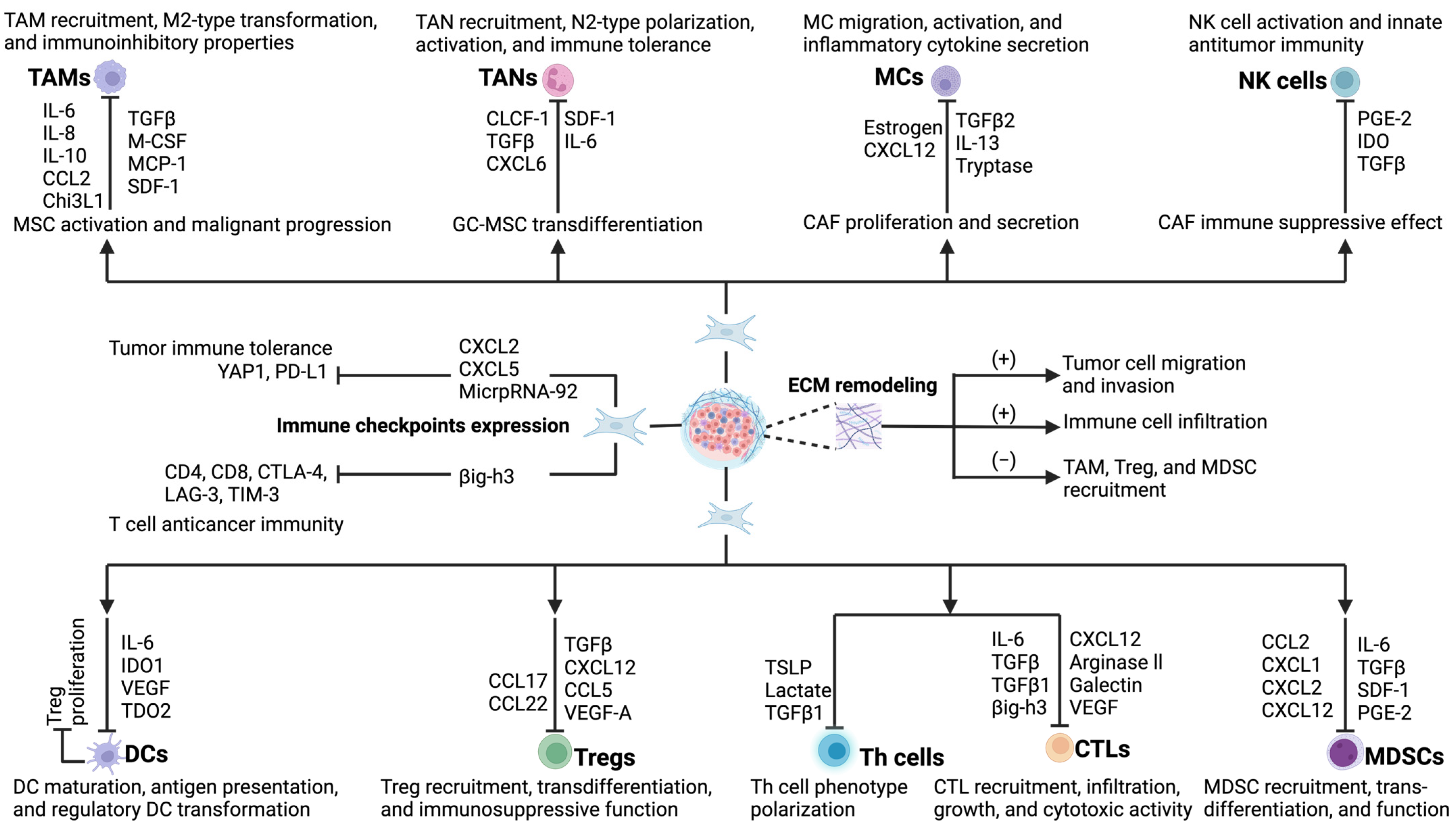
3. Tumor Microenvironment and Crosstalk Between CAFs and Tumor-Infiltrating Immune Cells
3.1. Interaction of CAFs and Tumor-Associated Macrophages (TAMs)
3.2. Interaction of CAFs and Tumor-Associated Neutrophils (TANs)
3.3. Interaction of CAFs and Mast Cells (MCs)
3.4. Interaction of CAFs and Natural Killer (NK) Cells
3.5. Interaction of CAFs and Dendritic Cells (DCs)
3.6. Interaction of CAFs and T Lymphocytes
3.7. Interaction of CAFs and Myeloid-Derived Suppressor Cells (MDSCs)
4. Interaction of CAFs and Immune Checkpoint Molecules
5. Role of CAFs in Remodeling the Extracellular Matrix (ECM)
6. CAF Signals to Tumor-Infiltrating Lymphocytes Towards Tumor Resistance
6.1. Secretion of Immunosuppressive Cytokines
6.2. Modulation of Immune Checkpoints
6.3. Metabolic Adaptation and Competition
6.4. Altered Trafficking by Secretion of Chemokines
6.5. Alterations in Apoptotic Pathways
6.6. Antigen Presentation
6.7. Remodelling of the Extracellular Matrix (ECM)
6.8. Expression of Inhibitory Ligands
6.9. Direct Cell–Cell Interaction
7. Immunotherapeutic Strategies for Tumors Based on Modulation of CAFs
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAFs | cancer-associated fibroblasts |
| TME | tumor microenvironment |
| ECM | extracellular matrix |
| Treg | T regulatory cells |
| MDSCs | myeloid-derived suppressor cells |
| FAP | fibroblast activation protein |
| SDF-1 | stromal-derived factor-1 |
| VEGF | vascular endothelial growth factor |
| PGE2 | prostaglandin E2 |
| C-C | chemokine ligand 2 |
| IDO | indoleamine 2,3-dioxygenase |
| TAMs | tumor-associated macrophages |
| TANs | tumor-associated neutrophils |
| DCs | dendritic cells |
| rDCs | regulatory dendritic cells |
| TILs | tumor-infiltrating lymphocytes |
| TIDCs | tumor-infiltrating DCs |
| MSCs | mesenchymal stem cells |
| BMSCs | bone marrow mesenchymal stem cells |
| EMT | epithelial–mesenchymal transition |
| TGFβ1 | transforming growth factor β1 |
| ADSCs | adipose tissue-derived mesenchymal stem cells |
| FSP-1 | fibroblast specific protein 1 |
| EndMT | endothelial–mesenchymal transition |
| α SMA | α smooth muscle actin |
| MMT | mesothelial-mesenchymal transition |
| PSCs | pancreatic stellate cells |
| PDGF | platelet-derived growth factor |
| FGF-2 | fibroblast growth factor 2 |
| HGF | hepatocyte growth factor |
| ROS | reactive oxygen species |
| CCL | C-C chemokine ligands |
| JAK/STAT3 | Janus kinase/TNF inducer and activator of transcription 3 |
| DAMPs | damage-associated molecular patterns |
| VIM | vimentin |
| CAV-1 | caveolin-1 |
| myCAFs | myofibroblast CAFs |
| iCAFs | inflammatory CAFs |
| apCAF | antigen-presenting CAFs |
| PDA | pancreatic ductal adenocarcinoma |
| scRNA-seq | single-cell RNA sequencing |
| MCP-1 | monocyte chemotactic protein-1 |
| Chi3L1 | chitinase 3-like protein 1 |
| PD-1 | programmed cell death protein 1 |
| NLR | neutrophil-to-lymphocyte ratio |
| ICAM1 | intercellular adhesion molecule 1 |
| VCAM-1 | vascular cell adhesion molecule-1 |
| MPO | myeloperoxidase |
| HCC | hepatocellular carcinoma |
| SCF | stem cell factor |
| TSLP | thymic stromal lymphopoietin |
| LSCC | lung squamous cell carcinoma |
| miR-21 | microRNA-21 |
| iICPs | immune checkpoints |
| MMPs | matrix metalloproteinases |
| FAKs | focal adhesion kinases |
| SHH | Sonic hedgehog |
| Smo | smoothened |
References
- Moura, T.; Laranjeira, P.; Caramelo, O.; Gil, A.M.; Paiva, A. Breast Cancer and Tumor Microenvironment: The Crucial Role of Immune Cells. Curr. Oncol. 2025, 32, 143. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Lei, Y.; Li, J.K.; Du, W.X.; Li, R.G.; Yang, J.; Li, J.; Li, F.; Tan, H.B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, J.; Peng, S.; Liu, Q.; Chen, D.; He, Z.; Xiang, J.; Wang, B. Tumor microenvironment: Nurturing cancer cells for immunoevasion and druggable vulnerabilities for cancer immunotherapy. Cancer Lett. 2024, 611, 217385. [Google Scholar] [CrossRef] [PubMed]
- Kwon, W.A.; Joung, J.Y. Immunotherapy in Prostate Cancer: From a “Cold” Tumor to a “Hot” Prospect. Cancers 2025, 17, 1064. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, C.; Tu, J.; Tang, M.; Ashrafizadeh, M.; Nabavi, N.; Sethi, G.; Zhao, P.; Liu, S. Advances in cancer immunotherapy: Historical perspectives, current developments, and future directions. Mol. Cancer 2025, 24, 136. [Google Scholar] [CrossRef]
- Wang, D.R.; Wu, X.L.; Sun, Y.L. Therapeutic targets and biomarkers of tumor immunotherapy: Response versus non-response. Signal Transduct. Target. Ther. 2022, 7, 331. [Google Scholar] [CrossRef]
- Schulz, M.; Salamero-Boix, A.; Niesel, K.; Alekseeva, T.; Sevenich, L. Microenvironmental Regulation of Tumor Progression and Therapeutic Response in Brain Metastasis. Front. Immunol. 2019, 10, 1713. [Google Scholar] [CrossRef]
- Xia, Z.; De Wever, O. The plasticity of cancer-associated fibroblasts. Trends Cancer 2025, S2405-8033. [Google Scholar] [CrossRef]
- Cheng, P.S.W.; Zaccaria, M.; Biffi, G. Functional heterogeneity of fibroblasts in primary tumors and metastases. Trends Cancer 2025, 11, 135–153. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Wang, Y.; Yang, J.; Li, Z.; Liu, F.; Wang, A.; Gao, Z.; Wu, C.; Yin, H. Overcoming cancer treatment resistance: Unraveling the role of cancer-associated fibroblasts. J. Natl. Cancer Cent. 2025, 5, 237–251. [Google Scholar] [CrossRef]
- Zheng, J.; Hao, H. The importance of cancer-associated fibroblasts in targeted therapies and drug resistance in breast cancer. Front. Oncol. 2023, 13, 1333839. [Google Scholar] [CrossRef]
- Sarkar, M.; Nguyen, T.; Gundre, E.; Ogunlusi, O.; El-Sobky, M.; Giri, B.; Sarkar, T.R. Cancer-associated fibroblasts: The chief architect in the tumor microenvironment. Front. Cell Dev. Biol. 2023, 11, 1089068. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, V.; Östman, A. Interactions between cancer-associated fibroblasts and T-cells: Functional crosstalk with targeting and biomarker potential. Ups. J. Med. Sci. 2024, 129, e10710. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Fang, L.; Liu, C.; Yang, M.; Yu, X.; Wang, L.; Zhang, W.; Sun, C.; Zhuang, J. Microenvironmental regulation in tumor progression: Interactions between cancer-associated fibroblasts and immune cells. Biomed. Pharmacother. 2023, 167, 115622. [Google Scholar] [CrossRef]
- Chen, M.; Chen, F.; Gao, Z.; Li, X.; Hu, L.; Yang, S.; Zhao, S.; Song, Z. CAFs and T cells interplay: The emergence of a new arena in cancer combat. Biomed. Pharmacother. 2024, 177, 117045. [Google Scholar] [CrossRef]
- Said, S.S.; Ibrahim, W.N. Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives. Pharmaceutics 2023, 15, 1143. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- van Elsas, M.J.; van Hall, T.; van der Burg, S.H. Future Challenges in Cancer Resistance to Immunotherapy. Cancers 2020, 12, 935. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.T.; Abuwarwar, M.H.; Poly, L.; Wilkins, S.; Fletcher, A.L. Cancer-Associated Fibroblasts and T Cells: From Mechanisms to Outcomes. J. Immunol. 2021, 206, 310–320. [Google Scholar] [CrossRef]
- Cao, Z.; Quazi, S.; Arora, S.; Osellame, L.D.; Burvenich, I.J.; Janes, P.W.; Scott, A.M. Cancer-associated fibroblasts as therapeutic targets for cancer: Advances, challenges, and future prospects. J. Biomed. Sci. 2025, 32, 7. [Google Scholar] [CrossRef]
- Lv, K.; He, T. Cancer-associated fibroblasts: Heterogeneity, tumorigenicity and therapeutic targets. Mol. Biomed. 2024, 5, 70. [Google Scholar] [CrossRef]
- Chhabra, Y.; Weeraratna, A.T. Fibroblasts in cancer: Unity in heterogeneity. Cell 2023, 186, 1580–1609. [Google Scholar] [CrossRef]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef]
- Forsthuber, A.; Aschenbrenner, B.; Korosec, A.; Jacob, T.; Annusver, K.; Krajic, N.; Kholodniuk, D.; Frech, S.; Zhu, S.; Purkhauser, K.; et al. Cancer-associated fibroblast subtypes modulate the tumor-immune microenvironment and are associated with skin cancer malignancy. Nat. Commun. 2024, 15, 9678. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. eLife 2020, 9, e57243. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, J.; Qian, H.; Zhuang, Q. Cancer-associated fibroblasts: From basic science to anticancer therapy. Exp. Mol. Med. 2023, 55, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Tsang, W.Y.; Li, Z.H.; Guan, X.Y. The Origin, Differentiation, and Functions of Cancer-Associated Fibroblasts in Gastrointestinal Cancer. Cell Mol. Gastroenterol. Hepatol. 2023, 16, 503–511. [Google Scholar] [CrossRef]
- Fotsitzoudis, C.; Koulouridi, A.; Messaritakis, I.; Konstantinidis, T.; Gouvas, N.; Tsiaoussis, J.; Souglakos, J. Cancer-Associated Fibroblasts: The Origin, Biological Characteristics and Role in Cancer-A Glance on Colorectal Cancer. Cancers 2022, 14, 4394. [Google Scholar] [CrossRef] [PubMed]
- Manoukian, P.; Bijlsma, M.; van Laarhoven, H. The Cellular Origins of Cancer-Associated Fibroblasts and Their Opposing Contributions to Pancreatic Cancer Growth. Front. Cell Dev. Biol. 2021, 9, 743907. [Google Scholar] [CrossRef]
- Kobayashi, H.; Gieniec, K.A.; Lannagan, T.R.M.; Wang, T.; Asai, N.; Mizutani, Y.; Iida, T.; Ando, R.; Thomas, E.M.; Sakai, A.; et al. The Origin and Contribution of Cancer-Associated Fibroblasts in Colorectal Carcinogenesis. Gastroenterology 2022, 162, 890–906. [Google Scholar] [CrossRef]
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y.; et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, S.E.; Peduto, L. The perivascular origin of pathological fibroblasts. J. Clin. Investig. 2018, 128, 54–63. [Google Scholar] [CrossRef]
- Tang, J.; Chen, Y.; Wang, C.; Xia, Y.; Yu, T.; Tang, M.; Meng, K.; Yin, L.; Yang, Y.; Shen, L.; et al. The role of mesenchymal stem cells in cancer and prospects for their use in cancer therapeutics. MedComm 2024, 5, e663. [Google Scholar] [CrossRef] [PubMed]
- Ghafoor, S.; Garcia, E.; Jay, D.J.; Persad, S. Molecular Mechanisms Regulating Epithelial Mesenchymal Transition (EMT) to Promote Cancer Progression. Int. J. Mol. Sci. 2025, 26, 4364. [Google Scholar] [CrossRef] [PubMed]
- Leggett, S.E.; Hruska, A.M.; Guo, M.; Wong, I.Y. The epithelial-mesenchymal transition and the cytoskeleton in bioengineered systems. Cell Commun. Signal 2021, 19, 32. [Google Scholar] [CrossRef]
- Zhu, H.; Guo, S.; Zhang, Y.; Yin, J.; Yin, W.; Tao, S.; Wang, Y.; Zhang, C. Proton-sensing GPCR-YAP Signalling Promotes Cancer-associated Fibroblast Activation of Mesenchymal Stem Cells. Int. J. Biol. Sci. 2016, 12, 389–396. [Google Scholar] [CrossRef]
- Jia, H.; Chen, X.; Zhang, L.; Chen, M. Cancer associated fibroblasts in cancer development and therapy. J. Hematol. Oncol. 2025, 18, 36. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, T.; Yuan, Y.; Zhu, Y. What is new in cancer-associated fibroblast biomarkers? Cell Commun. Signal 2023, 21, 96. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, Y.; Zhang, W.; Li, H.; Ma, W.; Ji, X.; Zhou, C. Molecular mechanisms of endothelial-mesenchymal transition and its pathophysiological feature in cerebrovascular disease. Cell Biosci. 2025, 15, 49. [Google Scholar] [CrossRef]
- Dudas, J.; Ladanyi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells 2020, 9, 428. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Tang, Y.; Xia, Z. Transformation of macrophages into myofibroblasts in fibrosis-related diseases: Emerging biological concepts and potential mechanism. Front. Immunol. 2024, 15, 1474688. [Google Scholar] [CrossRef]
- El Alaa, R.S.A.; Al-Mannai, W.; Darwish, N.; Al-Mansoori, L. Adipose-Derived Stromal Cells and Cancer-Associated Fibroblasts: Interactions and Implications in Tumor Progression. Int. J. Mol. Sci. 2024, 25, 11558. [Google Scholar] [CrossRef]
- Liu, X.; Gao, R.; Wu, Q.; Li, G.; Xu, X.; Li, W.; Liu, P.; Wang, X.; Cai, J.; Li, M.; et al. ITGA7 loss drives the differentiation of adipose-derived mesenchymal stem cells to cancer-associated fibroblasts. Mol. Carcinog. 2024, 63, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Bielczyk-Maczynska, E. White Adipocyte Plasticity in Physiology and Disease. Cells 2019, 8, 1507. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Giordanengo, L.; Proment, A.; Botta, V.; Picca, F.; Munir, H.M.W.; Tao, J.; Olivero, M.; Taulli, R.; Bersani, F.; Sangiolo, D.; et al. Shifting Shapes: The Endothelial-to-Mesenchymal Transition as a Driver for Cancer Progression. Int. J. Mol. Sci. 2025, 26, 6535. [Google Scholar] [CrossRef]
- Li, J.; Guo, T. Role of Peritoneal Mesothelial Cells in the Progression of Peritoneal Metastases. Cancers 2022, 14, 2856. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; He, C.; Hua, X.; Kan, A.; Mao, Y.; Sun, S.; Duan, F.; Wang, J.; Huang, P.; Li, S. Oxidative stress induces monocyte-to-myofibroblast transdifferentiation through p38 in pancreatic ductal adenocarcinoma. Clin. Transl. Med. 2020, 10, e41. [Google Scholar] [CrossRef]
- Peng, H.; Zhu, E.; Zhang, Y. Advances of cancer-associated fibroblasts in liver cancer. Biomark. Res. 2022, 10, 59. [Google Scholar] [CrossRef]
- Yang, M.; Mu, Y.; Yu, X.; Gao, D.; Zhang, W.; Li, Y.; Liu, J.; Sun, C.; Zhuang, J. Survival strategies: How tumor hypoxia microenvironment orchestrates angiogenesis. Biomed. Pharmacother. 2024, 176, 116783. [Google Scholar] [CrossRef]
- Fang, Z.; Meng, Q.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Zhao, Y.; Yu, X.; et al. Signaling pathways in cancer-associated fibroblasts: Recent advances and future perspectives. Cancer Commun. 2023, 43, 3–41. [Google Scholar] [CrossRef]
- Xuan, X.; Tian, C.; Zhao, M.; Sun, Y.; Huang, C. Mesenchymal stem cells in cancer progression and anticancer therapeutic resistance. Cancer Cell Int. 2021, 21, 595. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Huang, T.; Yu, X.; Tian, B. The role of cancer-associated fibroblasts in breast cancer metastasis. Front. Oncol. 2023, 13, 1194835. [Google Scholar] [CrossRef]
- Lillo, S.; Saleh, M. Inflammasomes in Cancer Progression and Anti-Tumor Immunity. Front. Cell Dev. Biol. 2022, 10, 839041. [Google Scholar] [CrossRef] [PubMed]
- Wieder, R. Fibroblasts as Turned Agents in Cancer Progression. Cancers 2023, 15, 2014. [Google Scholar] [CrossRef] [PubMed]
- Glabman, R.A.; Choyke, P.L.; Sato, N. Cancer-Associated Fibroblasts: Tumorigenicity and Targeting for Cancer Therapy. Cancers 2022, 14, 3906. [Google Scholar] [CrossRef] [PubMed]
- Bryce, A.S.; Dreyer, S.B.; Froeling, F.E.M.; Chang, D.K. Exploring the Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer. Cancers 2022, 14, 5302. [Google Scholar] [CrossRef]
- Nazemi, M.; Rainero, E. Cross-Talk Between the Tumor Microenvironment, Extracellular Matrix, and Cell Metabolism in Cancer. Front. Oncol. 2020, 10, 239. [Google Scholar] [CrossRef]
- Toledo, B.; Picon-Ruiz, M.; Marchal, J.A.; Perán, M. Dual Role of Fibroblasts Educated by Tumour in Cancer Behavior and Therapeutic Perspectives. Int. J. Mol. Sci. 2022, 23, 15576. [Google Scholar] [CrossRef]
- Mhaidly, R.; Mechta-Grigoriou, F. Fibroblast heterogeneity in tumor micro-environment: Role in immunosuppression and new therapies. Semin. Immunol. 2020, 48, 101417. [Google Scholar] [CrossRef]
- Kanzaki, R.; Pietras, K. Heterogeneity of cancer-associated fibroblasts: Opportunities for precision medicine. Cancer Sci. 2020, 111, 2708–2717. [Google Scholar] [CrossRef] [PubMed]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e410. [Google Scholar] [CrossRef]
- Givel, A.M.; Kieffer, Y.; Scholer-Dahirel, A.; Sirven, P.; Cardon, M.; Pelon, F.; Magagna, I.; Gentric, G.; Costa, A.; Bonneau, C.; et al. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat. Commun. 2018, 9, 1056. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef]
- Hornburg, M.; Desbois, M.; Lu, S.; Guan, Y.; Lo, A.A.; Kaufman, S.; Elrod, A.; Lotstein, A.; DesRochers, T.M.; Munoz-Rodriguez, J.L.; et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 2021, 39, 928–944. [Google Scholar] [CrossRef]
- Wu, S.Z.; Roden, D.L.; Wang, C.; Holliday, H.; Harvey, K.; Cazet, A.S.; Murphy, K.J.; Pereira, B.; Al-Eryani, G.; Bartonicek, N.; et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 2020, 39, e104063. [Google Scholar] [CrossRef]
- Boyd, L.N.C.; Andini, K.D.; Peters, G.J.; Kazemier, G.; Giovannetti, E. Heterogeneity and plasticity of cancer-associated fibroblasts in the pancreatic tumor microenvironment. Semin. Cancer Biol. 2022, 82, 184–196. [Google Scholar] [CrossRef]
- Lan, X.; Li, W.; Zhao, K.; Wang, J.; Li, S.; Zhao, H. Revisiting the role of cancer-associated fibroblasts in tumor microenvironment. Front. Immunol. 2025, 16, 1582532. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, Y.; Li, D.; Wei, J.; Chen, K.; Zhang, E.; Liu, G.; Chu, X.; Liu, X.; Liu, W.; et al. Cancer associated fibroblasts and metabolic reprogramming: Unraveling the intricate crosstalk in tumor evolution. J. Hematol. Oncol. 2024, 17, 80. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikoff, N.; Chen, C.H.; Samuel, M.S. Interrelationships between the extracellular matrix and the immune microenvironment that govern epithelial tumour progression. Clin. Sci. 2022, 136, 361–377. [Google Scholar] [CrossRef]
- Zhang, C.; Fei, Y.; Wang, H.; Hu, S.; Liu, C.; Hu, R.; Du, Q. CAFs orchestrates tumor immune microenvironment-A new target in cancer therapy? Front. Pharmacol. 2023, 14, 1113378. [Google Scholar] [CrossRef]
- Xue, J.D.; Gao, J.; Tang, A.F.; Feng, C. Shaping the immune landscape: Multidimensional environmental stimuli refine macrophage polarization and foster revolutionary approaches in tissue regeneration. Heliyon 2024, 10, e37192. [Google Scholar] [CrossRef] [PubMed]
- Hirano, R.; Okamoto, K.; Shinke, M.; Sato, M.; Watanabe, S.; Watanabe, H.; Kondoh, G.; Kadonosono, T.; Kizaka-Kondoh, S. Tissue-resident macrophages are major tumor-associated macrophage resources, contributing to early TNBC development, recurrence, and metastases. Commun. Biol. 2023, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, X.; Wei, S.; Jiang, P.; Xue, L.; Wang, J. Tumor-associated macrophages: Potential therapeutic strategies and future prospects in cancer. J. Immunother. Cancer 2021, 9, e001341. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Pinto, C.; Widawski, J.; Zahalka, S.; Thaler, B.; Schuster, L.C.; Lukowski, S.W.; Ramírez, F.; Tirapu, I. Cross-disease integration of single-cell RNA sequencing data from lung myeloid cells reveals TAM signature in in vitro model. Oncoimmunology 2025, 14, 2502278. [Google Scholar] [CrossRef] [PubMed]
- Gunaydin, G. CAFs Interacting with TAMs in Tumor Microenvironment to Enhance Tumorigenesis and Immune Evasion. Front. Oncol. 2021, 11, 668349. [Google Scholar] [CrossRef]
- Karimova, A.F.; Khalitova, A.R.; Suezov, R.; Markov, N.; Mukhamedshina, Y.; Rizvanov, A.A.; Huber, M.; Simon, H.U.; Brichkina, A. Immunometabolism of tumor-associated macrophages: A therapeutic perspective. Eur. J. Cancer 2025, 220, 115332. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, S.; To, K.K.W.; Zhu, S.; Wang, F.; Fu, L. Tumor-associated macrophages remodel the suppressive tumor immune microenvironment and targeted therapy for immunotherapy. J. Exp. Clin. Cancer Res. 2025, 44, 145. [Google Scholar] [CrossRef]
- Vecchiotti, D.; Clementi, L.; Cornacchia, E.; Di Vito Nolfi, M.; Verzella, D.; Capece, D.; Zazzeroni, F.; Angelucci, A. Evidence of the Link between Stroma Remodeling and Prostate Cancer Prognosis. Cancers 2024, 16, 3215. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.; Wang, R.; Yi, Z.; Luo, P.; Liu, W.; Xie, Y.; Liu, Z.; Xia, Z.; Zhang, H.; Cheng, Q. Tissue macrophages: Origin, heterogenity, biological functions, diseases and therapeutic targets. Signal Transduct. Target. Ther. 2025, 10, 93. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, M.; Ji, C.; Liu, X.; Gu, B.; Dong, T. Macrophage polarization in the tumor microenvironment: Emerging roles and therapeutic potentials. Biomed. Pharmacother. 2024, 177, 116930. [Google Scholar] [CrossRef]
- Yuan, Q.; Jia, L.; Yang, J.; Li, W. The role of macrophages in liver metastasis: Mechanisms and therapeutic prospects. Front. Immunol. 2025, 16, 1542197. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, X.; Zhang, X.; Li, Y.; Xu, R.; Li, H.J.; Zuo, D.; Chen, G. Unveiling the contribution of tumor-associated macrophages in driving epithelial-mesenchymal transition: A review of mechanisms and therapeutic Strategies. Front. Pharmacol. 2024, 15, 1404687. [Google Scholar] [CrossRef]
- Vitale, C.; Bottino, C.; Castriconi, R. Monocyte and Macrophage in Neuroblastoma: Blocking Their Pro-Tumoral Functions and Strengthening Their Crosstalk with Natural Killer Cells. Cells 2023, 12, 885. [Google Scholar] [CrossRef]
- Rakina, M.; Larionova, I.; Kzhyshkowska, J. Macrophage diversity in human cancers: New insight provided by single-cell resolution and spatial context. Heliyon 2024, 10, e28332. [Google Scholar] [CrossRef]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef]
- Selders, G.S.; Fetz, A.E.; Radic, M.Z.; Bowlin, G.L. An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regen. Biomater. 2017, 4, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Que, H.; Fu, Q.; Lan, T.; Tian, X.; Wei, X. Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochim. Et. Biophys. Acta. Rev. Cancer 2022, 1877, 188762. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; He, J.; Pan, Q.Z.; Yang, J.; Zhao, J.; Zhang, Y.J.; Huang, Y.; Tang, Y.; Wang, Q.; He, J.; et al. Cancer-Associated Fibroblast-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology 2021, 73, 1717–1735. [Google Scholar] [CrossRef] [PubMed]
- Akkız, H. Emerging Role of Cancer-Associated Fibroblasts in Progression and Treatment of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 3941. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, C.; Li, W.; Mao, Z.; Shi, Y.; Shi, H.; Ji, R.; Qian, H.; Xu, W.; Zhang, X. Tumor-Educated Neutrophils Activate Mesenchymal Stem Cells to Promote Gastric Cancer Growth and Metastasis. Front. Cell Dev. Biol. 2020, 8, 788. [Google Scholar] [CrossRef]
- Saxena, S.; Singh, A.; Singh, P. Tumor associated mast cells: Biological roles and therapeutic applications. Anat. Cell Biol. 2020, 53, 245–251. [Google Scholar] [CrossRef]
- Sobiepanek, A.; Kuryk, Ł.; Garofalo, M.; Kumar, S.; Baran, J.; Musolf, P.; Siebenhaar, F.; Fluhr, J.W.; Kobiela, T.; Plasenzotti, R.; et al. The Multifaceted Roles of Mast Cells in Immune Homeostasis, Infections and Cancers. Int. J. Mol. Sci. 2022, 23, 2249. [Google Scholar] [CrossRef]
- Turlej, E.; Domaradzka, A.; Radzka, J.; Drulis-Fajdasz, D.; Kulbacka, J.; Gizak, A. Cross-Talk Between Cancer and Its Cellular Environment-A Role in Cancer Progression. Cells 2025, 14, 403. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, R.; Tao, S.; Zhu, Y.; Luo, W.; Yang, Y.; Li, Y.; Zhou, K.; Zhao, Z. Stromal cell-derived chemokines modulate immune cells in inflammation: New findings and future perspectives. J. Immunol. 2025, vkaf136. [Google Scholar] [CrossRef] [PubMed]
- Segura-Villalobos, D.; Ramírez-Moreno, I.G.; Martínez-Aguilar, M.; Ibarra-Sánchez, A.; Muñoz-Bello, J.O.; Anaya-Rubio, I.; Padilla, A.; Macías-Silva, M.; Lizano, M.; González-Espinosa, C. Mast Cell-Tumor Interactions: Molecular Mechanisms of Recruitment, Intratumoral Communication and Potential Therapeutic Targets for Tumor Growth. Cells 2022, 11, 349. [Google Scholar] [CrossRef]
- Yang, F.C.; Chen, S.; Clegg, T.; Li, X.; Morgan, T.; Estwick, S.A.; Yuan, J.; Khalaf, W.; Burgin, S.; Travers, J.; et al. Nf1+/− mast cells induce neurofibroma like phenotypes through secreted TGF-beta signaling. Hum. Mol. Genet. 2006, 15, 2421–2437. [Google Scholar] [CrossRef]
- Chen, H.; Fang, S.; Zhu, X.; Liu, H. Cancer-associated fibroblasts and prostate cancer stem cells: Crosstalk mechanisms and implications for disease progression. Front. Cell Dev. Biol. 2024, 12, 1412337. [Google Scholar] [CrossRef] [PubMed]
- Perera Molligoda Arachchige, A.S. Human NK cells: From development to effector functions. Innate Immun. 2021, 27, 212–229. [Google Scholar] [CrossRef]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Hu, J.; Wang, Z.; Xu, F.; Liu, Y.; Cui, L.; Zhang, H.; Xie, C.; Yao, R.; Jin, H.; et al. RAC2 inhibition enhances tumor sensitivity to NK cell-mediated cytotoxicity. J. Immunother. Cancer 2025, 13, e010931. [Google Scholar] [CrossRef]
- Ielpo, S.; Barberini, F.; Dabbagh Moghaddam, F.; Pesce, S.; Cencioni, C.; Spallotta, F.; De Ninno, A.; Businaro, L.; Marcenaro, E.; Bei, R.; et al. Crosstalk and communication of cancer-associated fibroblasts with natural killer and dendritic cells: New frontiers and unveiled opportunities for cancer immunotherapy. Cancer Treat. Rev. 2024, 131, 102843. [Google Scholar] [CrossRef]
- Wang, D.; Dou, L.; Sui, L.; Xue, Y.; Xu, S. Natural killer cells in cancer immunotherapy. MedComm 2024, 5, e626. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, B.; Li, B.; Wu, H.; Jiang, M. Cold and hot tumors: From molecular mechanisms to targeted therapy. Signal Transduct. Target. Ther. 2024, 9, 274. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Wang, S.; Zhang, Y.; Tian, W.; Mai, G.; Xu, Y.; Xiao, W.; Graves, E.E.; Wu, F. Tumor-educated cells in tumor microenvironment: Key drivers of immunotherapy resistance. Chin. J. Cancer Res. 2025, 37, 446–465. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Tran Janco, J.M.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-infiltrating dendritic cells in cancer pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef]
- Ness, S.; Lin, S.; Gordon, J.R. Regulatory Dendritic Cells, T Cell Tolerance, and Dendritic Cell Therapy for Immunologic Disease. Front. Immunol. 2021, 12, 633436. [Google Scholar] [CrossRef]
- Chen, J.; Cui, L.; Lu, S.; Xu, S. Amino acid metabolism in tumor biology and therapy. Cell Death Dis. 2024, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target. Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Mutair, A.A.; Alawi, Z.A.; Alhumaid, S.; Dhama, K. Regulatory T cells (Tregs) and their therapeutic potential against autoimmune disorders—Advances and challenges. Hum. Vaccines Immunother. 2022, 18, 2035117. [Google Scholar] [CrossRef]
- Zhu, X.; Zhu, J. CD4 T Helper Cell Subsets and Related Human Immunological Disorders. Int. J. Mol. Sci. 2020, 21, 8011. [Google Scholar] [CrossRef]
- Wu, X.; Tian, J.; Wang, S. Insight Into Non-Pathogenic Th17 Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 1112. [Google Scholar] [CrossRef]
- Mentucci, F.M.; Ferrara, M.G.; Ercole, A.; Rumie Vittar, N.B.; Lamberti, M.J. Interplay between cancer-associated fibroblasts and dendritic cells: Implications for tumor immunity. Front. Immunol. 2025, 16, 1515390. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Montagnani, I.; Raspollini, M.R.; Serni, S.; Simeoni, L.; et al. Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, D.; Qian, H.; Shi, Y.; Tao, Z. CD8(+) T cell-based cancer immunotherapy. J. Transl. Med. 2024, 22, 394. [Google Scholar] [CrossRef] [PubMed]
- Freeman, P.; Mielgo, A. Cancer-Associated Fibroblast Mediated Inhibition of CD8+ Cytotoxic T Cell Accumulation in Tumours: Mechanisms and Therapeutic Opportunities. Cancers 2020, 12, 2687. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2019, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.Q.; Wang, S.B.; Liu, J.H.; Jin, L.; Liu, Y.; Li, C.Y.; Su, Y.R.; Liu, Y.R.; Sang, X.; Wan, Q.; et al. Modifying the tumour microenvironment and reverting tumour cells: New strategies for treating malignant tumours. Cell Prolif. 2020, 53, e12865. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Ma, W.; Wu, J.; Wu, Q.; Sun, C. Paracrine signaling in cancer-associated fibroblasts: Central regulators of the tumor immune microenvironment. J. Transl. Med. 2025, 23, 697. [Google Scholar] [CrossRef]
- Goehrig, D.; Nigri, J.; Samain, R.; Wu, Z.; Cappello, P.; Gabiane, G.; Zhang, X.; Zhao, Y.; Kim, I.S.; Chanal, M.; et al. Stromal protein βig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut 2019, 68, 693–707. [Google Scholar] [CrossRef]
- Qin, Q.; Yu, R.; Eriksson, J.E.; Tsai, H.I.; Zhu, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma therapy: Challenges and opportunities. Cancer Lett. 2024, 591, 216859. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Nakahata, S.; Iha, H. Complex Role of Regulatory T Cells (Tregs) in the Tumor Microenvironment: Their Molecular Mechanisms and Bidirectional Effects on Cancer Progression. Int. J. Mol. Sci. 2024, 25, 7346. [Google Scholar] [CrossRef]
- Sabit, H.; Adel, A.; Abdelfattah, M.M.; Ramadan, R.M.; Nazih, M.; Abdel-Ghany, S.; El-Hashash, A.; Arneth, B. The role of tumor microenvironment and immune cell crosstalk in triple-negative breast cancer (TNBC): Emerging therapeutic opportunities. Cancer Lett. 2025, 628, 217865. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; Deschoolmeester, V.; Zwaenepoel, K.; Flieswasser, T.; Deben, C.; Van den Bossche, J.; Hermans, C.; Rolfo, C.; Peeters, M.; De Wever, O.; et al. Unveiling a CD70-positive subset of cancer-associated fibroblasts marked by pro-migratory activity and thriving regulatory T cell accumulation. Oncoimmunology 2018, 7, e1440167. [Google Scholar] [CrossRef]
- Bourhis, M.; Palle, J.; Galy-Fauroux, I.; Terme, M. Direct and Indirect Modulation of T Cells by VEGF-A Counteracted by Anti-Angiogenic Treatment. Front. Immunol. 2021, 12, 616837. [Google Scholar] [CrossRef]
- Zhao, X.; Ding, L.; Lu, Z.; Huang, X.; Jing, Y.; Yang, Y.; Chen, S.; Hu, Q.; Ni, Y. Diminished CD68(+) Cancer-Associated Fibroblast Subset Induces Regulatory T-Cell (Treg) Infiltration and Predicts Poor Prognosis of Oral Squamous Cell Carcinoma Patients. Am. J. Pathol. 2020, 190, 886–899. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, W.; Lin, S.; Liu, B.; Wu, P.; Li, L. Fibroblast diversity and plasticity in the tumor microenvironment: Roles in immunity and relevant therapies. Cell Commun. Signal 2023, 21, 234. [Google Scholar] [CrossRef]
- Saúde-Conde, R.; Arçay Öztürk, A.; Stosic, K.; Azurmendi Senar, O.; Navez, J.; Bouchart, C.; Arsenijevic, T.; Flamen, P.; Van Laethem, J.L. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma or a Metaphor for Heterogeneity: From Single-Cell Analysis to Whole-Body Imaging. Biomedicines 2024, 12, 591. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Ramil, C.P.; Hai, J.; Zhang, C.; Wang, H.; Watkins, A.A.; Afshar, R.; Georgiev, P.; Sze, M.A.; Song, X.S.; et al. Cancer-Associated Fibroblasts Promote Immunosuppression by Inducing ROS-Generating Monocytic MDSCs in Lung Squamous Cell Carcinoma. Cancer Immunol. Res. 2020, 8, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Huang, L.; Qin, G.; Qiao, Y.; Ren, F.; Shen, C.; Wang, S.; Liu, S.; Lian, J.; Wang, D.; et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021, 518, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Eskandari-Malayeri, F.; Rezaei, M. Immune checkpoint inhibitors as mediators for immunosuppression by cancer-associated fibroblasts: A comprehensive review. Front. Immunol. 2022, 13, 996145. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Wu, J.; Shen, B.; Jiang, F.; Feng, J. Cancer-associated fibroblasts and resistance to anticancer therapies: Status, mechanisms, and countermeasures. Cancer Cell Int. 2022, 22, 166. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, J.; Zhang, J.; Li, S.; Wang, H.; Du, J. Cancer-associated fibroblasts promote PD-L1 expression in mice cancer cells via secreting CXCL5. Int. J. Cancer 2019, 145, 1946–1957. [Google Scholar] [CrossRef]
- Özdirik, B.; Jann, H.; Bischoff, P.; Fehrenbach, U.; Tacke, F.; Roderburg, C.; Wiedenmann, B. PD-L1—Inhibitors in neuroendocrine neoplasia: Results from a real-life study. Medicine 2021, 100, e23835. [Google Scholar] [CrossRef]
- Belhabib, I.; Zaghdoudi, S.; Lac, C.; Bousquet, C.; Jean, C. Extracellular Matrices and Cancer-Associated Fibroblasts: Targets for Cancer Diagnosis and Therapy? Cancers 2021, 13, 3466. [Google Scholar] [CrossRef]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef]
- Prakash, J.; Shaked, Y. The Interplay between Extracellular Matrix Remodeling and Cancer Therapeutics. Cancer Discov. 2024, 14, 1375–1388. [Google Scholar] [CrossRef]
- Dzobo, K.; Dandara, C. The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis. Biomimetics 2023, 8, 146. [Google Scholar] [CrossRef]
- Hu, Q.; Zhu, Y.; Mei, J.; Liu, Y.; Zhou, G. Extracellular matrix dynamics in tumor immunoregulation: From tumor microenvironment to immunotherapy. J. Hematol. Oncol. 2025, 18, 65. [Google Scholar] [CrossRef]
- Zou, L.; Xian, P.; Pu, Q.; Song, Y.; Ni, S.; Chen, L.; Hu, K. Nano-drug delivery strategies affecting cancer-associated fibroblasts to reduce tumor metastasis. Acta Pharm. Sin. B 2025, 15, 1841–1868. [Google Scholar] [CrossRef]
- Klabukov, I.; Kabakov, A.E.; Yakimova, A.; Baranovskii, D.; Sosin, D.; Atiakshin, D.; Ignatyuk, M.; Yatsenko, E.; Rybachuk, V.; Evstratova, E.; et al. Tumor-Associated Extracellular Matrix Obstacles for CAR-T Cell Therapy: Approaches to Overcoming. Curr. Oncol. 2025, 32, 79. [Google Scholar] [CrossRef]
- Mazumdar, A.; Urdinez, J.; Boro, A.; Migliavacca, J.; Arlt, M.J.E.; Muff, R.; Fuchs, B.; Snedeker, J.G.; Gvozdenovic, A. Osteosarcoma-Derived Extracellular Vesicles Induce Lung Fibroblast Reprogramming. Int. J. Mol. Sci. 2020, 21, 5451. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Qian, C.; Song, M.; Zhong, C.; Zhao, Y.; Lu, Y. Fibroblasts: Invigorated targets in pre-metastatic niche formation. Int. J. Biol. Sci. 2024, 20, 1110–1124. [Google Scholar] [CrossRef]
- Jiang, H.; Liu, X.; Knolhoff, B.L.; Hegde, S.; Lee, K.B.; Jiang, H.; Fields, R.C.; Pachter, J.A.; Lim, K.H.; DeNardo, D.G. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut 2020, 69, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Ng, K.Y.; Bakhtiar, A. Extracellular matrix: Unlocking new avenues in cancer treatment. Biomark. Res. 2025, 13, 78. [Google Scholar] [CrossRef]
- Feng, X.; Cao, F.; Wu, X.; Xie, W.; Wang, P.; Jiang, H. Targeting extracellular matrix stiffness for cancer therapy. Front. Immunol. 2024, 15, 1467602. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, T.; Sun, L.; Yuan, Y.; Zhu, Y. Potential mechanisms of cancer-associated fibroblasts in therapeutic resistance. Biomed. Pharmacother. 2023, 166, 115425. [Google Scholar] [CrossRef] [PubMed]
- Koppensteiner, L.; Mathieson, L.; O’Connor, R.A.; Akram, A.R. Cancer Associated Fibroblasts—An Impediment to Effective Anti-Cancer T Cell Immunity. Front. Immunol. 2022, 13, 887380. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.; Jungwirth, U.; Avgustinova, A.; Iravani, M.; Mills, A.; Haider, S.; Harper, J.; Isacke, C.M. Cancer-Associated Fibroblasts Suppress CD8+ T-cell Infiltration and Confer Resistance to Immune-Checkpoint Blockade. Cancer Res. 2022, 82, 2904–2917. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Lekan, A.A.; Weiner, L.M. The Role of Chemokines in Orchestrating the Immune Response to Pancreatic Ductal Adenocarcinoma. Cancers 2024, 16, 559. [Google Scholar] [CrossRef]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef]
- Yu, X.; Qian, J.; Ding, L.; Yin, S.; Zhou, L.; Zheng, S. Galectin-1: A Traditionally Immunosuppressive Protein Displays Context-Dependent Capacities. Int. J. Mol. Sci. 2023, 24, 6501. [Google Scholar] [CrossRef]
- Guo, T.; Xu, J. Cancer-associated fibroblasts: A versatile mediator in tumor progression, metastasis, and targeted therapy. Cancer Metastasis Rev. 2024, 43, 1095–1116. [Google Scholar] [CrossRef]
- Yamazaki, M.; Ishimoto, T. Targeting Cancer-Associated Fibroblasts: Eliminate or Reprogram? Cancer Sci. 2025, 116, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wei, R.; Liu, C.; Zhao, Z.; Liu, X.; Wang, Y.; Liu, F.; Liu, X. Antigen-presenting cancer associated fibroblasts enhance antitumor immunity and predict immunotherapy response. Nat. Commun. 2025, 16, 2175. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, R.; Chen, L. Drug Delivery System Targeting Cancer-Associated Fibroblast for Improving Immunotherapy. Int. J. Nanomed. 2025, 20, 483–503. [Google Scholar] [CrossRef]
- Fei, B.; Mo, Z.; Yang, J.; Wang, Z.; Li, S. Nanodrugs Reprogram Cancer-Associated Fibroblasts and Normalize Tumor Vasculatures for Sequentially Enhancing Photodynamic Therapy of Hepatocellular Carcinoma. Int. J. Nanomed. 2023, 18, 6379–6391. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huang, H.; Cheng, B.; Xie, H.; Peng, W.; Cui, H.; Liang, J.; Cao, M.; Yang, Y.; Chen, W.; et al. Revealing the role of cancer-associated fibroblast senescence in prognosis and immune landscape in pancreatic cancer. iScience 2025, 28, 111612. [Google Scholar] [CrossRef]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, G.; Tuveson, D.A. Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell 2023, 41, 434–449. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Strategy | Mechanism | Therapeutic Agents/Approach | Tumor Type(s) | Clinical Status | References |
|---|---|---|---|---|---|
| CAF reprogramming to antitumor phenotype | Converts tumor-promoting CAFs to tumor-suppressive phenotypes |
| Pancreatic, breast | Preclinical–Phase I | [168] |
| CAF-induced immune activation | Enhances CAF antigen presentation to stimulate T cell activity |
| Pancreatic, melanoma | Preclinical | [169] |
| CAF-mediated delivery of cytotoxic agents | Engineers CAFs to produce or deliver drugs locally |
| Solid tumors | Preclinical | [170] |
| Promotion of tumor vessel normalization | Targets CAF signaling to normalize vasculature |
| Breast, colorectal, pancreatic | Phase I–II | [171] |
| CAF-targeted senescence induction | Induces senescence in tumor-promoting CAFs |
| Breast, pancreatic | Preclinical | [172] |
| Restoring ECM homeostasis via CAFs | CAF modulation reduces excessive ECM stiffness and desmoplasia |
| Pancreatic, liver | Phase II–III | [37] |
| CAF–immune cell cotargeting | Concurrently modulates CAFs and immune cells for synergistic tumor killing |
| Pancreatic, lung, melanoma | Early clinical trials | [170] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawar, J.S.; Salam, M.A.; Dipto, M.S.U.; Al-Amin, M.Y.; Salam, M.T.; Sengupta, S.; Kumari, S.; Gujjari, L.; Yadagiri, G. Cancer-Associated Fibroblasts: Immunosuppressive Crosstalk with Tumor-Infiltrating Immune Cells and Implications for Therapeutic Resistance. Cancers 2025, 17, 2484. https://doi.org/10.3390/cancers17152484
Pawar JS, Salam MA, Dipto MSU, Al-Amin MY, Salam MT, Sengupta S, Kumari S, Gujjari L, Yadagiri G. Cancer-Associated Fibroblasts: Immunosuppressive Crosstalk with Tumor-Infiltrating Immune Cells and Implications for Therapeutic Resistance. Cancers. 2025; 17(15):2484. https://doi.org/10.3390/cancers17152484
Chicago/Turabian StylePawar, Jogendra Singh, Md. Abdus Salam, Md. Shalman Uddin Dipto, Md. Yusuf Al-Amin, Moushumi Tabassoom Salam, Sagnik Sengupta, Smita Kumari, Lohitha Gujjari, and Ganesh Yadagiri. 2025. "Cancer-Associated Fibroblasts: Immunosuppressive Crosstalk with Tumor-Infiltrating Immune Cells and Implications for Therapeutic Resistance" Cancers 17, no. 15: 2484. https://doi.org/10.3390/cancers17152484
APA StylePawar, J. S., Salam, M. A., Dipto, M. S. U., Al-Amin, M. Y., Salam, M. T., Sengupta, S., Kumari, S., Gujjari, L., & Yadagiri, G. (2025). Cancer-Associated Fibroblasts: Immunosuppressive Crosstalk with Tumor-Infiltrating Immune Cells and Implications for Therapeutic Resistance. Cancers, 17(15), 2484. https://doi.org/10.3390/cancers17152484