
Metabolic Adaptations in Cancer Progression: Optimization Strategies and Therapeutic Targets



Simple Summary
Abstract

1. Introduction
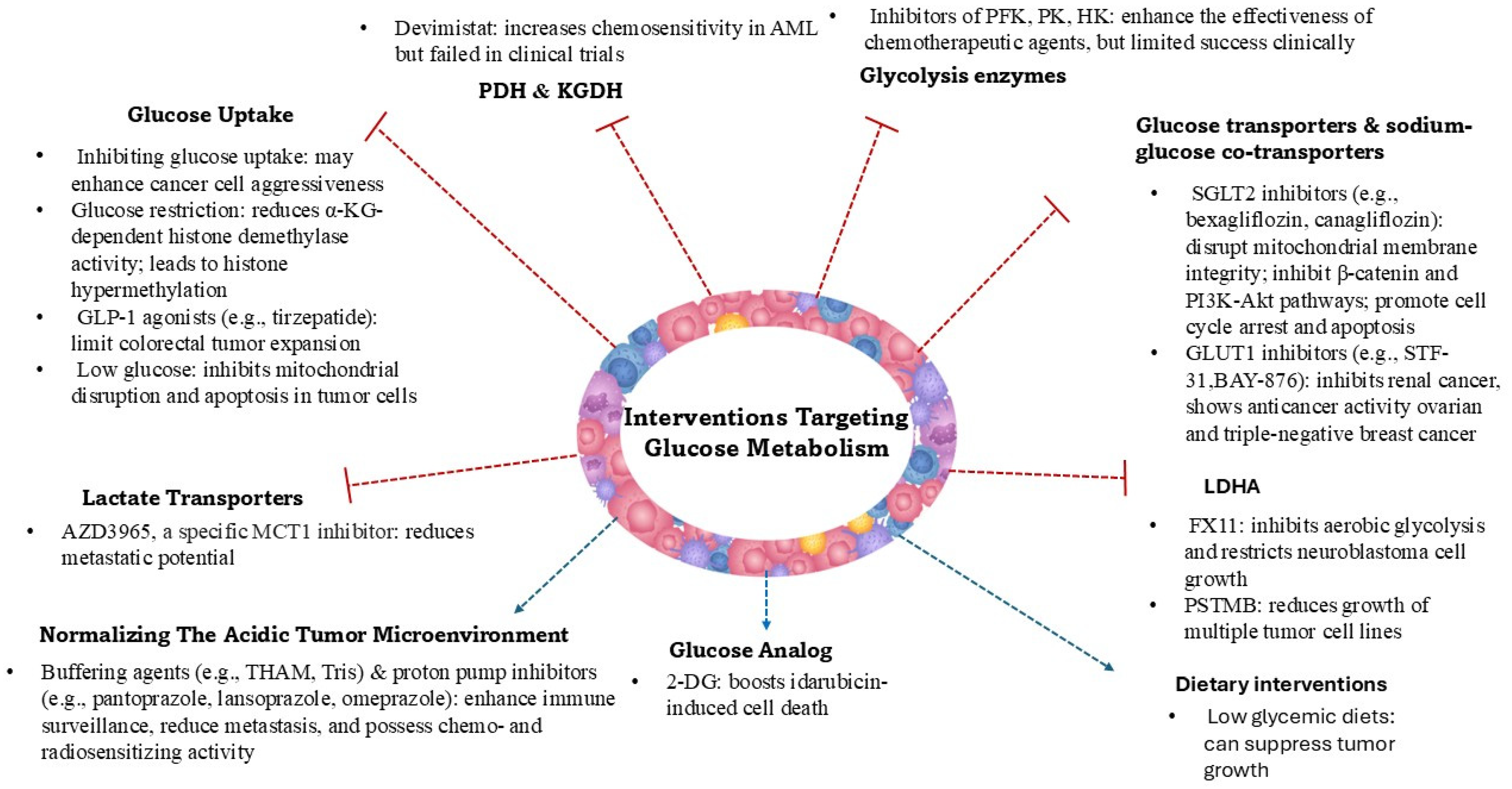
2. Glucose Metabolism Reprogramming in Cancer
- Immune Evasion: Elevated lactate levels suppress the function of immune cells, including T lymphocytes and natural killer cells, enabling tumor cells to evade immune surveillance [13].
- Angiogenesis: Lactate acts as a signaling molecule, inducing the expression of vascular endothelial growth factor (VEGF) and promoting the formation of new blood vessels, which supply nutrients and oxygen to metastatic tumors [14].
- Cell Migration and Invasion: Lactate influences the expression of matrix metalloproteinases (MMPs) and hyaluronan production, remodeling the extracellular matrix and facilitating cancer cell migration and invasion [15].
- Alternative Energy Source: Lactate can serve as an energy source for both tumor cells and endothelial cells, supporting the energy demands of metastatic growth [16].
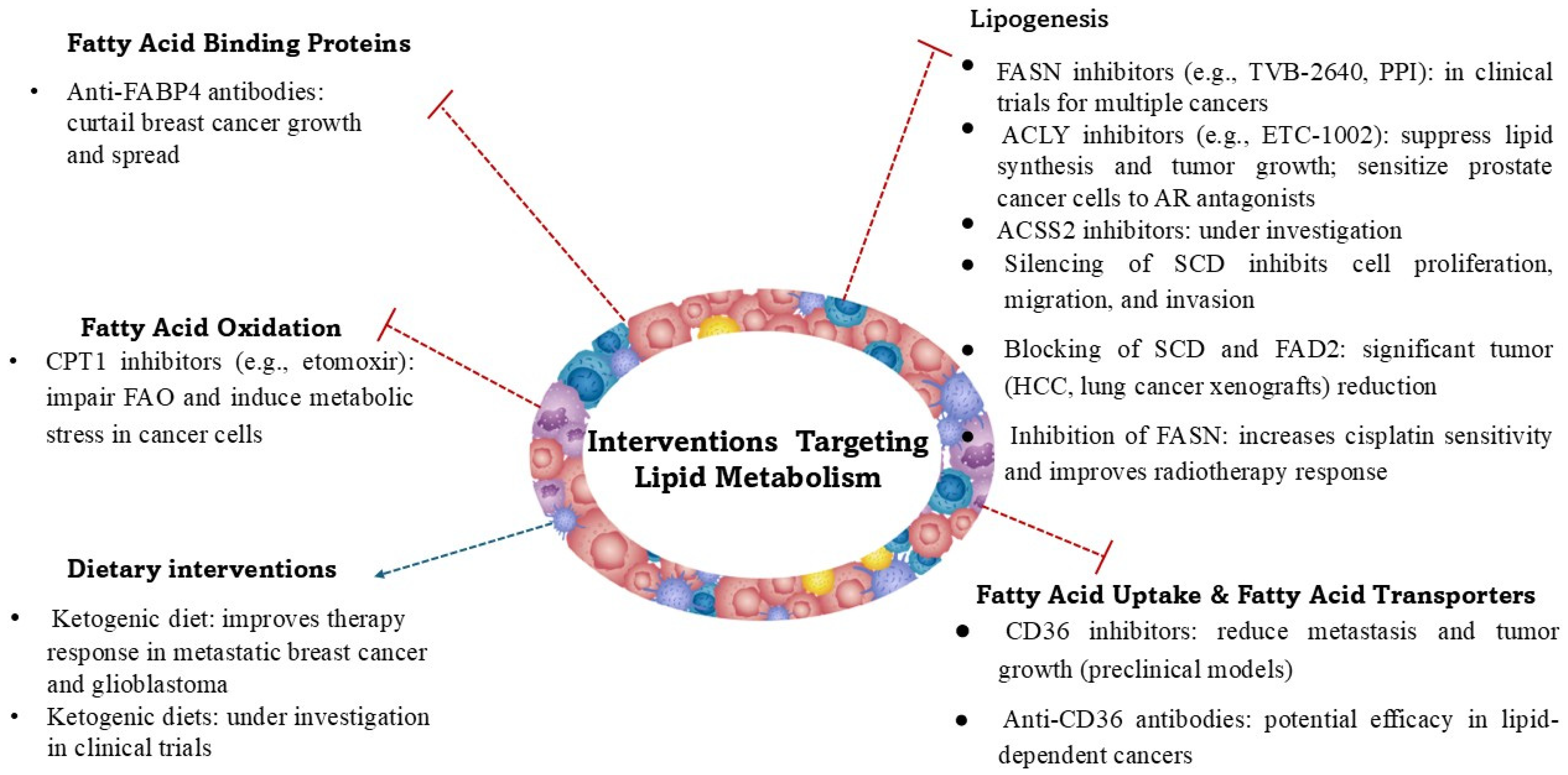
3. Fatty Acid Metabolism in Cancer
- Carnitine palmitoyltransferase 1 (CPT1) regulates the entry of FAs into mitochondria for oxidation. The overexpression of CPT1A has been observed in various cancers and is linked to resistance to metabolic stress [71].
- Acyl-CoA dehydrogenases are responsible for catalysis of the initial step in FA oxidation [72].
- Peroxisome proliferator-activated receptor (PPAR) signaling regulates FAO and lipid metabolism in cancer cells [73].
- Phase II trial in patients with metastatic pancreatic cancer receiving chemotherapy (NCT04631445),
- A ketogenic diet pilot study for overweight prostate cancer patients on active surveillance (NCT03194516),
- A pilot presurgical trial of insulin inhibition by a ketogenic diet in operable breast cancer to assess the effect of a low-fat diet with extra fiber, fruits, and vegetables, and a ketogenic diet low in carbohydrates, on breast tissue in women with ER+ or ER− breast cancer (NCT02744079),
- Single-center trial on ketogenic diet and immunotherapy in advanced cancer; this study evaluates the safety and effects of a ketogenic diet combined with immunotherapy in adults with advanced melanoma, cutaneous squamous cell carcinoma, or renal cell carcinoma (NCT06896552),
- A ketogenic diet therapy in patients with acromegaly (NCT06949891),
- Effectiveness of a ketogenic diet in MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) syndrome (NCT06013397),
- Impact of a ketogenic diet intervention during radiotherapy on body composition (NCT02516501),
- A phase II study on the ketogenic diet vs. standard anticancer diet guidance for patients with glioblastoma in combination with standard-of-care treatment (NCT05708352).
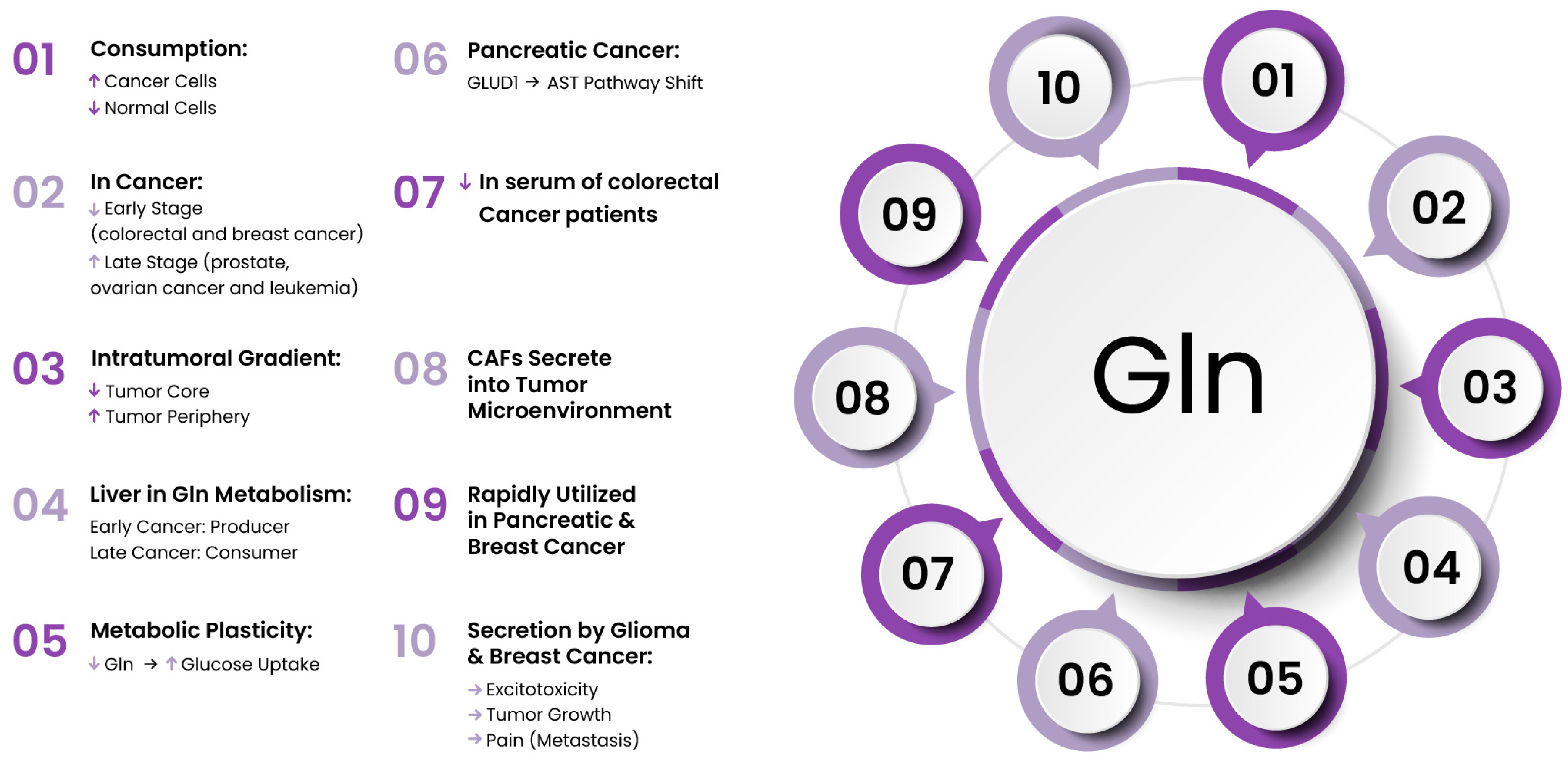
4. Amino Acid Metabolism in Tumors
4.1. Glutamine
4.2. Serine and Glycine
4.3. Branched-Chain Amino Acids
4.4. Arginine
4.5. Cystine and Cysteine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interventions Targeting Amino Acid Metabolism | |
|---|---|
| Amino Acids | Effects |
| Glutamine | -Alanyl-glutamine dipeptide reduces chemotherapy-related side effects -Glutamine antagonist suppress KEAP1-mutant tumors -Inhibition or genetic silencing of glutamine transporters (SLC7A5, SLC1A5) reduces tumor growth and migration -Glutaminase 1 inhibitors treat glutamine-dependent cancers |
| Serine and Glycine | -Inhibition of PHGDH reduces cancer cell survival -Increased serine consumption enhances DNA damage repair in colorectal cancer cells -Blocking sources of serine is associated with anticancer effects -Restriction of serine and glycine intake inhibits the proliferation of intestinal cancer and lymphoma -Sertraline: reduces SHMT1/2 activity, increases radiosensitivity, and enhances treatment effectiveness in non-small cell lung cancer |
| Branched-Chain Amino Acids | -BCAT1 knockdown inhibits melanoma cell proliferation and reduces growth and colony formation in breast cancer -SLC7A5 inhibitor suppresses growth of prostate cancer cells -High-BCAA diets reduce breast cancer growth and lung metastasis, showing positive outcomes in advanced liver cirrhosis -Diet rich in BCAA stimulates the progression of pancreatic intraepithelial neoplasia -BCAA supplementation increases event-free survival and reduces complications in hepatocellular carcinoma |
| Arginine | -Knockdown of arginine transporter reduces breast cancer cell viability -Pegylated recombinant human arginase I cobalt (HuArgI (Co)-PEG5000) induces cell cycle arrest at the G0/G1 phase -Arginine depletion in colorectal cancers decreases cell motility, invasion, and adhesion -Pegylated arginine deiminase (ADI-PEG20) and pegylated arginase exhibit anticancer properties. |
| Cysteine | -Cysteine enables cancer cells to survive in hypoxic conditions -N-acetylcysteine supplementation accelerates tumor progression and reduces survival in transgenic lung cancer models -Cysteine deficiency inhibits ovarian clear cell carcinoma -Inhibition of cysteine transporter induces ferroptosis and boosts cisplatin′s cytotoxic effects in ovarian cancer cells -Blocking xCT increases sensitivity of HeLa cells to chemotherapies and impedes the growth of colorectal cancer -Cysteine depletion induces death in pancreatic tumor cells |
5. Clinical Translation of Metabolic-Targeted Agents
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| a.a. | amino acid |
| ACC | Acetyl-CoA carboxylase |
| ACC1 | Acetyl-CoA carboxylase 1 |
| ACLY | ATP-citrate lyase |
| ACSS2 | Acetyl-CoA synthetase 2 |
| α-KG | Alpha-ketoglutarate |
| AML | Acute myeloid leukemia |
| AMPK | AMP-activated protein kinase |
| Arg | Arginine |
| ARG | Arginase |
| Asp | Aspartate |
| ASS1 | Argininosuccinate synthase 1 |
| BAK | Bcl-2 homologous antagonist/killer |
| BAX | Bcl-2-associated X protein |
| BCAA | Branched-chain amino acid |
| BCAT | Branched-chain amino acid transaminase |
| BCKDK | Branched-chain alpha-ketoacid dehydrogenase kinase |
| CAFs | Cancer-associated fibroblasts |
| CPT1 | Carnitine palmitoyltransferase 1 |
| DG | D-glucose |
| DHA | Docosahexaenoic acid (an omega-3 fatty acid) |
| EPA | Eicosapentaenoic acid (an omega-3 fatty acid) |
| FA | Fatty acid |
| FAO | Fatty acid oxidation |
| FASBP | Fatty acid binding protein |
| FASN | Fatty acid synthase |
| FATP | Fatty acid transporter protein |
| Gln | Glutamine |
| GLS | Glutaminase |
| Glu | Glutamate |
| GLUD | Glutamate dehydrogenase |
| GLUT | Glucose transporter |
| Gly | Glycine |
| GPX | Glutathione peroxidase |
| HIF1α | Hypoxia-inducible factor 1 alpha |
| HK | Hexokinase |
| HMG-CoA | 3-Hydroxy-3-methylglutaryl-coenzyme A |
| IDH | Isocitrate dehydrogenase |
| KGDH | α-ketoglutarate dehydrogenase |
| LDHA | Lactate dehydrogenase A |
| MDS | Myelodysplastic syndrome |
| MCT | Monocarboxylate transporter |
| MMP | Matrix metalloproteinase |
| MRI | Magnetic resonance imaging |
| MRS | Magnetic resonance spectroscopy |
| MUFA | Monounsaturated fatty acid |
| NO | Nitric oxide |
| PC | Pyruvate carboxylase |
| PDH | Pyruvate dehydrogenase |
| PET | Positron emission tomography |
| PFK | Phosphofructokinase |
| PHGDH | Phosphoglycerate dehydrogenase |
| PI3K | Phosphoinositide 3-kinase |
| PK | Pyruvate kinase |
| PPAR | Peroxisome proliferator-activated receptor |
| PPI | Proton pump inhibitor |
| PUFA | Polyunsaturated fatty acid |
| RCC | Renal cell carcinoma |
| SCD | Stearoyl-CoA desaturase |
| Ser | Serine |
| SGLT | Sodium/glucose co-transporter |
| SHMT | Serine hydroxymethyltransferase |
| T2D | Type 2 diabetes mellitus |
| TCA | Tricarboxylic acid cycle (Krebs cycle) |
| TME | Tumor microenvironment |
| VEGF | Vascular endothelial growth factor |
References
- Ribatti, D.; Mangialardi, G.; Vacca, A. Stephen Paget and the ‘seed and soil’ theory of metastatic dissemination. Clin. Exp. Med. 2006, 6, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Gu, W.; Su, H.; Wang, X.; Wang, X.; Zhang, J.; Xu, M.; Sheng, W. Unlocking the crucial role of cancer-associated fibroblasts in tumor metastasis: Mechanisms and therapeutic prospects. J. Adv. Res. 2025, 71, 399–413. [Google Scholar] [CrossRef]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef]
- Yang, J.; Shay, C.; Saba, N.F.; Teng, Y. Cancer metabolism and carcinogenesis. Exp. Hematol. Oncol. 2024, 13, 10. [Google Scholar] [CrossRef]
- Gao, T.; Yang, L.; Zhang, Y.; Bajinka, O.; Yuan, X. Cancer metabolic reprogramming and precision medicine-current perspective. Front. Pharmacol. 2024, 15, 1450441. [Google Scholar] [CrossRef] [PubMed]
- Nong, S.; Han, X.; Xiang, Y.; Qian, Y.; Wei, Y.; Zhang, T.; Tian, K.; Shen, K.; Yang, J.; Ma, X. Metabolic reprogramming in cancer: Mechanisms and therapeutics. MedComm 2023, 4, e218. [Google Scholar] [CrossRef]
- Mao, Y.; Xia, Z.; Xia, W.; Jiang, P. Metabolic reprogramming, sensing, and cancer therapy. Cell Rep. 2024, 43, 115064. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Fendt, S.-M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Bose, S.; Zhang, C.; Le, A. Glucose Metabolism in Cancer: The Warburg Effect and Beyond. Adv. Exp. Med. Biol. 2021, 1311, 3–15. [Google Scholar] [CrossRef]
- Mortazavi Farsani, S.S.; Verma, V. Lactate mediated metabolic crosstalk between cancer and immune cells and its therapeutic implications. Front. Oncol. 2023, 13, 1175532. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Chen, J.; Huang, Z.; Chen, Y.; Tian, H.; Chai, P.; Shen, Y.; Yao, Y.; Xu, S.; Ge, S.; Jia, R. Lactate and lactylation in cancer. Signal Transduct. Target. Ther. 2025, 10, 38. [Google Scholar] [CrossRef]
- Yuan, Z.; Li, Y.; Zhang, S.; Wang, X.; Dou, H.; Yu, X.; Zhang, Z.; Yang, S.; Xiao, M. Extracellular matrix remodeling in tumor progression and immune escape: From mechanisms to treatments. Mol. Cancer 2023, 22, 48. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.X.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Signal Transduct. Target. Ther. 2022, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Hendryx, M.; Dong, Y.; Ndeke, J.M.; Luo, J. Sodium-glucose cotransporter 2 (SGLT2) inhibitor initiation and hepatocellular carcinoma prognosis. PLoS ONE 2022, 17, e0274519. [Google Scholar] [CrossRef]
- Shin, E.; Koo, J.S. Glucose Metabolism and Glucose Transporters in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 728759. [Google Scholar] [CrossRef]
- Yang, S. Research Progress in Targeted Aerobic Glycolysis Pathway for the Treatment of Ovarian Cancer. Hans J. Biomed. 2023, 13, 354–358. [Google Scholar] [CrossRef]
- Guo, Y.; Lu, X.; Zhou, Y.; Chen, W.H.; Tam, K.Y. Combined inhibition of pyruvate dehydrogenase kinase 1 and hexokinase 2 induces apoptsis in non-small cell lung cancer cell models. Exp. Cell Res. 2023, 433, 113830. [Google Scholar] [CrossRef]
- Matsuo, T.; Konya, Y.; Hirayama, E.; Sadzuka, Y. 2-Deoxy-D-glucose enhances the anti-cancer effects of idarubicin on idarubicin-resistant P388 leukemia cells. Oncol. Lett. 2020, 20, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Ediriweera, M.K.; Jayasena, S. The Role of Reprogrammed Glucose Metabolism in Cancer. Metabolites 2023, 13, 345. [Google Scholar] [CrossRef] [PubMed]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef]
- Safarian, A.; Mirshahvalad, S.A.; Nasrollahi, H.; Jung, T.; Pirich, C.; Arabi, H.; Beheshti, M. Impact of [(18)F]FDG PET/CT Radiomics and Artificial Intelligence in Clinical Decision Making in Lung Cancer: Its Current Role. Semin. Nucl. Med. 2025, 55, 156–166. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Gori, S.S.; Tsukamoto, T.; Rais, R.; Slusher, B.S. Clinical development of metabolic inhibitors for oncology. J. Clin. Investig. 2022, 132, e148550. [Google Scholar] [CrossRef]
- Gonzalez-Flores, D.; Gripo, A.-A.; Rodríguez, A.-B.; Franco, L. Consequences of Glucose Enriched Diet on Oncologic Patients. Appl. Sci. 2023, 13, 2757. [Google Scholar] [CrossRef]
- Almouhanna, F.; Blagojevic, B.; Can, S.; Ghanem, A.; Wölfl, S. Pharmacological activation of pyruvate kinase M2 reprograms glycolysis leading to TXNIP depletion and AMPK activation in breast cancer cells. Cancer Metab. 2021, 9, 5. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Li, G.; Sun, L.; Zhang, W.; Jiang, J.; Ge, Q. Identification of a prognosis-associated signature associated with energy metabolism in triple-negative breast cancer. Oncol. Rep. 2020, 44, 819–837. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Yu, Z. Identification of a novel glycolysis-related gene signature for predicting metastasis and survival in patients with lung adenocarcinoma. J. Transl. Med. 2019, 17, 423. [Google Scholar] [CrossRef]
- Tan, J.; Le, A. The Heterogeneity of Breast Cancer Metabolism. Adv. Exp. Med. Biol. 2021, 1311, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Gaur, A.; Maity, R.; Dhali, A.; Biswas, J. Impact of poorly controlled type II diabetes mellitus on chemoresistance in colorectal cancer. World J. Gastroenterol. 2025, 31, 104065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, Y.; Xia, S.; Ge, X.; Li, J.; Liu, F.; Jia, F.; Wang, S.; Zhou, Q.; Gao, M.; et al. The Novel Dual GIP and GLP-1 Receptor Agonist Tirzepatide Attenuates Colon Cancer Development by Regulating Glucose Metabolism. Adv. Sci. 2025, 12, e2411980. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Otero, D.; González-Vidal, T.; Agüeria-Cabal, P.; Ramos-Ruiz, G.; Jimenez-Fonseca, P.; Lambert, C.; Alarcón, P.P.; Torre, E.M.; García-Villarino, M.; Álvarez, E.D. The Influence of Performance Status, Inflammation, and Nutrition on the Impact of SGLT2 Inhibitors on Cancer Outcomes. Cancer Med. 2025, 14, e70807. [Google Scholar] [CrossRef]
- Nam, M.; Xia, W.; Mir, A.H.; Jerrett, A.; Spinelli, J.B.; Huang, T.T.; Possemato, R. Glucose limitation protects cancer cells from apoptosis induced by pyrimidine restriction and replication inhibition. Nat. Metab. 2024, 6, 2338–2353. [Google Scholar] [CrossRef]
- Saggese, P.; Pandey, A.; Alcaraz, M., Jr.; Fung, E.; Hall, A.; Yanagawa, J.; Rodriguez, E.F.; Grogan, T.R.; Giurato, G.; Nassa, G.; et al. Glucose Deprivation Promotes Pseudohypoxia and Dedifferentiation in Lung Adenocarcinoma. Cancer Res. 2024, 84, 305–327. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Plas, D.R.; Rathmell, J.C.; Fox, C.J.; Harris, M.H.; Thompson, C.B. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol. Cell Biol. 2001, 21, 5899–5912. [Google Scholar] [CrossRef]
- Pardee, T.S.; Anderson, R.G.; Pladna, K.M.; Isom, S.; Ghiraldeli, L.P.; Miller, L.D.; Chou, J.W.; Jin, G.; Zhang, W.; Ellis, L.R.; et al. A Phase I Study of CPI-613 in Combination with High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 2060–2073. [Google Scholar] [CrossRef]
- Pardee, T.S.; Powell, B.L.; Larson, R.A.; Maly, J.; Keng, M.; Foster, M.; Choi, E.-J.; Sill, H.; Cluzeau, T.; Jeyakumar, D.; et al. Devimistat plus chemotherapy vs chemotherapy alone for older relapsed or refractory patients with AML: Results of the ARMADA trial. Blood Neoplasia 2024, 1, 100009. [Google Scholar] [CrossRef]
- Philip, P.A.; Sahai, V.; Bahary, N.; Mahipal, A.; Kasi, A.; Rocha Lima, C.M.S.; Alistar, A.T.; Oberstein, P.E.; Golan, T.; Metges, J.P.; et al. Devimistat (CPI-613) with Modified Fluorouarcil, Oxaliplatin, Irinotecan, and Leucovorin (FFX) Versus FFX for Patients with Metastatic Adenocarcinoma of the Pancreas: The Phase III AVENGER 500 Study. J. Clin. Oncol. 2024, 42, 3692–3701. [Google Scholar] [CrossRef] [PubMed]
- García-Cañaveras, J.C.; Chen, L.; Rabinowitz, J.D. The Tumor Metabolic Microenvironment: Lessons from Lactate. Cancer Res. 2019, 79, 3155–3162. [Google Scholar] [CrossRef] [PubMed]
- Beloueche-Babari, M.; Wantuch, S.; Casals Galobart, T.; Koniordou, M.; Parkes, H.G.; Arunan, V.; Chung, Y.L.; Eykyn, T.R.; Smith, P.D.; Leach, M.O. MCT1 Inhibitor AZD3965 Increases Mitochondrial Metabolism, Facilitating Combination Therapy and Noninvasive Magnetic Resonance Spectroscopy. Cancer Res. 2017, 77, 5913–5924. [Google Scholar] [CrossRef]
- Ooi, A.T.; Gomperts, B.N. Molecular Pathways: Targeting Cellular Energy Metabolism in Cancer via Inhibition of SLC2A1 and LDHA. Clin. Cancer Res. 2015, 21, 2440–2444. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-Y.; Chung, T.-W.; Han, C.W.; Park, S.Y.; Park, K.H.; Jang, S.B.; Ha, K.-T. A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth through Apoptotic Cell Death. Sci. Rep. 2019, 9, 3969. [Google Scholar] [CrossRef]
- Ibrahim-Hashim, A.; Cornnell, H.H.; Abrahams, D.; Lloyd, M.; Bui, M.; Gillies, R.J.; Gatenby, R.A. Systemic buffers inhibit carcinogenesis in TRAMP mice. J. Urol. 2012, 188, 624–631. [Google Scholar] [CrossRef]
- Ibrahim-Hashim, A.; Abrahams, D.; Enriquez-Navas, P.M.; Luddy, K.; Gatenby, R.A.; Gillies, R.J. Tris-base buffer: A promising new inhibitor for cancer progression and metastasis. Cancer Med. 2017, 6, 1720–1729. [Google Scholar] [CrossRef]
- Monaco, M.E. Fatty acid metabolism in breast cancer subtypes. Oncotarget 2017, 8, 29487–29500. [Google Scholar] [CrossRef]
- Corn, K.C.; Windham, M.A.; Rafat, M. Lipids in the tumor microenvironment: From cancer progression to treatment. Prog. Lipid Res. 2020, 80, 101055. [Google Scholar] [CrossRef]
- Lin, Z.; Hua, G.; Hu, X. Lipid metabolism associated crosstalk: The bidirectional interaction between cancer cells and immune/stromal cells within the tumor microenvironment for prognostic insight. Cancer Cell Int. 2024, 24, 295. [Google Scholar] [CrossRef]
- Han, L.; Dai, W.; Luo, W.; Ye, L.; Fang, H.; Mo, S.; Li, Q.; Xu, Y.; Wang, R.; Cai, G. Enhanced De Novo Lipid Synthesis Mediated by FASN Induces Chemoresistance in Colorectal Cancer. Cancers 2023, 15, 562. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.B.; Singh, S.V. Fatty Acid Synthesis Intermediates Represent Novel Noninvasive Biomarkers of Prostate Cancer Chemoprevention by Phenethyl Isothiocyanate. Cancer Prev. Res. 2017, 10, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Chai, Y.D.; Hu, S. Fatty Acid Metabolism and Cancer. Adv. Exp. Med. Biol. 2021, 1280, 231–241. [Google Scholar] [CrossRef]
- Vanauberg, D.; Schulz, C.; Lefebvre, T. Involvement of the pro-oncogenic enzyme fatty acid synthase in the hallmarks of cancer: A promising target in anti-cancer therapies. Oncogenesis 2023, 12, 16. [Google Scholar] [CrossRef]
- Szutowicz, A.; Kwiatkowski, J.; Angielski, S. Lipogenetic and glycolytic enzyme activities in carcinoma and nonmalignant diseases of the human breast. Br. J. Cancer 1979, 39, 681–687. [Google Scholar] [CrossRef]
- Granchi, C. ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 2018, 157, 1276–1291. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef]
- Batchuluun, B.; Pinkosky, S.L.; Steinberg, G.R. Lipogenesis inhibitors: Therapeutic opportunities and challenges. Nat. Rev. Drug Discov. 2022, 21, 283–305. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, Y.; Tie, Y. Fatty acid metabolism influences the immune microenvironment in papillary thyroid cancer and identifies SCD as a novel biomarker. Front. Endocrinol. 2025, 16, 1534393. [Google Scholar] [CrossRef]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Guerrero-Rodríguez, S.L.; Mata-Cruz, C.; Pérez-Tapia, S.M.; Velasco-Velázquez, M.A. Role of CD36 in cancer progression, stemness, and targeting. Front. Cell Dev. Biol. 2022, 10, 1079076. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Su, M.; Lu, J.; Li, D.; Niu, X.; Wang, Y. CD36: The Bridge between Lipids and Tumors. Molecules 2024, 29, 531. [Google Scholar] [CrossRef]
- Yang, P.; Qin, H.; Li, Y.; Xiao, A.; Zheng, E.; Zeng, H.; Su, C.; Luo, X.; Lu, Q.; Liao, M.; et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat. Commun. 2022, 13, 5782. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Sp, N.; Kang, D.Y.; Kim, D.H.; Park, J.H.; Lee, H.G.; Kim, H.J.; Darvin, P.; Park, Y.M.; Yang, Y.M. Nobiletin Inhibits CD36-Dependent Tumor Angiogenesis, Migration, Invasion, and Sphere Formation Through the Cd36/Stat3/Nf-Κb Signaling Axis. Nutrients 2018, 10, 772. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Li, Y.; Xing, L.; Tan, Y.; Sun, J.; Zeng, B.; Xiang, T.; Tan, J.; Ren, G.; Wang, Y. Utilization of adipocyte-derived lipids and enhanced intracellular trafficking of fatty acids contribute to breast cancer progression. Cell Commun. Signal 2018, 16, 32. [Google Scholar] [CrossRef]
- Jiang, X.; Xiong, Y.; Yu, J.; Avellino, A.; Liu, S.; Han, X.; Wang, Z.; Shilyansky, J.S.; Curry, M.A.; Hao, J.; et al. Expression profiles of FABP4 and FABP5 in breast cancer: Clinical implications and perspectives. Discov. Oncol. 2025, 16, 357. [Google Scholar] [CrossRef]
- Smolková, K.; Gotvaldová, K. Fatty Acid Trafficking Between Lipid Droplets and Mitochondria: An Emerging Perspective. Int. J. Biol. Sci. 2025, 21, 1863–1873. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, W.; Wang, W.; Ma, Y.; Wang, Y.; Drum, D.L.; Cai, J.; Blevins, H.; Lee, E.; Shah, S.; et al. CPT1A-mediated fatty acid oxidation confers cancer cell resistance to immune-mediated cytolytic killing. Proc. Natl. Acad. Sci. USA 2023, 120, e2302878120. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xian, H.-c.; Tang, Y.-J.; Liang, X.-h.; Tang, Y.-l. Fatty acid oxidation: Driver of lymph node metastasis. Cancer Cell Int. 2021, 21, 339. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Cai, S.; Zhang, J.-K.; Ding, S.-Q.; Zhang, Z.-H.; Zhang, C.-D.; Dai, D.-Q.; Li, Y.-S. The role and mechanism of fatty acid oxidation in cancer drug resistance. Cell Death Discov. 2025, 11, 277. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, Q.; Dong, C. Metabolic reprogramming in triple-negative breast cancer. Cancer Biol. Med. 2020, 17, 44–59. [Google Scholar] [CrossRef]
- Eltayeb, K.; La Monica, S.; Tiseo, M.; Alfieri, R.; Fumarola, C. Reprogramming of Lipid Metabolism in Lung Cancer: An Overview with Focus on EGFR-Mutated Non-Small Cell Lung Cancer. Cells 2022, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm 2021, 2, 27–59. [Google Scholar] [CrossRef]
- Mikhael, S.; Daoud, G. Navigating Metabolic Challenges in Ovarian Cancer: Insights and Innovations in Drug Repurposing. Cancer Med. 2025, 14, e70681. [Google Scholar] [CrossRef]
- Menard, J.A.; Christianson, H.C.; Kucharzewska, P.; Bourseau-Guilmain, E.; Svensson, K.J.; Lindqvist, E.; Indira Chandran, V.; Kjellén, L.; Welinder, C.; Bengzon, J.; et al. Metastasis Stimulation by Hypoxia and Acidosis-Induced Extracellular Lipid Uptake Is Mediated by Proteoglycan-Dependent Endocytosis. Cancer Res. 2016, 76, 4828–4840. [Google Scholar] [CrossRef]
- Gouw, A.M.; Eberlin, L.S.; Margulis, K.; Sullivan, D.K.; Toal, G.G.; Tong, L.; Zare, R.N.; Felsher, D.W. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 4300–4305. [Google Scholar] [CrossRef]
- Lin, H.M.; Mahon, K.L.; Weir, J.M.; Mundra, P.A.; Spielman, C.; Briscoe, K.; Gurney, H.; Mallesara, G.; Marx, G.; Stockler, M.R.; et al. A distinct plasma lipid signature associated with poor prognosis in castration-resistant prostate cancer. Int. J. Cancer 2017, 141, 2112–2120. [Google Scholar] [CrossRef]
- Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Li, J.; Xu, H.; Zhao, Y.; Yu, X.; Shi, S. Functional significance of cholesterol metabolism in cancer: From threat to treatment. Exp. Mol. Med. 2023, 55, 1982–1995. [Google Scholar] [CrossRef]
- Liu, C.; Chen, H.; Hu, B.; Shi, J.; Chen, Y.; Huang, K. New insights into the therapeutic potentials of statins in cancer. Front. Pharmacol. 2023, 14, 1188926. [Google Scholar] [CrossRef]
- Kim, M.K.; Myung, S.K.; Tran, B.T.; Park, B. Statins and risk of cancer: A meta-analysis of randomized, double-blind, placebo-controlled trials. Indian J. Cancer 2017, 54, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Papaevangelou, E.; Almeida, G.S.; Box, C.; deSouza, N.M.; Chung, Y.L. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int. J. Cancer 2018, 143, 992–1002. [Google Scholar] [CrossRef]
- Rae, C.; Haberkorn, U.; Babich, J.W.; Mairs, R.J. Inhibition of Fatty Acid Synthase Sensitizes Prostate Cancer Cells to Radiotherapy. Radiat. Res. 2015, 184, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Sardesai, S.D.; Thomas, A.; Gallagher, C.; Lynce, F.; Ottaviano, Y.L.; Ballinger, T.J.; Schneider, B.P.; Storniolo, A.M.; Bauchle, A.; Althouse, S.K.; et al. Inhibiting Fatty Acid Synthase with Omeprazole to Improve Efficacy of Neoadjuvant Chemotherapy in Patients with Operable TNBC. Clin. Cancer Res. 2021, 27, 5810–5817. [Google Scholar] [CrossRef] [PubMed]
- Velez, B.C.; Petrella, C.P.; DiSalvo, K.H.; Cheng, K.; Kravtsov, R.; Krasniqi, D.; Krucher, N.A. Combined inhibition of ACLY and CDK4/6 reduces cancer cell growth and invasion. Oncol. Rep. 2023, 49, 32. [Google Scholar] [CrossRef]
- Shah, S.; Carriveau, W.J.; Li, J.; Campbell, S.L.; Kopinski, P.K.; Lim, H.W.; Daurio, N.; Trefely, S.; Won, K.J.; Wallace, D.C.; et al. Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget 2016, 7, 43713–43730. [Google Scholar] [CrossRef]
- Wu, Y.; Pu, X.; Wang, X.; Xu, M. Reprogramming of lipid metabolism in the tumor microenvironment: A strategy for tumor immunotherapy. Lipids Health Dis. 2024, 23, 35. [Google Scholar] [CrossRef]
- Mei, J.; Qian, M.; Hou, Y.; Liang, M.; Chen, Y.; Wang, C.; Zhang, J. Association of saturated fatty acids with cancer risk: A systematic review and meta-analysis. Lipids Health Dis. 2024, 23, 32. [Google Scholar] [CrossRef]
- van der Meij, B.; Parsons, S.; Mazurak, V. The impact of n-3 polyunsaturated fatty acids in patients with cancer: Emerging themes. Curr. Opin. Clin. Nutr. Metab. Care 2025, 28, 75–85. [Google Scholar] [CrossRef]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated Fatty acids. J. Nutr. Metab. 2012, 2012, 539426. [Google Scholar] [CrossRef]
- Srilaxmi, V.; Kanteti, K.; Vityala, Y.; Zhumabekova, A.; Vipin. Effects of Supplementation with Omega-3 Polyunsaturated Fatty Acids as an Adjuvant Therapy in the Treatment of Patients with Breast Cancer: A Systematic Review: Effect of omega-3 as adjuvant therapy. Arch. Breast Cancer 2023, 10, 323–330. [Google Scholar] [CrossRef]
- Luo, J.; Peng, S.; Jiang, Z.; Wang, Q.; Zhang, M.; Zeng, Y.; Yuan, Y.; Xia, M.; Hong, Z.; Yan, Y.; et al. Roles and therapeutic opportunities of ω-3 long-chain polyunsaturated fatty acids in lung cancer. iScience 2025, 28, 111601. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Solé, C.; Torres-Herrera, B.; Gelerstein-Claro, S.; Medina-Pérez, D.; Gómez-Venegas, H.; Alzolay-Sepúlveda, J.; Chichiarelli, S.; Saso, L.; Rodrigo, R. Cellular Basis of Adjuvant Role of n-3 Polyunsaturated Fatty Acids in Cancer Therapy: Molecular Insights and Therapeutic Potential against Human Melanoma. Appl. Sci. 2024, 14, 4548. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Nekoufar, S.; Ghorbani, M.; Safaei, S.; Khosroushahi, G.A.; Shirian, F.I.; Baradaran, B.; Tavakoli-Yaraki, M. Exploring the potential of gemcitabine-metal-organic frameworks in combating pancreatic cancer under ketogenic conditions. BMC Cancer 2025, 25, 53. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.; Yoo, J.Y.; Nelson, E.R.; Sikora, M.J.; Riggins, R.B.; Madak-Erdogan, Z. Co-targeting of metabolism using dietary and pharmacologic approaches reduces breast cancer metastatic burden. npj Breast Cancer 2025, 11, 3. [Google Scholar] [CrossRef]
- Duraj, T.; Kalamian, M.; Zuccoli, G.; Maroon, J.C.; D’Agostino, D.P.; Scheck, A.C.; Poff, A.; Winter, S.F.; Hu, J.; Klement, R.J.; et al. Clinical research framework proposal for ketogenic metabolic therapy in glioblastoma. BMC Med. 2024, 22, 578. [Google Scholar] [CrossRef]
- Allen, B.G.; Bhatia, S.K.; Anderson, C.M.; Eichenberger-Gilmore, J.M.; Sibenaller, Z.A.; Mapuskar, K.A.; Schoenfeld, J.D.; Buatti, J.M.; Spitz, D.R.; Fath, M.A. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biol. 2014, 2, 963–970. [Google Scholar] [CrossRef]
- Talib, W.H.; Mahmod, A.I.; Kamal, A.; Rashid, H.M.; Alashqar, A.M.D.; Khater, S.; Jamal, D.; Waly, M. Ketogenic Diet in Cancer Prevention and Therapy: Molecular Targets and Therapeutic Opportunities. Curr. Issues Mol. Biol. 2021, 43, 558–589. [Google Scholar] [CrossRef]
- Liang, Z.; Deng, L.; Zhou, X.; Zhang, Z.; Zhao, W. Comprehensive Overview of Ketone Bodies in Cancer Metabolism: Mechanisms and Application. Biomedicines 2025, 13, 210. [Google Scholar] [CrossRef]
- Crosby, L.; Davis, B.; Joshi, S.; Jardine, M.; Paul, J.; Neola, M.; Barnard, N.D. Ketogenic Diets and Chronic Disease: Weighing the Benefits Against the Risks. Front. Nutr. 2021, 8, 702802. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Kalisz, J. Ketogenic diet and cardiovascular risk—State of the art review. Curr. Probl. Cardiol. 2024, 49, 102402. [Google Scholar] [CrossRef] [PubMed]
- Hersant, H.; Grossberg, G. The Ketogenic Diet and Alzheimer’s Disease. J. Nutr. Health Aging 2022, 26, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Eagle, H. The minimum vitamin requirements of the L and HeLa cells in tissue culture, the production of specific vitamin deficiencies, and their cure. J. Exp. Med. 1955, 102, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Shao, N.; Liu, J.; Zhao, J.; Chen, C.; Li, Q.; He, Z.; Zhao, X.; Xu, L. Amino acid metabolism in tumor: New shine in the fog? Clin. Nutr. 2023, 42, 1521–1530. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino Acid Depletion Therapies: Starving Cancer Cells to Death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef]
- Chen, J.; Cui, L.; Lu, S.; Xu, S. Amino acid metabolism in tumor biology and therapy. Cell Death Dis. 2024, 15, 42. [Google Scholar] [CrossRef]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New aspects of amino acid metabolism in cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Tra-Ly, N.; Raúl, V.D. Glutamine metabolism in cancer therapy. Cancer Drug Resist. 2018, 1, 126–138. [Google Scholar] [CrossRef]
- Lai, H.S.; Lee, J.C.; Lee, P.H.; Wang, S.T.; Chen, W.J. Plasma free amino acid profile in cancer patients. Semin. Cancer Biol. 2005, 15, 267–276. [Google Scholar] [CrossRef]
- Erb, H.H.H.; Polishchuk, N.; Stasyk, O.; Kahya, U.; Weigel, M.M.; Dubrovska, A. Glutamine Metabolism and Prostate Cancer. Cancers 2024, 16, 2871. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, M.; Yang, Q. Glutamine and leukemia research: Progress and clinical prospects. Discov. Oncol. 2024, 15, 391. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, C.; Zheng, Y.; Liu, C.; Wang, X.; Cong, X. Glutamine deficiency promotes recurrence and metastasis in colorectal cancer through enhancing epithelial-mesenchymal transition. J. Transl. Med. 2022, 20, 330. [Google Scholar] [CrossRef]
- Qiu, J.; Qian, D.; Jiang, Y.; Meng, L.; Huang, L. Circulating tumor biomarkers in early-stage breast cancer: Characteristics, detection, and clinical developments. Front. Oncol. 2023, 13, 1288077. [Google Scholar] [CrossRef]
- Reitzer, L.J.; Wice, B.M.; Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [Google Scholar] [CrossRef]
- Bode, B.P.; Kaminski, D.L.; Souba, W.W.; Li, A.P. Glutamine transport in isolated human hepatocytes and transformed liver cells. Hepatology 1995, 21, 511–520. [Google Scholar]
- Ling, H.H.; Pan, Y.P.; Fan, C.W.; Tseng, W.K.; Huang, J.S.; Wu, T.H.; Chou, W.C.; Wang, C.H.; Yeh, K.Y.; Chang, P.H. Clinical Significance of Serum Glutamine Level in Patients with Colorectal Cancer. Nutrients 2019, 11, 898. [Google Scholar] [CrossRef]
- Sirniö, P.; Väyrynen, J.P.; Klintrup, K.; Mäkelä, J.; Karhu, T.; Herzig, K.H.; Minkkinen, I.; Mäkinen, M.J.; Karttunen, T.J.; Tuomisto, A. Alterations in serum amino-acid profile in the progression of colorectal cancer: Associations with systemic inflammation, tumour stage and patient survival. Br. J. Cancer 2019, 120, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Djukovic, D.; Deng, L.; Gu, H.; Himmati, F.; Chiorean, E.G.; Raftery, D. Colorectal cancer detection using targeted serum metabolic profiling. J. Proteome Res. 2014, 13, 4120–4130. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human Pancreatic Cancer Tumors Are Nutrient Poor and Tumor Cells Actively Scavenge Extracellular Protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Sasayama, T.; Nagashima, H.; Irino, Y.; Takahashi, M.; Izumi, Y.; Uno, T.; Satoh, N.; Kitta, A.; Kyotani, K.; et al. Glioma cells require one-carbon metabolism to survive glutamine starvation. Acta Neuropathol. Commun. 2021, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Menon, D.; Bernfeld, E.; Mroz, V.; Kalan, S.; Loayza, D.; Foster, D.A. Aspartate Rescues S-phase Arrest Caused by Suppression of Glutamine Utilization in KRas-driven Cancer Cells. J. Biol. Chem. 2016, 291, 9322–9329. [Google Scholar] [CrossRef]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 2016, 36, 540–549. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Son, J.; Cantley, L.C.; Kimmelman, A.C. Pancreatic cancers rely on a novel glutamine metabolism pathway to maintain redox balance. Cell Cycle 2013, 12, 1987–1988. [Google Scholar] [CrossRef]
- Chen, M.K.; Salloum, R.M.; Austgen, T.R.; Bland, J.B.; Bland, K.I.; Copeland, E.M., 3rd; Souba, W.W. Tumor regulation of hepatic glutamine metabolism. JPEN J. Parenter. Enteral Nutr. 1991, 15, 159–164. [Google Scholar] [CrossRef]
- Pacitti, A.J.; Inoue, Y.; Souba, W.W. Characterization of Na(+)-independent glutamine transport in rat liver. Am. J. Physiol.-Gastrointest. Liver Physiol. 1993, 265, G90–G98. [Google Scholar] [CrossRef]
- Pacitti, A.J.; Inoue, Y.; Souba, W.W. Tumor necrosis factor stimulates amino acid transport in plasma membrane vesicles from rat liver. J. Clin. Investig. 1993, 91, 474–483. [Google Scholar] [CrossRef]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X.; et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Oosterveer, M.H.; Stein, S.; Demagny, H.; Ryu, D.; Moullan, N.; Wang, X.; Can, E.; Zamboni, N.; Comment, A.; et al. LRH-1-dependent programming of mitochondrial glutamine processing drives liver cancer. Genes. Dev. 2016, 30, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Vié, N.; Copois, V.; Bascoul-Mollevi, C.; Denis, V.; Bec, N.; Robert, B.; Fraslon, C.; Conseiller, E.; Molina, F.; Larroque, C.; et al. Overexpression of phosphoserine aminotransferase PSAT1 stimulates cell growth and increases chemoresistance of colon cancer cells. Mol. Cancer 2008, 7, 14. [Google Scholar] [CrossRef]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meininger, C.J.; Bazer, F.W.; Wu, G. Intracellular sources of ornithine for polyamine synthesis in endothelial cells. Amino Acids 2016, 48, 2401–2410. [Google Scholar] [CrossRef]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef]
- Kanai, Y. Amino acid transporter LAT1 (SLC7A5) as a molecular target for cancer diagnosis and therapeutics. Pharmacol. Ther. 2022, 230, 107964. [Google Scholar] [CrossRef]
- Kawakami, I.; Yoshino, H.; Fukumoto, W.; Tamai, M.; Okamura, S.; Osako, Y.; Sakaguchi, T.; Inoguchi, S.; Matsushita, R.; Yamada, Y.; et al. Targeting of the glutamine transporter SLC1A5 induces cellular senescence in clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2022, 611, 99–106. [Google Scholar] [CrossRef]
- Reinfeld, B.I.; Madden, M.Z.; Wolf, M.M.; Chytil, A.; Bader, J.E.; Patterson, A.R.; Sugiura, A.; Cohen, A.S.; Ali, A.; Do, B.T.; et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 2021, 593, 282–288. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Sontheimer, H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999, 59, 4383–4391. [Google Scholar]
- Fazzari, J.; Lin, H.; Murphy, C.; Ungard, R.; Singh, G. Inhibitors of glutamate release from breast cancer cells; new targets for cancer-induced bone-pain. Sci. Rep. 2015, 5, 8380. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Talmon, G.; Wang, J. Glutamate in cancers: From metabolism to signaling. J. Biomed. Res. 2019, 34, 260–270. [Google Scholar] [CrossRef]
- Ahluwalia, G.S.; Grem, J.L.; Hao, Z.; Cooney, D.A. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol. Ther. 1990, 46, 243–271. [Google Scholar] [CrossRef]
- Pillai, R.; LeBoeuf, S.E.; Hao, Y.; New, C.; Blum, J.L.E.; Rashidfarrokhi, A.; Huang, S.M.; Bahamon, C.; Wu, W.L.; Karadal-Ferrena, B.; et al. Glutamine antagonist DRP-104 suppresses tumor growth and enhances response to checkpoint blockade in KEAP1 mutant lung cancer. bioRxiv 2023. [Google Scholar] [CrossRef]
- Curthoys, N.P.; Watford, M. Regulation of glutaminase activity and glutamine metabolism. Annu. Rev. Nutr. 1995, 15, 133–159. [Google Scholar] [CrossRef]
- Suzuki, S.; Venkatesh, D.; Kanda, H.; Nakayama, A.; Hosokawa, H.; Lee, E.; Miki, T.; Stockwell, B.R.; Yokote, K.; Tanaka, T.; et al. GLS2 Is a Tumor Suppressor and a Regulator of Ferroptosis in Hepatocellular Carcinoma. Cancer Res. 2022, 82, 3209–3222. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Soth, M.J.; Le, K.; Di Francesco, M.E.; Hamilton, M.M.; Liu, G.; Burke, J.P.; Carroll, C.L.; Kovacs, J.J.; Bardenhagen, J.P.; Bristow, C.A.; et al. Discovery of IPN60090, a Clinical Stage Selective Glutaminase-1 (GLS-1) Inhibitor with Excellent Pharmacokinetic and Physicochemical Properties. J. Med. Chem. 2020, 63, 12957–12977. [Google Scholar] [CrossRef]
- Yu, W.; Yang, X.; Zhang, Q.; Sun, L.; Yuan, S.; Xin, Y. Targeting GLS1 to cancer therapy through glutamine metabolism. Clin. Transl. Oncol. 2021, 23, 2253–2268. [Google Scholar] [CrossRef]
- Robinson, M.M.; McBryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.V.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 2007, 406, 407–414. [Google Scholar] [CrossRef]
- Huang, Q.; Stalnecker, C.; Zhang, C.; McDermott, L.A.; Iyer, P.; O’Neill, J.; Reimer, S.; Cerione, R.A.; Katt, W.P. Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. J. Biol. Chem. 2018, 293, 3535–3545. [Google Scholar] [CrossRef]
- Li, Y.; Ping, X.; Yu, B.; Liu, F.; Ni, X.; Li, J. Clinical trial: Prophylactic intravenous alanyl-glutamine reduces the severity of gastrointestinal toxicity induced by chemotherapy--a randomized crossover study. Aliment. Pharmacol. Ther. 2009, 30, 452–458. [Google Scholar] [CrossRef]
- Wang, W.S.; Lin, J.K.; Lin, T.C.; Chen, W.S.; Jiang, J.K.; Wang, H.S.; Chiou, T.J.; Liu, J.H.; Yen, C.C.; Chen, P.M. Oral glutamine is effective for preventing oxaliplatin-induced neuropathy in colorectal cancer patients. Oncologist 2007, 12, 312–319. [Google Scholar] [CrossRef]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Castillo, A.; Kampen, K.R. Understanding serine and glycine metabolism in cancer: A path towards precision medicine to improve patient’s outcomes. Discov. Oncol. 2024, 15, 652. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wu, J.L.; Qin, B.; Fan, Z.; Tang, Q.; Lu, W.; Zhang, H.; Xing, F.; Meng, M.; Zou, S.; et al. ILF3 is a substrate of SPOP for regulating serine biosynthesis in colorectal cancer. Cell Res. 2020, 30, 163–178. [Google Scholar] [CrossRef]
- Zhao, E.; Hou, J.; Cui, H. Serine-glycine-one-carbon metabolism: Vulnerabilities in MYCN-amplified neuroblastoma. Oncogenesis 2020, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Locasale, J.W.; Reid, M.A. Serine and Methionine Metabolism: Vulnerabilities in Lethal Prostate Cancer. Cancer Cell 2019, 35, 339–341. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef]
- Kampen, K.R.; Fancello, L.; Girardi, T.; Rinaldi, G.; Planque, M.; Sulima, S.O.; Loayza-Puch, F.; Verbelen, B.; Vereecke, S.; Verbeeck, J.; et al. Translatome analysis reveals altered serine and glycine metabolism in T-cell acute lymphoblastic leukemia cells. Nat. Commun. 2019, 10, 2542. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef]
- Pacold, M.E.; Brimacombe, K.R.; Chan, S.H.; Rohde, J.M.; Lewis, C.A.; Swier, L.J.; Possemato, R.; Chen, W.W.; Sullivan, L.B.; Fiske, B.P.; et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 2016, 12, 452–458. [Google Scholar] [CrossRef]
- Furuya, S. An essential role for de novo biosynthesis of L-serine in CNS development. Asia Pac. J. Clin. Nutr. 2008, 17 (Suppl. S1), 312–315. [Google Scholar] [PubMed]
- Yin, K. Positive correlation between expression level of mitochondrial serine hydroxymethyltransferase and breast cancer grade. Onco Targets Ther. 2015, 8, 1069–1074. [Google Scholar] [CrossRef]
- Pranzini, E.; Pardella, E.; Muccillo, L.; Leo, A.; Nesi, I.; Santi, A.; Parri, M.; Zhang, T.; Uribe, A.H.; Lottini, T.; et al. SHMT2-mediated mitochondrial serine metabolism drives 5-FU resistance by fueling nucleotide biosynthesis. Cell Rep. 2022, 40, 111233. [Google Scholar] [CrossRef]
- Tajan, M.; Hennequart, M.; Cheung, E.C.; Zani, F.; Hock, A.K.; Legrave, N.; Maddocks, O.D.K.; Ridgway, R.A.; Athineos, D.; Suárez-Bonnet, A.; et al. Serine synthesis pathway inhibition cooperates with dietary serine and glycine limitation for cancer therapy. Nat. Commun. 2021, 12, 366. [Google Scholar] [CrossRef] [PubMed]
- Geeraerts, S.L.; Kampen, K.R.; Rinaldi, G.; Gupta, P.; Planque, M.; Louros, N.; Heylen, E.; De Cremer, K.; De Brucker, K.; Vereecke, S.; et al. Repurposing the Antidepressant Sertraline as SHMT Inhibitor to Suppress Serine/Glycine Synthesis-Addicted Breast Tumor Growth. Mol. Cancer Ther. 2021, 20, 50–63. [Google Scholar] [CrossRef]
- Li, J.T.; Yin, M.; Wang, D.; Wang, J.; Lei, M.Z.; Zhang, Y.; Liu, Y.; Zhang, L.; Zou, S.W.; Hu, L.P.; et al. BCAT2-mediated BCAA catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2020, 22, 167–174. [Google Scholar] [CrossRef]
- Mayers, J.R.; Wu, C.; Clish, C.B.; Kraft, P.; Torrence, M.E.; Fiske, B.P.; Yuan, C.; Bao, Y.; Townsend, M.K.; Tworoger, S.S.; et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014, 20, 1193–1198. [Google Scholar] [CrossRef]
- Zhang, L.; Han, J. Branched-chain amino acid transaminase 1 (BCAT1) promotes the growth of breast cancer cells through improving mTOR-mediated mitochondrial biogenesis and function. Biochem. Biophys. Res. Commun. 2017, 486, 224–231. [Google Scholar] [CrossRef]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef]
- Tönjes, M.; Barbus, S.; Park, Y.J.; Wang, W.; Schlotter, M.; Lindroth, A.M.; Pleier, S.V.; Bai, A.H.C.; Karra, D.; Piro, R.M.; et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat. Med. 2013, 19, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Xu, E.; Ji, B.; Jin, K.; Chen, Y. Branched-chain amino acids catabolism and cancer progression: Focus on therapeutic interventions. Front. Oncol. 2023, 13, 1220638. [Google Scholar] [CrossRef] [PubMed]
- Chi, R.; Yao, C.; Chen, S.; Liu, Y.; He, Y.; Zhang, J.; Ellies, L.G.; Wu, X.; Zhao, Q.; Zhou, C.; et al. Elevated BCAA Suppresses the Development and Metastasis of Breast Cancer. Front. Oncol. 2022, 12, 887257. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Shi, X.; Zhou, M.; Bao, L.; Hatanpaa, K.J.; Patel, T.; DeBerardinis, R.J.; Wang, Y.; Luo, W. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell Mol. Life Sci. 2021, 78, 195–206. [Google Scholar] [CrossRef]
- Shafei, M.A.; Flemban, A.; Daly, C.; Kendrick, P.; White, P.; Dean, S.; Qualtrough, D.; Conway, M.E. Differential expression of the BCAT isoforms between breast cancer subtypes. Breast Cancer 2021, 28, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.; Mora, S.; Madu, G.; Adegoke, O.A.J. Branched-chain Amino Acids: Catabolism in Skeletal Muscle and Implications for Muscle and Whole-body Metabolism. Front. Physiol. 2021, 12, 702826. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, F.; Wang, K.; Liu, M.; Li, J.; Zhao, Q.; Jiang, L.; Zhang, Z.; Li, Y.; Chen, H.; et al. BCAT1 knockdown-mediated suppression of melanoma cell proliferation and migration is associated with reduced oxidative phosphorylation. Am. J. Cancer Res. 2021, 11, 2670–2683. [Google Scholar]
- Wang, Z.Q.; Faddaoui, A.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.C.; Sebastianelli, A.; Guillemette, C.; Gobeil, S.; Macdonald, E.; et al. BCAT1 expression associates with ovarian cancer progression: Possible implications in altered disease metabolism. Oncotarget 2015, 6, 31522–31543. [Google Scholar] [CrossRef]
- Zheng, Y.H.; Hu, W.J.; Chen, B.C.; Grahn, T.H.; Zhao, Y.R.; Bao, H.L.; Zhu, Y.F.; Zhang, Q.Y. BCAT1, a key prognostic predictor of hepatocellular carcinoma, promotes cell proliferation and induces chemoresistance to cisplatin. Liver Int. 2016, 36, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.B.; Reiche, W.S.; Fifelski, N.A.; Schultz, A.J.; Stanford, S.J.; Martin, A.A.; Nack, D.L.; Radlwimmer, B.; Boyer, M.P.; Ananieva, E.A. Leucine and branched-chain amino acid metabolism contribute to the growth of bone sarcomas by regulating AMPK and mTORC1 signaling. Biochem. J. 2020, 477, 1579–1599. [Google Scholar] [CrossRef]
- Wang, P.; Wu, S.; Zeng, X.; Zhang, Y.; Zhou, Y.; Su, L.; Lin, W. BCAT1 promotes proliferation of endometrial cancer cells through reprogrammed BCAA metabolism. Int. J. Clin. Exp. Pathol. 2018, 11, 5536–5546. [Google Scholar] [PubMed]
- Ananieva, E.A.; Wilkinson, A.C. Branched-chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64–70. [Google Scholar] [CrossRef]
- Rii, J.; Sakamoto, S.; Mizokami, A.; Xu, M.; Fujimoto, A.; Saito, S.; Koike, H.; Tamura, T.; Arai, T.; Yamada, Y.; et al. L-type amino acid transporter 1 inhibitor JPH203 prevents the growth of cabazitaxel-resistant prostate cancer by inhibiting cyclin-dependent kinase activity. Cancer Sci. 2024, 115, 937–953. [Google Scholar] [CrossRef]
- Rossi, M.; Mascaretti, F.; Parpinel, M.; Serraino, D.; Crispo, A.; Celentano, E.; Giacosa, A.; La Vecchia, C. Dietary intake of branched-chain amino acids and colorectal cancer risk. Br. J. Nutr. 2021, 126, 22–27. [Google Scholar] [CrossRef]
- Long, L.; Yang, W.; Liu, L.; Tobias, D.K.; Katagiri, R.; Wu, K.; Jin, L.; Zhang, F.F.; Luo, X.; Liu, X.; et al. Dietary intake of branched-chain amino acids and survival after colorectal cancer diagnosis. Int. J. Cancer 2021, 148, 2471–2480. [Google Scholar] [CrossRef] [PubMed]
- Ericksen, R.E.; Lim, S.L.; McDonnell, E.; Shuen, W.H.; Vadiveloo, M.; White, P.J.; Ding, Z.; Kwok, R.; Lee, P.; Radda, G.K.; et al. Loss of BCAA Catabolism during Carcinogenesis Enhances mTORC1 Activity and Promotes Tumor Development and Progression. Cell Metab. 2019, 29, 1151–1165.e1156. [Google Scholar] [CrossRef]
- Takegoshi, K.; Honda, M.; Okada, H.; Takabatake, R.; Matsuzawa-Nagata, N.; Campbell, J.S.; Nishikawa, M.; Shimakami, T.; Shirasaki, T.; Sakai, Y.; et al. Branched-chain amino acids prevent hepatic fibrosis and development of hepatocellular carcinoma in a non-alcoholic steatohepatitis mouse model. Oncotarget 2017, 8, 18191–18205. [Google Scholar] [CrossRef]
- Cha, J.H.; Bae, S.H.; Kim, H.L.; Park, N.R.; Choi, E.S.; Jung, E.S.; Choi, J.Y.; Yoon, S.K. Branched-chain amino acids ameliorate fibrosis and suppress tumor growth in a rat model of hepatocellular carcinoma with liver cirrhosis. PLoS ONE 2013, 8, e77899. [Google Scholar] [CrossRef]
- Li, J.T.; Li, K.Y.; Su, Y.; Shen, Y.; Lei, M.Z.; Zhang, F.; Yin, M.; Chen, Z.J.; Wen, W.Y.; Hu, W.G.; et al. Diet high in branched-chain amino acid promotes PDAC development by USP1-mediated BCAT2 stabilization. Natl. Sci. Rev. 2022, 9, nwab212. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, S.; Usui, T.; Kuhara, K.; Tsuchiya, A.; Miyauchi, T.; Kono, T.; Asaka, S.; Yamaguchi, K.; Yokomizo, H.; Shimakawa, T.; et al. Impact of Branched-Chain Amino Acid-Enriched Nutrient on liver Cirrhosis with Hepatocellular Carcinoma Undergoing Transcatheter Arterial Chemoembolization in Barcelona Clinic Liver Cancer Stage B: A Prospective Study. J. Nippon. Med. Sch. 2016, 83, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Nojiri, S.; Fujiwara, K.; Shinkai, N.; Iio, E.; Joh, T. Effects of branched-chain amino acid supplementation after radiofrequency ablation for hepatocellular carcinoma: A randomized trial. Nutrition 2017, 33, 20–27. [Google Scholar] [CrossRef]
- Park, J.G.; Tak, W.Y.; Park, S.Y.; Kweon, Y.O.; Jang, S.Y.; Lee, Y.R.; Bae, S.H.; Jang, J.Y.; Kim, D.Y.; Lee, J.S.; et al. Effects of branched-chain amino acids (BCAAs) on the progression of advanced liver disease: A Korean nationwide, multicenter, retrospective, observational, cohort study. Medicine 2017, 96, e6580. [Google Scholar] [CrossRef]
- Ji, J.X.; Cochrane, D.R.; Tessier-Cloutier, B.; Chen, S.Y.; Ho, G.; Pathak, K.V.; Alcazar, I.N.; Farnell, D.; Leung, S.; Cheng, A.; et al. Arginine Depletion Therapy with ADI-PEG20 Limits Tumor Growth in Argininosuccinate Synthase-Deficient Ovarian Cancer, Including Small-Cell Carcinoma of the Ovary, Hypercalcemic Type. Clin. Cancer Res. 2020, 26, 4402–4413. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.M.; Sheaff, M.T.; Szlosarek, P.W. Targeting arginine-dependent cancers with arginine-degrading enzymes: Opportunities and challenges. Cancer Res. Treat. 2013, 45, 251–262. [Google Scholar] [CrossRef]
- Yang, J.S.; Wang, C.C.; Qiu, J.D.; Ren, B.; You, L. Arginine metabolism: A potential target in pancreatic cancer therapy. Chin Med. J. 2020, 134, 28–37. [Google Scholar] [CrossRef]
- Alexandrou, C.; Al-Aqbi, S.S.; Higgins, J.A.; Boyle, W.; Karmokar, A.; Andreadi, C.; Luo, J.L.; Moore, D.A.; Viskaduraki, M.; Blades, M.; et al. Sensitivity of Colorectal Cancer to Arginine Deprivation Therapy is Shaped by Differential Expression of Urea Cycle Enzymes. Sci. Rep. 2018, 8, 12096. [Google Scholar] [CrossRef]
- Cendan, J.C.; Souba, W.W.; Copeland, E.M., 3rd; Lind, D.S. Characterization and growth factor stimulation of L-arginine transport in a human colon cancer cell line. Ann. Surg. Oncol. 1995, 2, 257–265. [Google Scholar] [CrossRef]
- Cendan, J.C.; Souba, W.W.; Copeland, E.M., 3rd; Lind, D.S. Increased L-arginine transport in a nitric oxide-producing metastatic colon cancer cell line. Ann. Surg. Oncol. 1996, 3, 501–508. [Google Scholar] [CrossRef]
- Abdelmagid, S.A.; Rickard, J.A.; McDonald, W.J.; Thomas, L.N.; Too, C.K. CAT-1-mediated arginine uptake and regulation of nitric oxide synthases for the survival of human breast cancer cell lines. J. Cell Biochem. 2011, 112, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, A.S.; Geerts, D. Polyamine synthesis as a target of MYC oncogenes. J. Biol. Chem. 2018, 293, 18757–18769. [Google Scholar] [CrossRef] [PubMed]
- Pilanc, P.; Wojnicki, K.; Roura, A.J.; Cyranowski, S.; Ellert-Miklaszewska, A.; Ochocka, N.; Gielniewski, B.; Grzybowski, M.M.; Błaszczyk, R.; Stańczak, P.S.; et al. A Novel Oral Arginase 1/2 Inhibitor Enhances the Antitumor Effect of PD-1 Inhibition in Murine Experimental Gliomas by Altering the Immunosuppressive Environment. Front. Oncol. 2021, 11, 703465. [Google Scholar] [CrossRef]
- Gannon, P.O.; Godin-Ethier, J.; Hassler, M.; Delvoye, N.; Aversa, M.; Poisson, A.O.; Péant, B.; Alam Fahmy, M.; Saad, F.; Lapointe, R.; et al. Androgen-regulated expression of arginase 1, arginase 2 and interleukin-8 in human prostate cancer. PLoS ONE 2010, 5, e12107. [Google Scholar] [CrossRef]
- Sousa, M.S.; Latini, F.R.; Monteiro, H.P.; Cerutti, J.M. Arginase 2 and nitric oxide synthase: Pathways associated with the pathogenesis of thyroid tumors. Free Radic. Biol. Med. 2010, 49, 997–1007. [Google Scholar] [CrossRef]
- Porembska, Z.; Luboiński, G.; Chrzanowska, A.; Mielczarek, M.; Magnuska, J.; Barańczyk-Kuźma, A. Arginase in patients with breast cancer. Clin. Chim. Acta 2003, 328, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Kus, K.; Kij, A.; Zakrzewska, A.; Jasztal, A.; Stojak, M.; Walczak, M.; Chlopicki, S. Alterations in arginine and energy metabolism, structural and signalling lipids in metastatic breast cancer in mice detected in plasma by targeted metabolomics and lipidomics. Breast Cancer Res. 2018, 20, 148. [Google Scholar] [CrossRef]
- Singh, R.; Pervin, S.; Karimi, A.; Cederbaum, S.; Chaudhuri, G. Arginase activity in human breast cancer cell lines: N(omega)-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000, 60, 3305–3312. [Google Scholar]
- Tabe, Y.; Lorenzi, P.L.; Konopleva, M. Amino acid metabolism in hematologic malignancies and the era of targeted therapy. Blood 2019, 134, 1014–1023. [Google Scholar] [CrossRef]
- Massi, D.; Marconi, C.; Franchi, A.; Bianchini, F.; Paglierani, M.; Ketabchi, S.; Miracco, C.; Santucci, M.; Calorini, L. Arginine metabolism in tumor-associated macrophages in cutaneous malignant melanoma: Evidence from human and experimental tumors. Hum. Pathol. 2007, 38, 1516–1525. [Google Scholar] [CrossRef]
- Mielczarek-Puta, M.; Graboń, W.; Chrzanowska, A.; Barańczyk-Kuźma, A. Arginase and arginine in diagnostics of patients with colorectal cancer and patients with colorectal cancer liver metastases. Contemp. Oncol./Współczesna Onkol. 2008, 12, 51–55. [Google Scholar]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef]
- Nasreddine, G.; El-Sibai, M.; Abi-Habib, R.J. Cytotoxicity of [HuArgI (co)-PEG5000]-induced arginine deprivation to ovarian Cancer cells is autophagy dependent. Investig. New Drugs 2020, 38, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Tanios, R.; Bekdash, A.; Kassab, E.; Stone, E.; Georgiou, G.; Frankel, A.E.; Abi-Habib, R.J. Human recombinant arginase I(Co)-PEG5000 [HuArgI(Co)-PEG5000]-induced arginine depletion is selectively cytotoxic to human acute myeloid leukemia cells. Leuk. Res. 2013, 37, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Khalil, N.; Abi-Habib, R.J. [HuArgI (co)-PEG5000]-induced arginine deprivation leads to autophagy dependent cell death in pancreatic cancer cells. Investig. New Drugs 2020, 38, 1236–1246. [Google Scholar] [CrossRef]
- Al-Koussa, H.; Al-Haddad, M.; Abi-Habib, R.; El-Sibai, M. Human Recombinant Arginase I [HuArgI (Co)-PEG5000]-Induced Arginine Depletion Inhibits Colorectal Cancer Cell Migration and Invasion. Int. J. Mol. Sci. 2019, 20, 6018. [Google Scholar] [CrossRef]
- Lam, S.K.; Li, Y.Y.; Xu, S.; Leung, L.L.; U, K.P.; Zheng, Y.F.; Cheng, P.N.; Ho, J.C. Growth suppressive effect of pegylated arginase in malignant pleural mesothelioma xenografts. Respir. Res. 2017, 18, 80. [Google Scholar] [CrossRef]
- Ott, P.A.; Carvajal, R.D.; Pandit-Taskar, N.; Jungbluth, A.A.; Hoffman, E.W.; Wu, B.W.; Bomalaski, J.S.; Venhaus, R.; Pan, L.; Old, L.J.; et al. Phase I/II study of pegylated arginine deiminase (ADI-PEG 20) in patients with advanced melanoma. Investig. New Drugs 2013, 31, 425–434. [Google Scholar] [CrossRef]
- Mussai, F.; Egan, S.; Higginbotham-Jones, J.; Perry, T.; Beggs, A.; Odintsova, E.; Loke, J.; Pratt, G.; U, K.P.; Lo, A.; et al. Arginine dependence of acute myeloid leukemia blast proliferation: A novel therapeutic target. Blood 2015, 125, 2386–2396. [Google Scholar] [CrossRef]
- Abou-Alfa, G.; Qin, S.; Ryoo, B.Y.; Lu, S.-N.; Yen, C.J.; Feng, Y.H.; Lim, H.; Izzo, F.; Colombo, M.; Sarker, D.; et al. ADI-PEG 20 Plus Best Supportive Care versus Placebo Plus Best Supportive Care in Patients with Advanced Hepatocellular Carcinoma. Ann. Oncol. 2018, 29, 1402–1408. [Google Scholar] [CrossRef]
- Daher, B.; Vučetić, M.; Pouysségur, J. Cysteine Depletion, a Key Action to Challenge Cancer Cells to Ferroptotic Cell Death. Front. Oncol. 2020, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine metabolic circuitries: Druggable targets in cancer. Br. J. Cancer 2021, 124, 862–879. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Yang, S.; Qiu, Q.; Cui, G.; Zhang, Y.; Yao, M.; Li, X.; Chen, C.; Gu, J.; Wang, T.; et al. Hypoxia-induced cysteine metabolism reprogramming is crucial for the tumorigenesis of colorectal cancer. Redox Biol. 2024, 75, 103286. [Google Scholar] [CrossRef]
- Nunes, S.C.; Ramos, C.; Lopes-Coelho, F.; Sequeira, C.O.; Silva, F.; Gouveia-Fernandes, S.; Rodrigues, A.; Guimarães, A.; Silveira, M.; Abreu, S.; et al. Cysteine allows ovarian cancer cells to adapt to hypoxia and to escape from carboplatin cytotoxicity. Sci. Rep. 2018, 8, 9513. [Google Scholar] [CrossRef]
- Lin, J.; Lee, I.M.; Song, Y.; Cook, N.R.; Selhub, J.; Manson, J.E.; Buring, J.E.; Zhang, S.M. Plasma homocysteine and cysteine and risk of breast cancer in women. Cancer Res. 2010, 70, 2397–2405. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, Z.; Xie, Z.; Chen, Y.; Zheng, Z.; Wei, X.; Huang, B.; Shan, Z.; Liu, J.; Fan, S.; et al. Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic. Biol. Med. 2020, 160, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Novera, W.; Lee, Z.W.; Nin, D.S.; Dai, M.Z.; Binte Idres, S.; Wu, H.; Damen, J.M.A.; Tan, T.Z.; Sim, A.Y.L.; Long, Y.C.; et al. Cysteine Deprivation Targets Ovarian Clear Cell Carcinoma Via Oxidative Stress and Iron-Sulfur Cluster Biogenesis Deficit. Antioxid. Redox Signal 2020, 33, 1191–1208. [Google Scholar] [CrossRef]
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am. J. Cancer Res. 2020, 10, 3106–3126. [Google Scholar]
- Tang, X.; Chen, W.; Liu, H.; Liu, N.; Chen, D.; Tian, D.; Wang, J. Research progress on SLC7A11 in the regulation of cystine/cysteine metabolism in tumors. Oncol. Lett. 2022, 23, 47. [Google Scholar] [CrossRef]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system x(c)- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef]
- Liu, J.; Liu, M.; Zhang, H.; Wei, X.; Wang, J.; Xian, M.; Guo, W. Exploring cysteine regulation in cancer cell survival with a highly specific “Lock and Key” fluorescent probe for cysteine. Chem. Sci. 2019, 10, 10065–10071. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Tian, W.; Zhang, W.; Wang, Y.; Jin, R.; Wang, Y.; Guo, H.; Tang, Y.; Yao, X. Recent advances of IDH1 mutant inhibitor in cancer therapy. Front. Pharmacol. 2022, 13, 982424. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Norsworthy, K.J.; Wang, X.; Vallejo, J.; Chiu Yuen Chow, E.; Li, R.J.; Sun, J.; Charlab, R.; Jiang, X.; Pazdur, R.; et al. FDA Approval Summary: Ivosidenib in Combination with Azacitidine for Treatment of Patients with Newly Diagnosed Acute Myeloid Leukemia with an IDH1 Mutation. Clin. Cancer Res. 2024, 30, 1226–1231. [Google Scholar] [CrossRef]
- Gouda, M.A.; Voss, M.H.; Tawbi, H.; Gordon, M.; Tykodi, S.S.; Lam, E.T.; Vaishampayan, U.; Tannir, N.M.; Chaves, J.; Nikolinakos, P.; et al. A phase I/II study of the safety and efficacy of telaglenastat (CB-839) in combination with nivolumab in patients with metastatic melanoma, renal cell carcinoma, and non-small-cell lung cancer. ESMO Open 2025, 10, 104536. [Google Scholar] [CrossRef]
- Tannir, N.M.; Agarwal, N.; Porta, C.; Lawrence, N.J.; Motzer, R.; McGregor, B.; Lee, R.J.; Jain, R.K.; Davis, N.; Appleman, L.J.; et al. Efficacy and Safety of Telaglenastat Plus Cabozantinib vs Placebo Plus Cabozantinib in Patients with Advanced Renal Cell Carcinoma: The CANTATA Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.K.; Choi, S.; Yoon, S.J.; Choi, R.J.; Park, J.; Lee, E.H.; Cho, H.J.; Lee, S.; Teo, W.Y.; Moon, J.H.; et al. Etomoxir, a carnitine palmitoyltransferase 1 inhibitor, combined with temozolomide reduces stemness and invasiveness in patient-derived glioblastoma tumorspheres. Cancer Cell Int. 2022, 22, 309. [Google Scholar] [CrossRef]
- Zhou, P.; Xiao, Y.; Zhou, X.; Fang, J.; Zhang, J.; Liu, J.; Guo, L.; Zhang, J.; Zhang, N.; Chen, K.; et al. Mapping Spatiotemporal Heterogeneity in Multifocal Breast Tumor Progression by Noninvasive Ultrasound Elastography-Guided Mass Spectrometry Imaging Strategy. JACS Au 2024, 4, 465–475. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminazdeh-Gohari, S.; Kofler, B. Ketogenic diet in cancer therapy. Aging 2018, 10, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Nong, S.; Qian, Y.; Zhang, T.; Zhou, X.; Wei, Y.; Yin, X.; Ma, X. Mechanism and application of nonessential amino acid deprivation associated with tumor therapy. MedComm—Future Med. 2022, 1, e12. [Google Scholar] [CrossRef]
- Abene, J.; Tyburski, S.; Kral, T.V.E.; Quinn, R.; Deng, J. Diet as an Adjunct Therapy in Reducing Chemotherapy Toxicities and Improving Patients Quality of Life: A Systematic Review and Meta-Analysis. Nutr. Cancer 2025, 77, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, G.; Andreeva, N.; MacDonald, M.H.; Garrett, W.S. Interactions between diet and gut microbiota in cancer. Nat. Microbiol. 2024, 9, 1644–1654. [Google Scholar] [CrossRef]
| Part A | |||||||
| Target/Enzyme/Transporter | Inhibitor/Compound | Associated Cancer Types | Clinical Trial Phase/Status | ||||
| Hexokinase (HK) | 2-Deoxy-D-glucose (2-DG) | Leukemia (P388), solid tumors | Phase I (NCT00096707), limited efficacy | ||||
| GLUT1/GLUT family | Under investigation | Various solid tumors | Preclinical/exploratory | ||||
| Sodium-Glucose Cotransporter2 (SGLT2) | SGLT2 inhibitors (e.g., dapagliflozin, empagliflozin) | Hepatocellular carcinoma (HCC), T2D-associated cancers | Observational/real-world studies; no defined clinical trial phase | ||||
| Pyruvate Kinase M2 (PKM2) | Not specified | Lung adenocarcinoma, triple-negative breast cancer (TNBC) | Preclinical; prognostic marker | ||||
| Lactate Dehydrogenase A (LDHA) | FX11, oxamate, dichloroacetate, PSTMB | Neuroblastoma, breast (MCF-7), liver (Hep3B), colon (HT29), others cancers | Preclinical | ||||
| Monocarboxylate Transporter 1 (MCT1) | AZD3965 | Lung, breast, metabolic symbiotic tumors | Phase I/II (NCT01791595) | ||||
| Monocarboxylate Transporter 4 (MCT4) | Under investigation | Hypoxic/metastatic tumors | Preclinical | ||||
| Pyruvate Dehydrogenase (PDH) | Devimistat (CPI-613) | Acute myeloid leukemia (AML), pancreatic cancer | Phase III (NCT03504410, failed) | ||||
| α-Ketoglutarate Dehydrogenase (KGDH) | Devimistat | AML, pancreatic adenocarcinoma | Phase III (AVENGER 500, no clinical benefit) | ||||
| Pyruvate Carboxylase (PC) | Not specified | Metastatic breast cancer | Preclinical; linked to metastasis | ||||
| Acetyl-CoA Carboxylase 1 (ACC1) | Not specified | Breast cancer (mesenchymal phenotype) | Preclinical; epigenetic target | ||||
| Proton pumps/Tumor pH regulation | TRIS buffer (Tris–base) | Various solid tumors | Experimental; limited clinical feasibility | ||||
| Lactate (as a target) | Alkaline buffers (e.g., TRIS), MCT inhibition | Multiple cancer types | Preclinical/early clinical | ||||
| Part B | |||||||
| Target/Enzyme/Transporter | Function/Mechanism | Associated Cancer Types | Inhibitor/Compound | Clinical Status/Notes | |||
| Fatty Acid Synthase (FASN) | De novo fatty acid synthesis (palmitate) | Breast (esp. TNBC), lung, prostate, colorectal, ovarian cancers | TVB-2640 (denifanstat), Orlistat, PPIs | Multiple Phase I/II trials (NCT02223247, NCT03808558); sensitizes to chemo/radiotherapy | |||
| ATP-Citrate Lyase (ACLY) | Citrate → Acetyl-CoA, key in lipogenesis | Breast, prostate, pancreatic cancers | Bempedoic acid (ETC-1002) | Preclinical/early clinical; sensitizes prostate CA to AR antagonism | |||
| Acetyl-CoA Carboxylase (ACC) | Acetyl-CoA → Malonyl-CoA (rate-limiting step in FA synthesis) | Breast cancer (mesenchymal subtypes) | ND (not detailed) | Preclinical | |||
| Acetyl-CoA Synthetase 2 (ACSS2) | Acetate → Acetyl-CoA (supports lipid synthesis under stress) | Various tumors under hypoxia | ND | Preclinical | |||
| Stearoyl-CoA Desaturase (SCD) | Converts saturated FA to MUFA (for membrane fluidity, signaling) | Breast, thyroid cancer and others | SCD inhibitors (unspecified) | Preclinical; silencing inhibits proliferation/migration | |||
| Fatty Acid Desaturase 2 (FADS2) | Palmitate → sapienate (alternative desaturation) | SCD-resistant tumors (HCC, lung) | ND | Dual inhibition (SCD + FADS2) shows tumor reduction | |||
| Fatty Acid Transporters (FATPs) | Long-chain FA uptake | Breast, ovarian, metastatic tumors | ND | Preclinical; key in FA scavenging from microenvironment | |||
| CD36 (Fatty Acid Translocase) | FA uptake + signaling receptor | Breast, ovarian (esp. metastatic) | Anti-CD36 antibodies (experimental) | Target for blocking metastasis | |||
| Fatty Acid Binding Proteins (FABP4/5) | Intracellular FA transport | Breast cancer, macrophages, adipocytes in TME | FABP4/5 inhibitors (unspecified) | Preclinical; prognostic markers; hypoxia-induced expression | |||
| Carnitine Palmitoyltransferase 1 (CPT1A) | Controls FA entry into mitochondria for β-oxidation | Various solid tumors (esp. metastatic) | Etomoxir (experimental) | Preclinical; associated with resistance to metabolic stress; granted orphan drug status for malignant glioma | |||
| Acyl-CoA Dehydrogenases | Initiates fatty acid β-oxidation | Not specified | ND | Preclinical | |||
| PPARs (esp. PPARα/δ) | Regulate FAO, lipid metabolism, and immune modulation | Breast, prostate, colorectal cancers | Agonists/antagonists | Experimental; role in immune evasion and metabolic flexibility | |||
| Lipid Droplets (LD) Formation | Store excess FA; source of NADPH and energy | Breast, prostate, metastatic cancers | ND | Protective against lipotoxicity and ROS; linked to aggressiveness | |||
| Oncogenic Signaling & Lipid Metabolism | EGFR/HER1/2 → PI3K-Akt-mTOR → FASN upregulation | Breast (esp. HER2+), lung (KRAS-mutant) | Indirect targeting via FASN/PI3K/mTOR inhibitors | Supports autocrine loop; contributes to resistance and metastasis | |||
| ROS Resistance via Lipid Remodeling | FA saturation status alters membrane permeability/resistance | Castration-resistant prostate cancer | ND | Lipidomic signatures correlate with poor prognosis | |||
| FA Metabolism in Immune Evasion | Alters T cell and dendritic cell function in TME | Solid tumors | Lipid metabolism modulators (e.g., FASN inhibitors) | Enhances efficacy of immunotherapy (checkpoint blockade etc.) | |||
| Part C | |||||||
| Tatget/Enzyme/Transporter | Inhibitor/Compound | Associated Cancer Types | Clinical Trial Phase/Status | ||||
| Glutaminase (GLS1) | Telaglenastat (CB-839) | Triple-negative breast cancer, hematological malignance, glioblastoma, renal carcinoma, colorectal, lung and cervical cancers | Phase I—completed in hematologic cancers (NCT02071862) Phase I/II + nivolumab—not demonstrated therapeutic efficacy in advanced solid tumors (NCT02861300) Phase Ib/II + talazoparib—completed in solid tumors (NCT03875313) Phase II in NSCLC—terminated (NCT02771626) Phase II in RCC + everolimus—completed (NCT03163667) Phase II in RCC + cabozantinib—did not meet primary endpoint (NCT03428217) Fast Track status + cabozantinib in metastatic RCC—currently ongoing | ||||
| Glutaminase (GLS1) | IPN60090 (IACS-6274) | Advanced solid tumors | Phase I in advanced solid tumors—completed (NCT04771783) Phase I + pembrolizumab, paclitaxel, bevacizumab—ongoing (NCT05340835) | ||||
| Glutaminase (GLS1) | bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) | Pancreatic, ovarian cancers, alveolar adenocarcinoma, non-small cell lung cancer, hepatocellular carcinoma | Preclinical | ||||
| Glutaminase C (GAC) | UPGL00004 | Triple-negative breast cancer (TNBC) cells | Preclinical | ||||
| Glutamine transporter ASCT2 (SLC1A5) | V-9302 | Renal carcinoma, breast, colon cancers | Preclinical | ||||
| Glutamine transporter ASCT2 (SLC1A5) | MEDI7247 (antibody) | Hematological malignancies, advance or metastatic solid tumors | Phase I (NCT03811652, NCT03106428)—completed | ||||
| Glutamine transporter ASCT2 (SLC1A5) | L-γ-Glutamyl-p-nitroanilide (GPNA) | Lung, stomach, colon and prostate cancers | Preclinical | ||||
| Glutamine antagonist | 6-Diazo-5-oxo-L-norleucine (DON) | Glioblastoma, pancreatic cancer, sarcomas, leukemias, medulloblastoma | Phase I/II—halted due to toxicity and discontinued | ||||
| Glutamine antagonist | JHU-083 (pro-drug of DON) | Prostate, bladder and colon cancers, lymphoma, melanoma, medulloblastoma | Preclinical | ||||
| Glutamine antagonist | Sirpiglenastat (pro-drug of DRP-104) | Advanced solid tumor | Phase I/II—ongoing (NCT04265534); | ||||
| Glutamine amidotransferases/Glutamine antagonist | azaserine | Pancreatic and colorectal cancers, solid tumors, breast cancer, sarcoma | In solid tumor—halted due to toxicity, preclinical | ||||
| γ-Glutamyl transferase (γ-GT)/Glutamine analog | Acivicin | Leukemia, breast cancer, and ovarian carcinoma, lung cancer | Phase II; halted due to toxicity | ||||
| 3-phosphoglycerate dehydrogenase (PHGDH) | NCT-502/NCT-503 | Breast cancer, melanoma and cervical cancer | Preclinical | ||||
| 3-phosphoglycerate dehydrogenase (PHGDH) | PKUMDL-WQ-2101/-2201 | Breast cancer | Preclinical | ||||
| 3-phosphoglycerate dehydrogenase (PHGDH) | CBR-5884 | Ovarian and breast cancers | Prelinical | ||||
| 3-phosphoglycerate dehydrogenase (PHGDH) | BI-4916 | Cervical, breast cancer and acute myeloid leukemia | Preclinical | ||||
| Serine hydroxymethyltransferase 1 and 2 (SHMT1/2) | sertraline (antidepressant repurposed) | Breast cancer, lung cancer, glioblastoma, leukemia | Phase I + cytosine Arabinoside in leukemia (NCT02891278)—completed Phase II + metronomic temozolomide in glioblastoma (NCT02770378)—completed Phase II in breast cancer (NCT00667121) completed | ||||
| L-type amino acid transporter 1 (SLC7A5) | JPH203 (KYT-0353) | Solid tumors | Phase I (NCT02028403)—completed Phase I/II ongoing (NCT04671432) | ||||
| Branched-Chain Aminotransferase 1 (BCAT1) | ERG240 | Lung cancer | preclinical | ||||
| Branched-Chain Ketoacid Dehydrogenase Kinase (BCKDK) | BT2 (3,6-dichlorobenzo[b]thiophene-2-carboxylic acid) | Lung cancer, triple-negative breast cancer, colorectal cancer | Preclinical | ||||
| Arginase 1/2 (ARG1/2) | CB-1158 (INCB001158) | Solid tumor | Phase I/II ongoing (NCT02903914) | ||||
| Arginase (cobalt-substituted human arginase) | HuArgI (Co)-PEG5000 (Pegylated cobalt-substituted human arginase) | Hepatocellular carcinoma, melanoma | Preclinical | ||||
| Arginine Deiminase (enzyme degrading arginine) | ADI-PEG20 (Pegylated Arginine Deiminase) | Hepatocellular carcinoma, mesothelioma and metastatic melanoma | Phase I ongoing (NCT02732184) Phase I/II completed (NCT02353318, NCT03455140, NCT01092091, NCT00988195, NCT02285101) Phase III (NCT01287585) in hepatocellular carcinoma—failed | ||||
| Arginase (recombinant human arginase) | PEG-BCT-100 (Pegylated recombinant human arginase) | solid tumors | |||||
| xCT (SLC7A11/SLC3A2; cystine/glutamate antiporter) | Erastin, imidazole ketone erastin | Solid tumors | Preclinical | ||||
| xCT (SLC7A11 cystine/glutamate antiporter) | Sulfasalazine | Glioma, pancreatic, lung and colorectal cancers, leukemia | Phase I/II in gliomas (ISRCTN45828668)—failed | ||||
| Glutamate-cysteine ligase (GCL) | Buthionine sulfoximine (BSO) | Multiple cancer types | Phase I in neuroblastoma—completed | ||||
| Isocitrate dehydrogenase 1 (IDH1) | Ivosidenib (AG-120) | AML (mut. IDH1), cholangiocarcinoma (IDH1 mut.), glioma | Phase I in solid tumor, including glioma (IDH1-mutant) (NCT02073994) Phase Ib in Relapsed/Refractory MDS (IDH1-mutant)—FDA approved October 2023 (NCT02074839) Phase Ib/II in newly diagnosed AML (IDH1-mutant in patient who are not candidates to receive intensive induction chemotherapy, FDA approved July 2022 (NCT02677922) Phase III + azacitidine in AML with an IDH1 Mutation, FDA approved July 2022 (AGILE trial) (NCT03173248) Phase III in Cholangiocarcinoma (advanced/metastatic, IDH1-mutant) FDA approved August 2021 (NCT02989857) | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dominiak, A.; Chełstowska, B.; Nowicka, G. Metabolic Adaptations in Cancer Progression: Optimization Strategies and Therapeutic Targets. Cancers 2025, 17, 2341. https://doi.org/10.3390/cancers17142341
Dominiak A, Chełstowska B, Nowicka G. Metabolic Adaptations in Cancer Progression: Optimization Strategies and Therapeutic Targets. Cancers. 2025; 17(14):2341. https://doi.org/10.3390/cancers17142341
Chicago/Turabian StyleDominiak, Agnieszka, Beata Chełstowska, and Grażyna Nowicka. 2025. "Metabolic Adaptations in Cancer Progression: Optimization Strategies and Therapeutic Targets" Cancers 17, no. 14: 2341. https://doi.org/10.3390/cancers17142341
APA StyleDominiak, A., Chełstowska, B., & Nowicka, G. (2025). Metabolic Adaptations in Cancer Progression: Optimization Strategies and Therapeutic Targets. Cancers, 17(14), 2341. https://doi.org/10.3390/cancers17142341