
Targeting Triple-Negative Breast Cancer with Momordicine-I for Therapeutic Gain in Preclinical Models




Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Momordicine-I (M-I) Treatment
2.2. Cell Viability Assay
2.3. RNA Isolation and Quantitative Real-Time PCR
2.4. Western Blot Analysis
2.5. Cell Cycle Analysis
2.6. Animal Model
2.7. Flow Cytometry for Tumor-Infiltrated Immune Cells
2.8. Statistical Analysis
3. Results
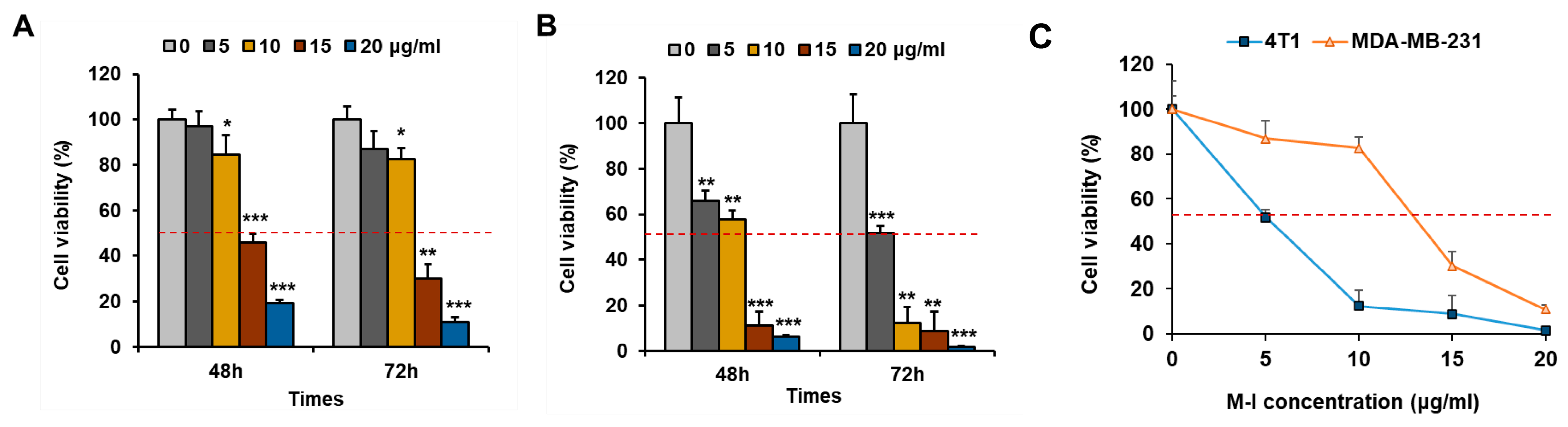
3.1. M-I Treatment Reduces Proliferation of Triple-Negative Breast Cancer Cells In Vitro
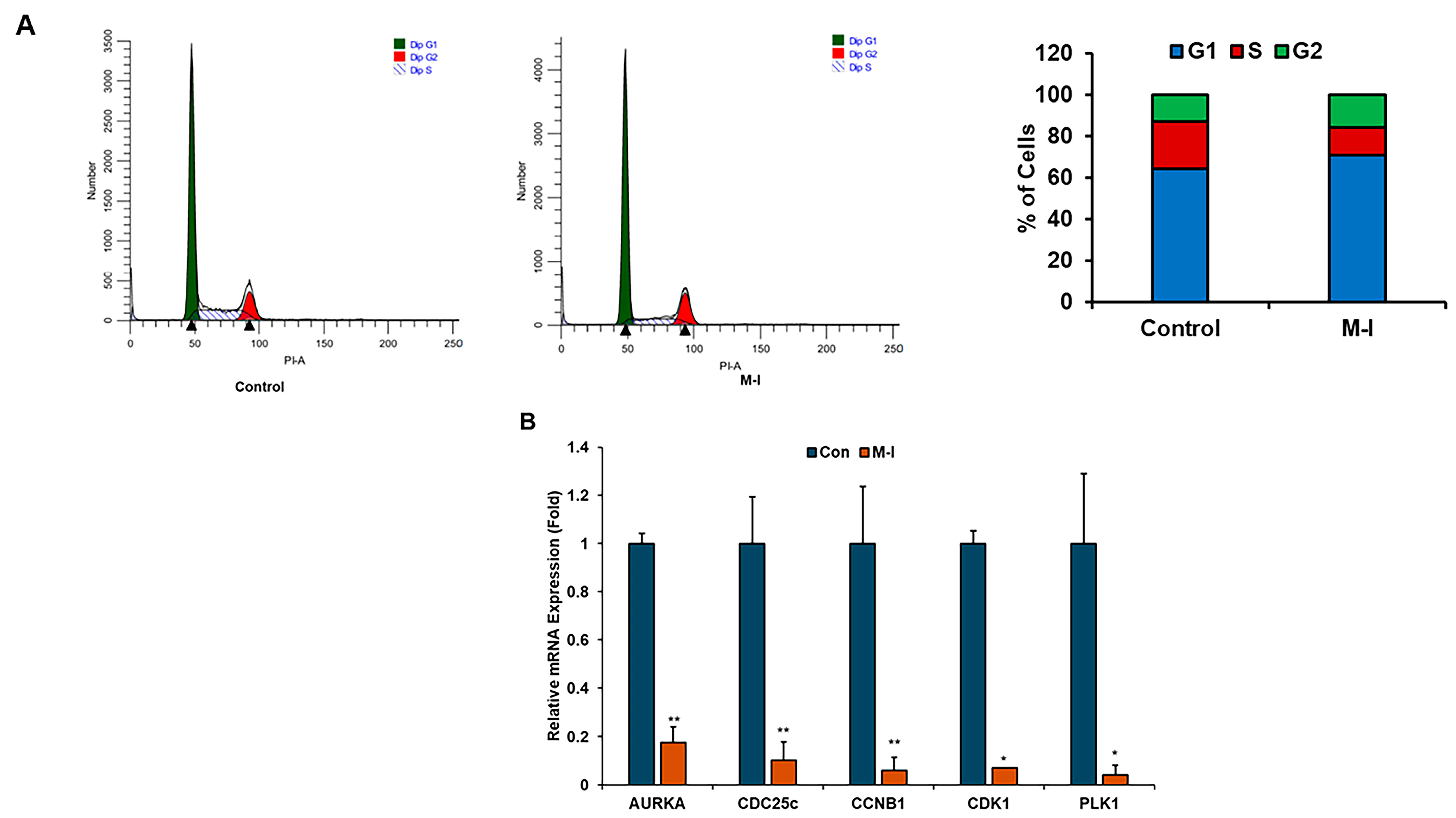
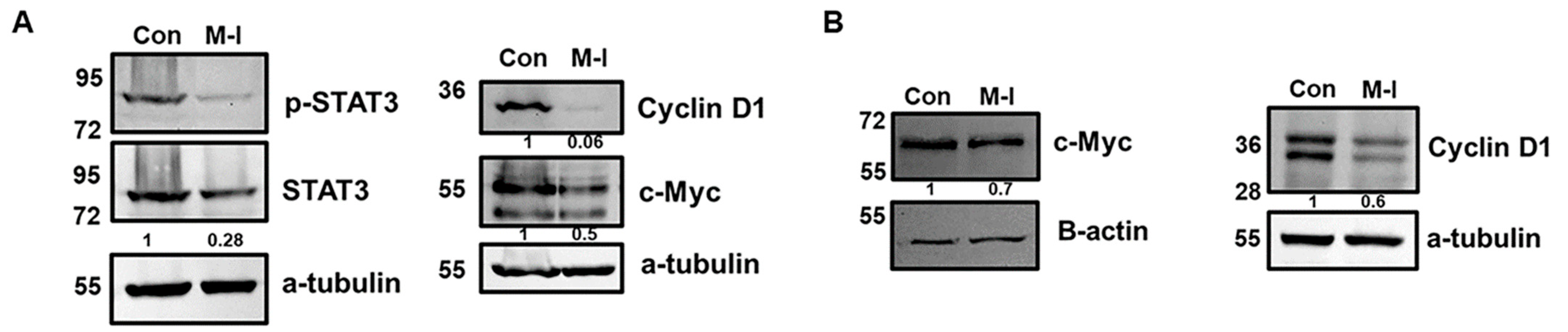
3.2. M-I Decreases Breast Cancer Growth Through Regulation of Cell Cycle Genes
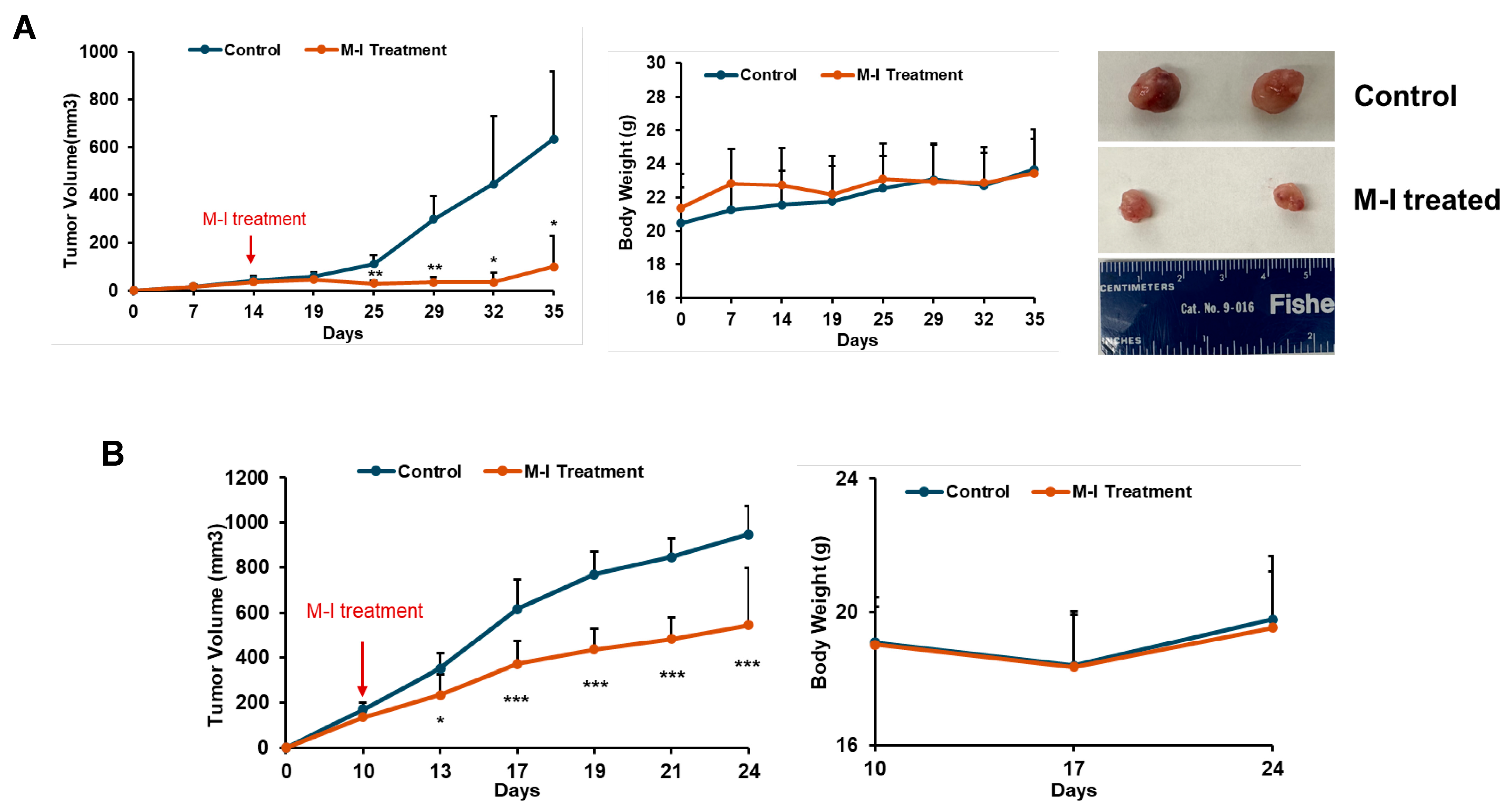
3.3. M-I Treatment Suppresses Tumor Progression in Orthotropic TNBC Mouse Models
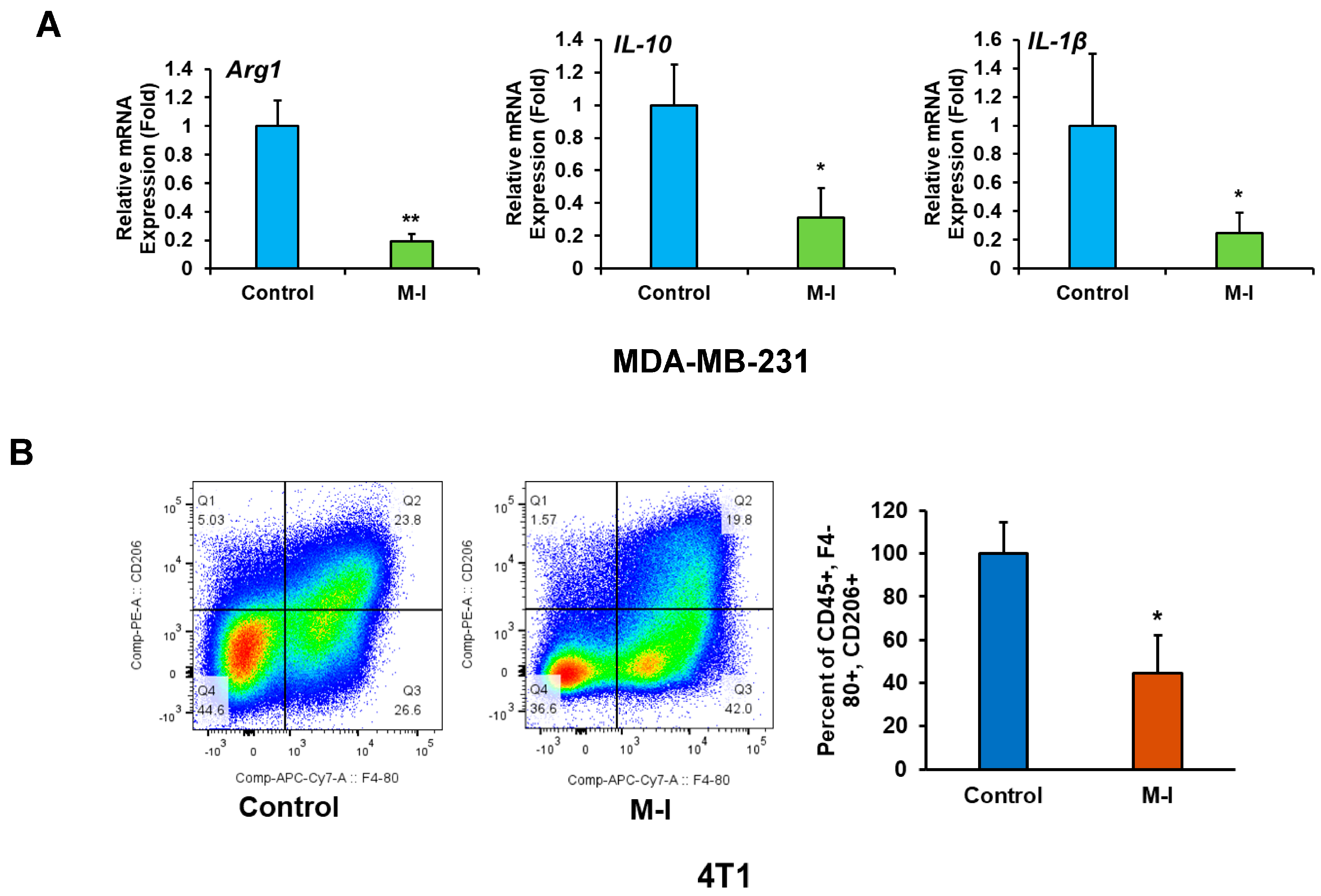
3.4. M-I Plays an Immunomodulatory Role in TNBC Tumors
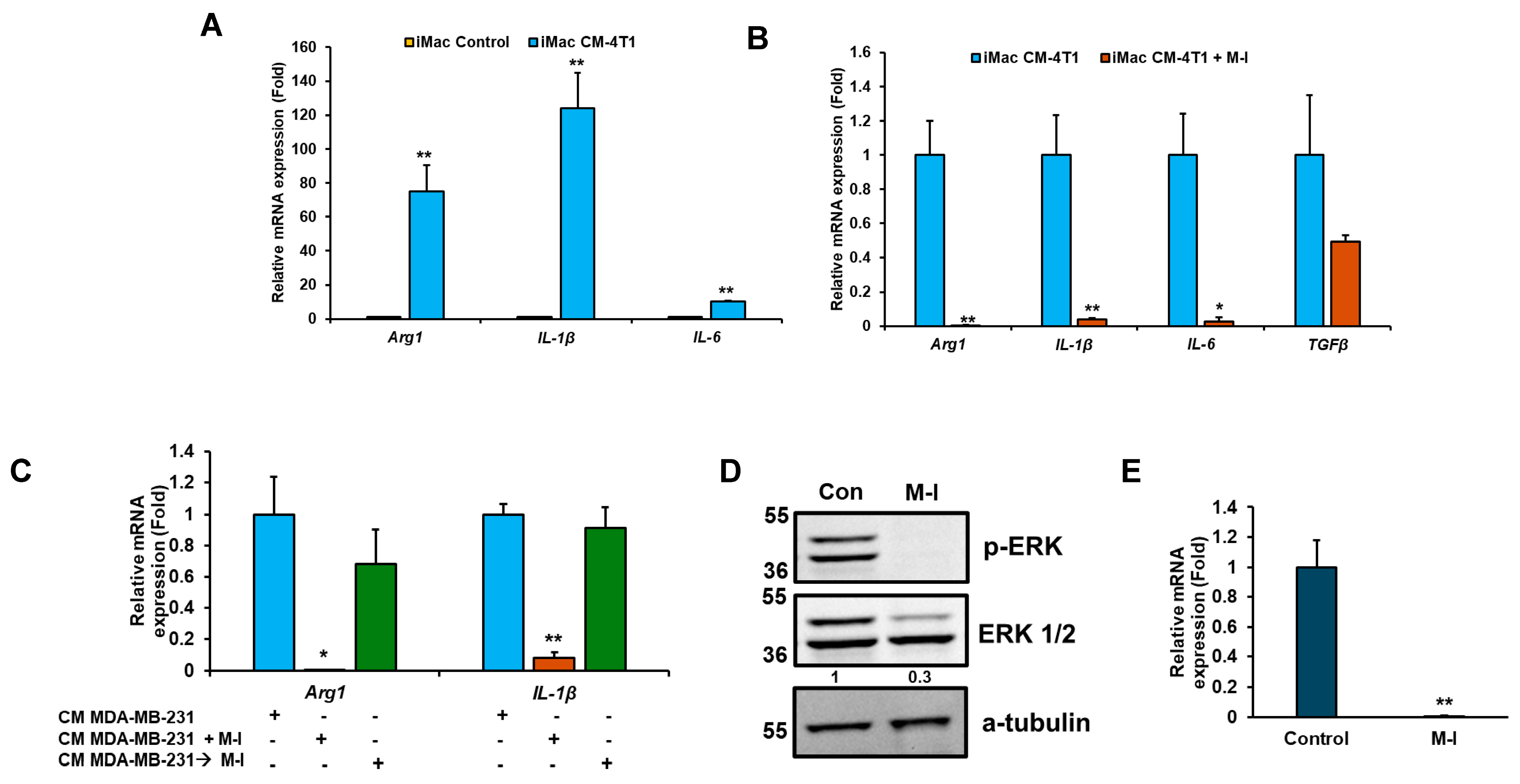
3.5. M-I Treatment Suppresses M2 Macrophage Polarization In Vitro
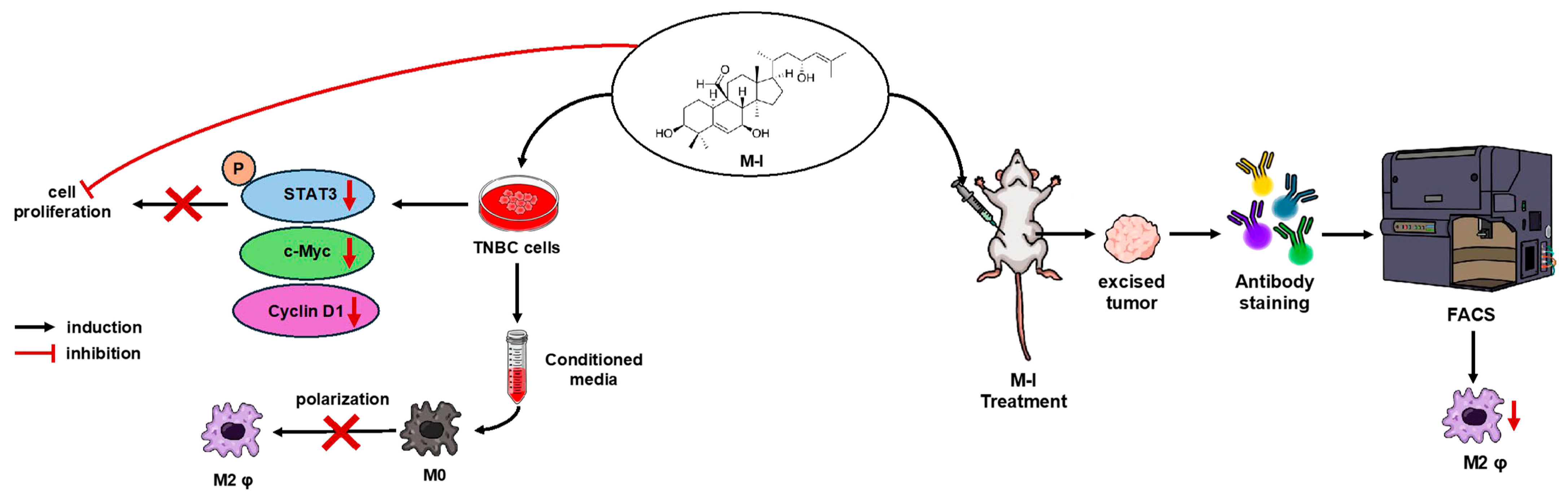
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13 Pt 1, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Penn, A.; Bhandari, N.; Petrovich, R.; DeLiberto, L.K.; Burcher, J.T.; Barbalho, S.M.; Nagini, S. Dietary plants for oral cancer prevention and therapy: A review of preclinical and clinical studies. Phytother. Res. 2024, 38, 5225–5263. [Google Scholar] [CrossRef] [PubMed]
- Nerurkar, P.; Ray, R.B. Bitter melon: Antagonist to cancer. Pharm. Res. 2010, 27, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Gano, C.A.; Fatima, S.; Failes, T.W.; Arndt, G.M.; Sajinovic, M.; Mahns, D.; Saedisomeolia, A.; Coorssen, J.R.; Bucci, J.; de Souza, P.; et al. Anti-cancer potential of synergistic phytochemical combinations is influenced by the genetic profile of prostate cancer cell lines. Front. Nutr. 2023, 10, 1119274. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in Cancer Treatment: From Preclinical Studies to Clinical Practice. Front. Pharmacol. 2019, 10, 1614. [Google Scholar]
- Wang, H.; Khor, T.O.; Shu, L.; Su, Z.Y.; Fuentes, F.; Lee, J.H.; Kong, A.N. Plants vs. cancer: A review on natural phytochemicals in preventing and treating cancers and their druggability. Anti-Cancer Agents Med. Chem. 2012, 12, 1281–1305. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Ray, R.B. Bitter Melon (Momordica Charantia), a Nutraceutical Approach for Cancer Prevention and Therapy. Cancers 2020, 12, 2064. [Google Scholar] [CrossRef] [PubMed]
- Dandawate, P.R.; Subramaniam, D.; Padhye, S.B.; Anant, S. Bitter melon: A panacea for inflammation and cancer. Chin. J. Nat. Med. 2016, 14, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Raina, K.; Kumar, D.; Agarwal, R. Promise of bitter melon (Momordica charantia) bioactives in cancer prevention and therapy. Semin. Cancer Biol. 2016, 40–41, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Bhartiya, P.; Steele, R.; Brennan, M.; DiPaolo, R.J.; Ray, R.B. Momordicine-I Suppresses Head and Neck Cancer Growth by Reprogrammimg Immunosuppressive Effect of the Tumor-Infiltrating Macrophages and B Lymphocytes. Mol. Cancer Ther. 2024, 23, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Steele, R.; Isbell, T.S.; Venkata, K.N.; Rateb, M.E.; Ray, R.B. Momordicine-I, a Bitter Melon Bioactive Metabolite, Displays Anti-Tumor Activity in Head and Neck Cancer Involving c-Met and Downstream Signaling. Cancers 2021, 13, 1432. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Tran, E.T.; Patel, R.A.; Luetzen, M.A.; Cho, K.; Shriver, L.P.; Patti, G.J.; Varvares, M.A.; Ford, D.A.; McCommis, K.S.; et al. Momordicine-I suppresses head and neck cancer growth by modulating key metabolic pathways. Cell Commun. Signal. 2024, 22, 597. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Harris, M.B.; Rothman, P. IL-4/IL-13 signaling beyond JAK/STAT. J. Allergy Clin. Immunol. 2000, 105 Pt 1, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sioud, M. Tumor-Associated Macrophage Subsets: Shaping Polarization and Targeting. Int. J. Mol. Sci. 2023, 24, 7493. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Triple-negative breast cancer therapeutic resistance: Where is the Achilles’ heel? Cancer Lett. 2021, 497, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Hashem, S.; Ali, T.A.; Akhtar, S.; Nisar, S.; Sageena, G.; Ali, S.; Al-Mannai, S.; Therachiyil, L.; Mir, R.; Elfaki, I.; et al. Targeting cancer signaling pathways by natural products: Exploring promising anti-cancer agents. Biomed. Pharmacother. 2022, 150, 113054. [Google Scholar] [CrossRef] [PubMed]
- Bailon-Moscoso, N.; Cevallos-Solorzano, G.; Romero-Benavides, J.C.; Orellana, M.I. Natural Compounds as Modulators of Cell Cycle Arrest: Application for Anticancer Chemotherapies. Curr. Genom. 2017, 18, 106–131. [Google Scholar] [CrossRef] [PubMed]
- Kwatra, D.; Subramaniam, D.; Ramamoorthy, P.; Standing, D.; Moran, E.; Velayutham, R.; Mitra, A.; Umar, S.; Anant, S. Methanolic extracts of bitter melon inhibit colon cancer stem cells by affecting energy homeostasis and autophagy. Evid.-Based Complement. Altern. Med. 2013, 2013, 702869. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; He, S. The Characteristics of Tumor Microenvironment in Triple Negative Breast Cancer. Cancer Manag. Res. 2022, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Monkkonen, J.; Kellokumpu-Lehtinen, P.L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 101. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, T.; Xia, R.; Wei, Y.; Wei, X. Targeting the tumor stroma for cancer therapy. Mol. Cancer 2022, 21, 208. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schroder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Baumann, D.; Drebant, J.; Hagele, T.; Burger, L.; Serger, C.; Lauenstein, C.; Dudys, P.; Erdmann, G.; Offringa, R. p38 MAPK signaling in M1 macrophages results in selective elimination of M2 macrophages by MEK inhibition. J. Immunother. Cancer 2021, 9, e002319. [Google Scholar] [CrossRef] [PubMed]
- Metge, B.J.; Williams, L.; Swain, C.A.; Hinshaw, D.C.; Elhamamsy, A.R.; Chen, D.; Samant, R.S.; Shevde, L.A. Ribosomal RNA Biosynthesis Functionally Programs Tumor-Associated Macrophages to Support Breast Cancer Progression. Cancer Res. 2025, 85, 1459–1478. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, X.; Sun, Q.; Zhang, W.; Liu, C.; Ma, W.; Sun, C. Natural compounds: A new perspective on targeting polarization and infiltration of tumor-associated macrophages in lung cancer. Biomed. Pharmacother. 2022, 151, 113096. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Prasannan, A.; Venkatachalam, K.; Binesh, A. Exploring the mechanism and crosstalk between IL-6 and IL-1β on M2 macrophages under metabolic stress conditions. Cytokine 2025, 186, 156852. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Hui, X.; Wang, J.; Zeng, H.; Yan, Y.; Hu, Q.; Ge, G.; Lei, T. Matrine suppresses lung cancer metastasis via targeting M2-like tumour-associated-macrophages polarization. Am. J. Cancer Res. 2021, 11, 4308–4328. [Google Scholar] [PubMed]
- Sanmarco, L.M.; Ponce, N.E.; Visconti, L.M.; Eberhardt, N.; Theumer, M.G.; Minguez, A.R.; Aoki, M.P. IL-6 promotes M2 macrophage polarization by modulating purinergic signaling and regulates the lethal release of nitric oxide during Trypanosoma cruzi infection. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, Z.; Song, Y.; Zhang, B.; Fan, C. Resveratrol-driven macrophage polarization: Unveiling mechanisms and therapeutic potential. Front. Pharmacol. 2024, 15, 1516609. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, S.; Nemati, M.; Zandvakili, R.; Jafarzadeh, A. Modulation of M1 and M2 macrophage polarization by metformin: Implications for inflammatory diseases and malignant tumors. Int. Immunopharmacol. 2025, 151, 114345. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Garcia, L.; Herranz, S.; Luque, A.; Hortelano, S. Critical role of p38 MAPK in IL-4-induced alternative activation of peritoneal macrophages. Eur. J. Immunol. 2015, 45, 273–286. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Species | Primer Type | Sequence |
|---|---|---|---|
| AURKA | Hs | FP | 5′-TCC TAA TAA GGT ACC TGG AGG GAT G-3′ |
| RP | 5′-AGC TGC AAG ATC TCC GTA CGC TTA C-3′ | ||
| CDC25c | Hs | FP | 5′-GAA GAG GAC AGG TCT CTG AA-3′ |
| RP | 5′-CTC AGT CTT GTG GTC CTG AT-3′ | ||
| CCNB1 | Hs | FP | 5′-CCT GAG CCT GTT AAA GAA GA-3′ |
| RP | 5′-TTC TGC ATC CAC ATC ATT TA-3′ | ||
| CDK1 | Hs | FP | 5′-CTC CCA ATA ATG AAG TGT GG-3′ |
| RP | 5′-GTT TGG CTG GAT CAT AGA TT-3′ | ||
| PLK1 | Hs | FP | 5′-CGA TAC TAC CTA CGG CAA AT-3′ |
| RP | 5′-CGG GAG CTA TGT AAT TAG GA-3′ | ||
| Arg1 | Mm | FP | 5′-CTC CAA GCC AAA GTC CTT AGA G-3′ |
| RP | 5′-AGG AGC TGT CAT TAG GGA CAT C-3′ | ||
| IL-10 | Mm | FP | 5′-GCT CTT ACT GAC TGG CAT GAG-3′ |
| RP | 5′-CGC AGC TCT AGG AGC ATG TG-3′ | ||
| IL-1β | Mm | FP | 5′-GCA ACT GTT CCT GAA CTC AAC T-3′ |
| RP | 5′-ATC TTT TGG GGT CCG TCA ACT-3′ | ||
| IL-6 | Mm | FP | 5′-TGT GCA ATG GCA ATT CTG AT-3′ |
| RP | 5′-GGA AAT TGG GGT AGG AAG GA-3′ | ||
| IL-4 | Mm | FP | 5′-TCT GCA TCC CGT TGT TTT GC-3′ |
| RP | 5′-GCA CCT GTG CAT CCT GAA TG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kesh, K.; Tran, E.T.; Patel, R.A.; Ma, C.X.; Ray, R.B. Targeting Triple-Negative Breast Cancer with Momordicine-I for Therapeutic Gain in Preclinical Models. Cancers 2025, 17, 2342. https://doi.org/10.3390/cancers17142342
Kesh K, Tran ET, Patel RA, Ma CX, Ray RB. Targeting Triple-Negative Breast Cancer with Momordicine-I for Therapeutic Gain in Preclinical Models. Cancers. 2025; 17(14):2342. https://doi.org/10.3390/cancers17142342
Chicago/Turabian StyleKesh, Kousik, Ellen T. Tran, Ruchi A. Patel, Cynthia X. Ma, and Ratna B. Ray. 2025. "Targeting Triple-Negative Breast Cancer with Momordicine-I for Therapeutic Gain in Preclinical Models" Cancers 17, no. 14: 2342. https://doi.org/10.3390/cancers17142342
APA StyleKesh, K., Tran, E. T., Patel, R. A., Ma, C. X., & Ray, R. B. (2025). Targeting Triple-Negative Breast Cancer with Momordicine-I for Therapeutic Gain in Preclinical Models. Cancers, 17(14), 2342. https://doi.org/10.3390/cancers17142342