


Mesothelin-Associated Anti-Senescence Through P53 in Pancreatic Ductal Adenocarcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Antibodies, and Cell Line Generation
2.2. Pathway Enrichment Analysis
2.3. Senescence-Associated β-Galactosidase Assay (SA-β-Gal) and Cell Viability Assay
2.4. Western Blot Assay
2.5. Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Statistical Analysis
3. Results
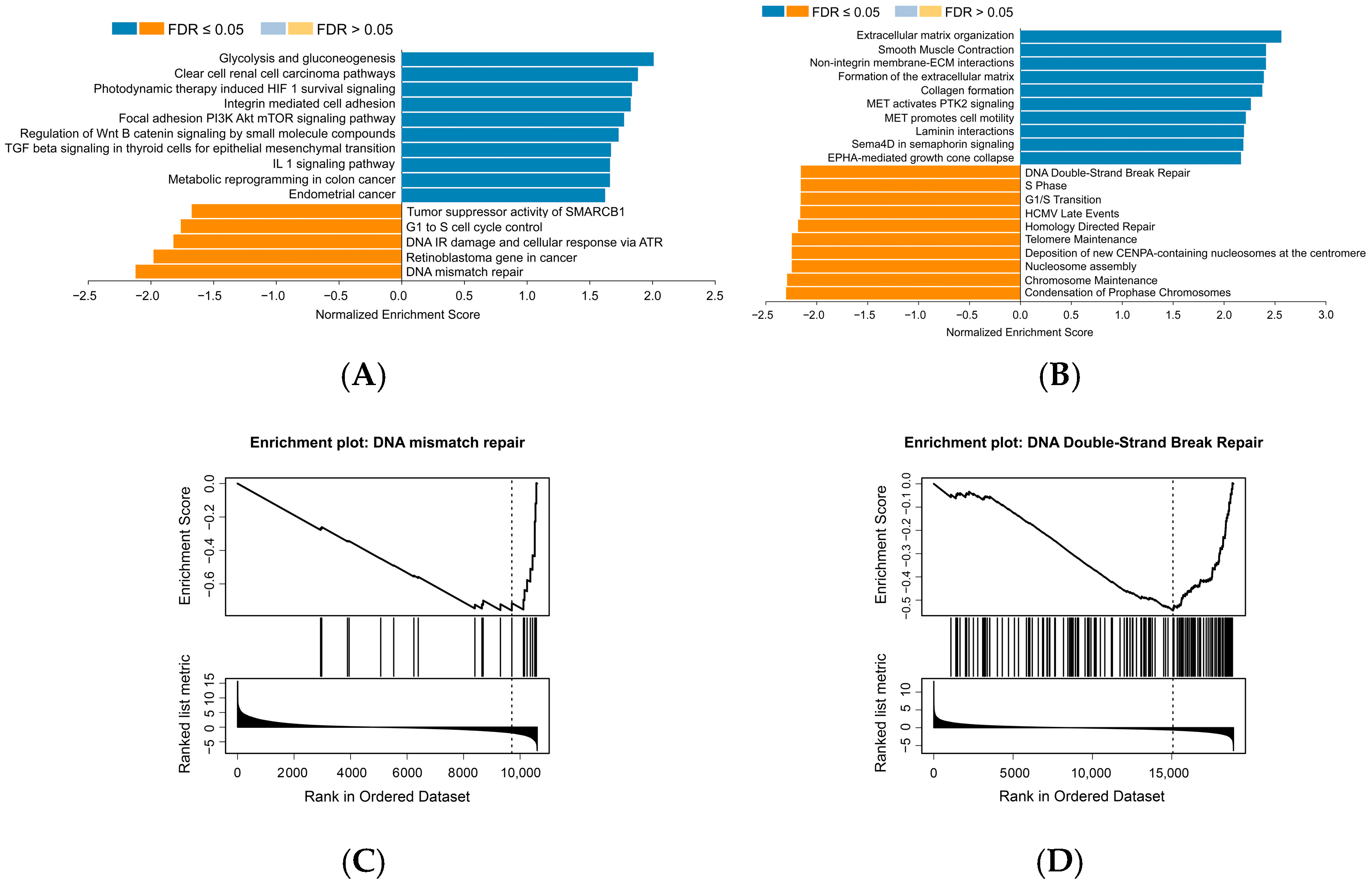
3.1. MSLN Negatively Correlates with Cell Senescence-Related Signaling Pathways in Human PDAC Samples
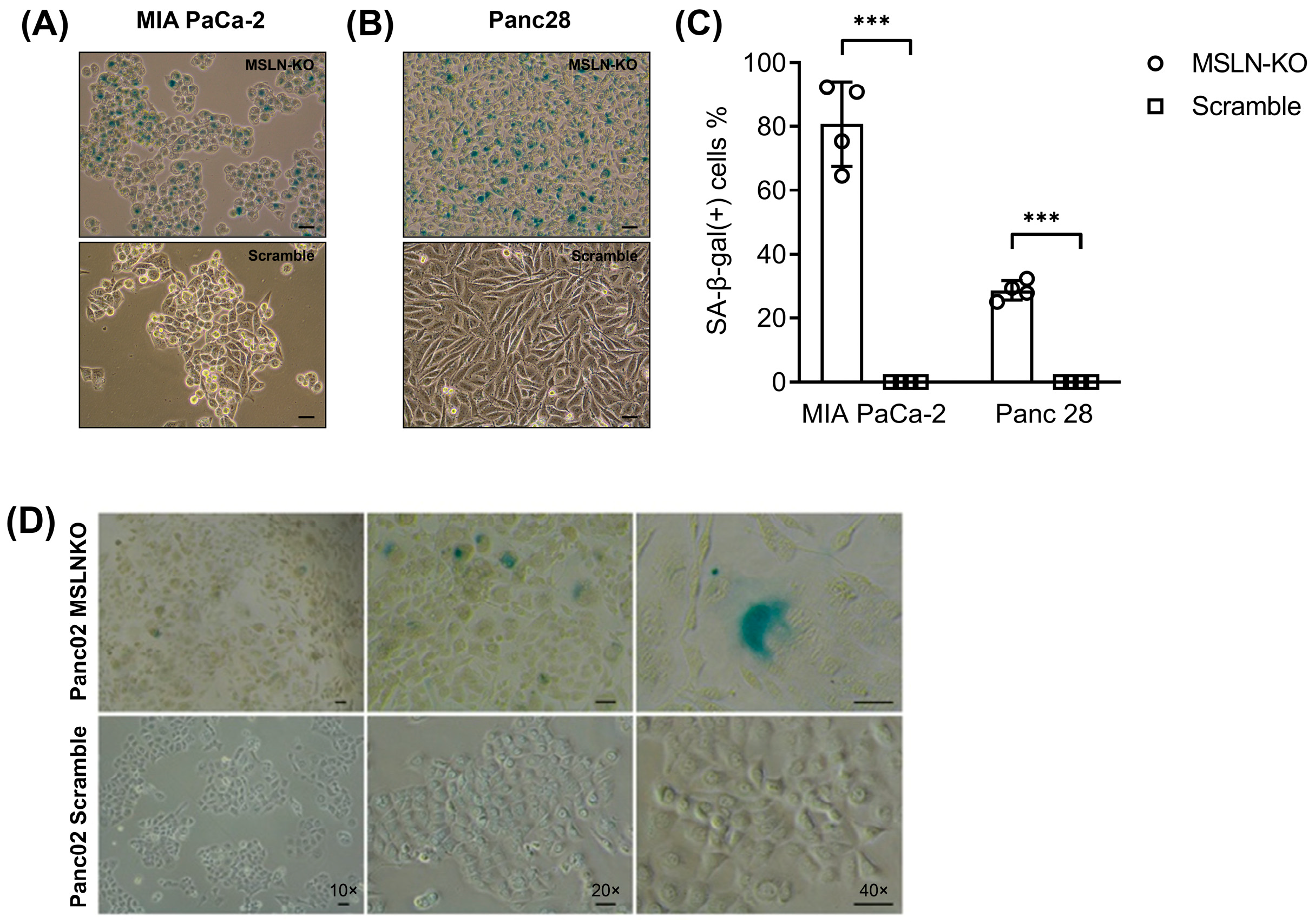
3.2. MSLN Loss Leads to Morphological Changes and Cell Senescence in PDAC Cells
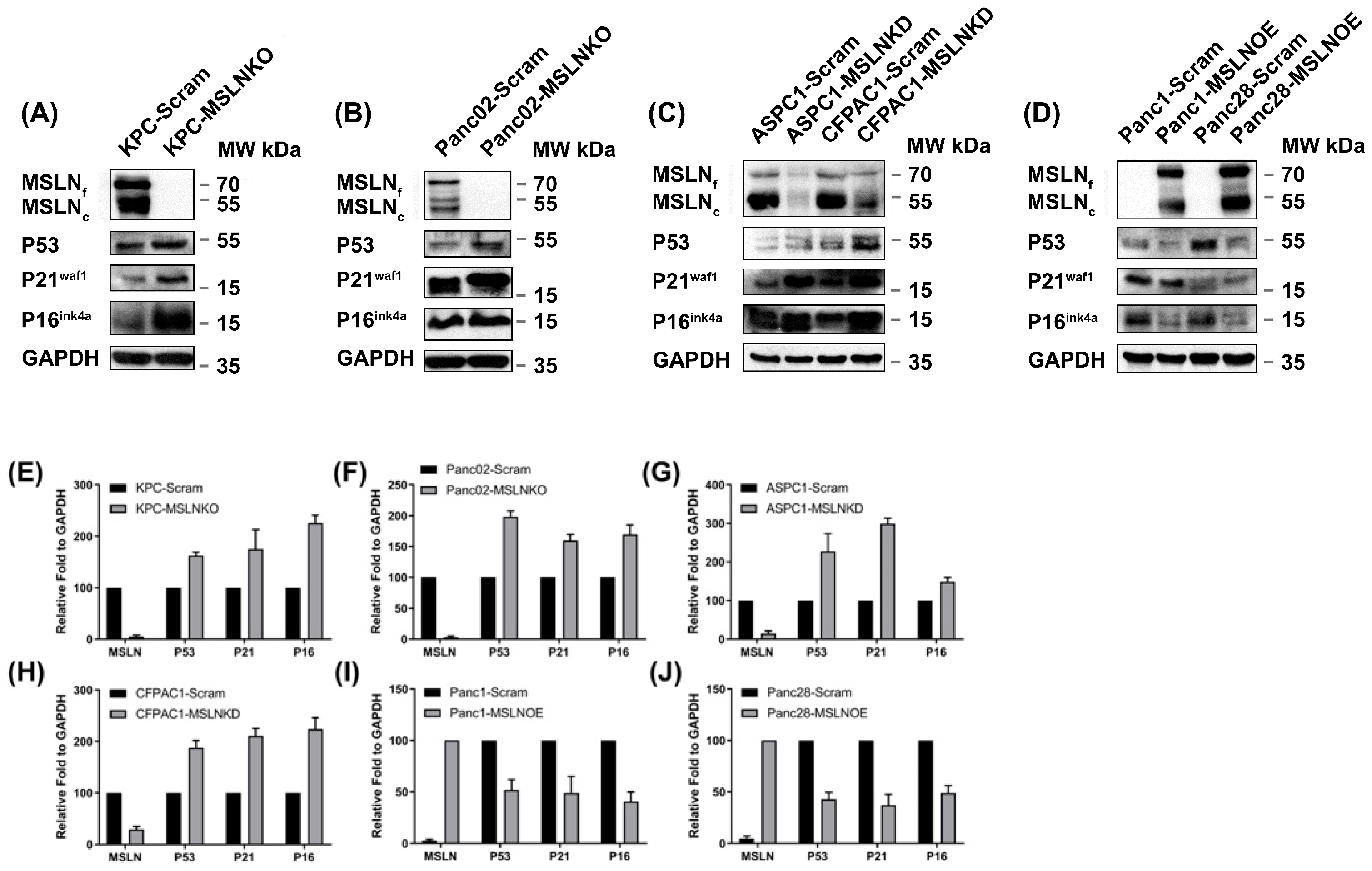
3.3. MSLN Is Negatively Correlated with Senescence Regulators
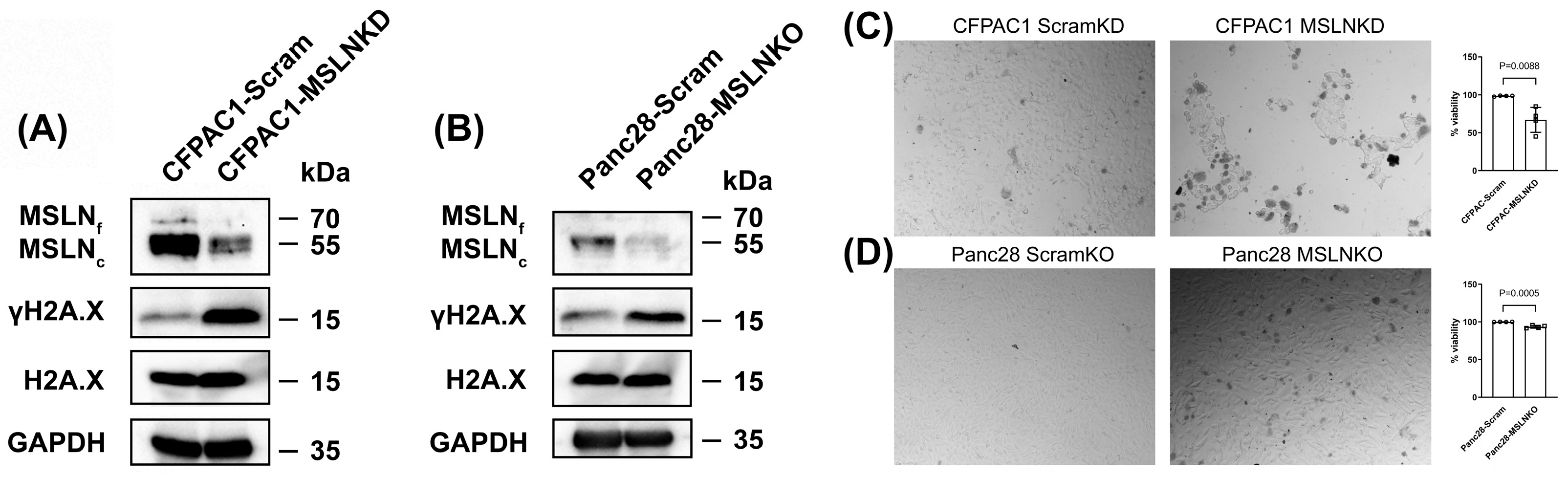
3.4. MSLN Deficiency Induces DNA Damage and Reduces Cell Viability in PDAC Cells
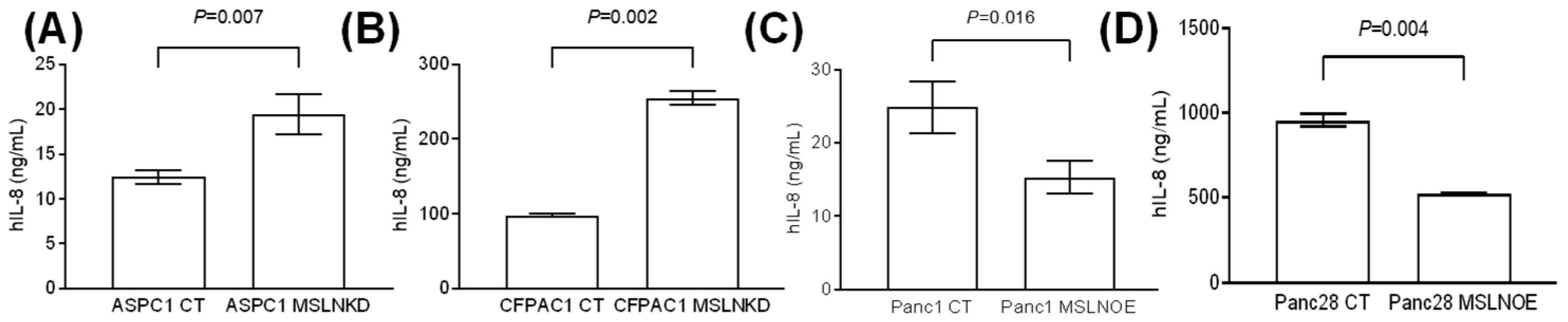
3.5. MSLN Suppresses the Production of the Senescence-Associated Secretory Phenotype
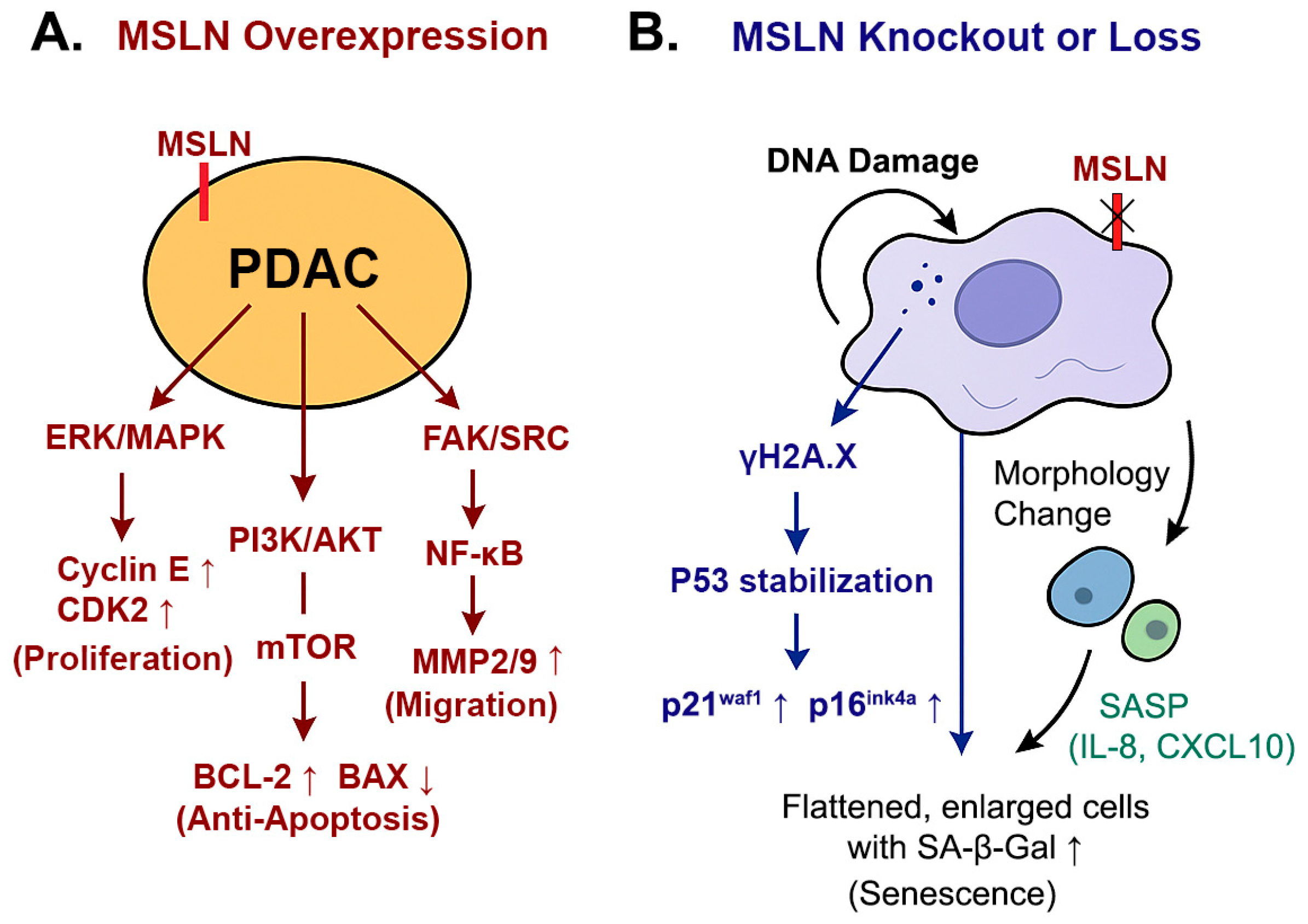
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic cancer: Pathogenesis, screening, diagnosis, and treatment. Gastroenterology 2022, 163, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Deng, G.; Ma, Y.; Si, H.; Wang, Z.; Dai, G. Survival outcome of different treatment sequences in patients with locally advanced and metastatic pancreatic cancer. BMC Cancer 2024, 24, 67. [Google Scholar] [CrossRef]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated projection of US cancer incidence and death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Jat, P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Dong, Z.; Luo, Y.; Yuan, Z.; Tian, Y.; Jin, T.; Xu, F. Cellular senescence and SASP in tumor progression and therapeutic opportunities. Mol. Cancer 2024, 23, 181. [Google Scholar] [CrossRef]
- Yamauchi, S.; Takahashi, A. Cellular senescence: Mechanisms and relevance to cancer and aging. J. Biochem. 2025, 177, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Marstrand-Daucé, L.; Lorenzo, D.; Chassac, A.; Nicole, P.; Couvelard, A.; Haumaitre, C. Acinar-to-ductal metaplasia (ADM): On the road to pancreatic intraepithelial neoplasia (PanIN) and pancreatic cancer. Int. J. Mol. Sci. 2023, 24, 9946. [Google Scholar] [CrossRef]
- Kolodkin-Gal, D.; Roitman, L.; Ovadya, Y.; Azazmeh, N.; Assouline, B.; Schlesinger, Y.; Kalifa, R.; Horwitz, S.; Khalatnik, Y.; Hochner-Ger, A.; et al. Senolytic elimination of Cox2-expressing senescent cells inhibits the growth of premalignant pancreatic lesions. Gut 2022, 71, 345–355. [Google Scholar] [CrossRef]
- Yang, K.; Li, X.; Xie, K. Senescence program and its reprogramming in pancreatic premalignancy. Cell Death Dis. 2023, 14, 528. [Google Scholar] [CrossRef]
- Jaber, S.; Warnier, M.; Leers, C.; Vernier, M.; Goehrig, D.; Médard, J.J.; Vindrieux, D.; Ziegler, D.V.; Bernard, D. Targeting chemoresistant senescent pancreatic cancer cells improves conventional treatment efficacy. Mol. Biomed. 2023, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Li, M.; Chen, C.Y.; Yao, Q.Z. Mesothelin-induced pancreatic cancer cell proliferation involves alteration of cyclin E via activation of signal transducer and activator of transcription protein 3. Mol. Cancer Res. 2008, 6, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Marin-Muller, C.; Li, M.; Chen, C.Y.; Yao, Q.Z. Mesothelin confers pancreatic cancer cell resistance to TNF-α-induced apoptosis through Akt/PI3K/NF-κB activation and IL-6/Mcl-1 overexpression. Mol. Cancer 2011, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zeng, X.; Zhu, Y.; Yang, D.; Zhao, X. Mesothelin-based CAR-T cells exhibit potent antitumor activity against ovarian cancer. J. Transl. Med. 2024, 22, 367. [Google Scholar] [CrossRef]
- Rupert, P.B.; Buerger, M.; Friend, D.J.; Strong, R.K. Structural elucidation of the mesothelin-mucin-16/CA125 interaction. Structure 2024, 32, 1049–1054. [Google Scholar] [CrossRef]
- Li, M.; Bharadwaj, U.; Zhang, R.; Zhang, S.; Mu, H.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.Y.; Yao, Q.Z. Mesothelin is a malignant factor and therapeutic vaccine target for pancreatic cancer. Mol. Cancer Ther. 2008, 7, 286–296. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Marin-Muller, C.; Zhang, Y.; Li, M.; Chen, C.Y.; Yao, Q.Z. Mesothelin overexpression promotes autocrine IL-6/sIL-6R trans-signaling to stimulate pancreatic cancer cell proliferation. J. Surg. Res. 2010, 158, 335. [Google Scholar] [CrossRef]
- Zhang, S.; Yong, L.K.; Li, D.; Cubas, R.; Chen, C.Y.; Yao, Q.Z. Mesothelin virus-like particle immunization controls pancreatic cancer growth through CD8+ T cell induction and reduction in the frequency of CD4+ foxp3+ ICOS−regulatory T cells. PLoS ONE 2013, 8, e68303. [Google Scholar] [CrossRef]
- Nichetti, F.; Marra, A.; Corti, F.; Guidi, A.; Raimondi, A.; Prinzi, N.; de Braud, F.; Pusceddu, S. The role of mesothelin as a diagnostic and therapeutic target in pancreatic ductal adenocarcinoma: A comprehensive review. Target. Oncol. 2018, 13, 333–351. [Google Scholar] [CrossRef]
- Cubas, R.; Zhang, S.; Li, M.; Chen, C.Y.; Yao, Q.Z. Trop2 expression contributes to tumor pathogenesis by activating the ERK MAPK pathway. Mol. Cancer 2010, 9, 1–13. [Google Scholar] [CrossRef]
- Cao, L.; Huang, C.; Zhou, D.C.; Hu, Y.; Lih, T.M.; Savage, S.R.; Krug, K.; Clark, D.J.; Schnaubelt, M.; Chen, L.; et al. Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 2021, 184, 5031–5052. [Google Scholar] [CrossRef]
- Liao, Y.; Savage, S.R.; Dou, Y.; Shi, Z.; Yi, X.; Jiang, W.; Lei, J.T.; Zhang, B. A proteogenomics data-driven knowledge base of human cancer. Cell Syst. 2023, 14, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Elizarraras, J.M.; Liao, Y.; Shi, Z.; Zhu, Q.; Pico, A.R.; Zhang, B. WebGestalt 2024: Faster gene set analysis and new support for metabolomics and multi-omics. Nucleic Acids Res. 2024, 52, W415–W421. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Santiago, F.E.; Grassi, D.; Ling, Y.; Niedernhofer, L.J.; Robbins, P.D. SA-β-galactosidase-based screening assay for the identification of senotherapeutic drugs. J. Vis. Exp. 2019, 148, e58133. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Wang, B.; Han, J.; Elisseeff, J.H.; Demaria, M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat. Rev. Mol. Cell Biol. 2024, 25, 958–978. [Google Scholar] [CrossRef]
- GraphPad Prism for Windows; Version 8.4.0; GraphPad Software: San Diego, CA, USA, 2020; Available online: https://www.graphpad.com (accessed on 5 February 2025).
- Li, Y.; Dou, Y.; Leprevost, F.D.V.; Geffen, Y.; Calinawan, A.P.; Aguet, F.; Akiyama, Y.; Anand, S.; Birger, C.; Cao, S.; et al. Proteogenomic data and resources for pan-cancer analysis. Cancer Cell 2023, 41, 1397–1406. [Google Scholar] [CrossRef]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Kutmon, M.; Riutta, A.; Nunes, N.; Hanspers, K.; Willighagen, E.L.; Bohler, A.; Mélius, J.; Waagmeester, A.; Sinha, S.R.; Miller, R.; et al. WikiPathways: Capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [Google Scholar] [CrossRef]
- Sulli, G.; Di Micco, R.; di Fagagna, F.D.A. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef]
- Yamauchi, S.; Sugiura, Y.; Yamaguchi, J.; Zhou, X.; Takenaka, S.; Odawara, T.; Fukaya, S.; Fujisawa, T.; Naguro, I.; Uchiyama, Y.; et al. Mitochondrial fatty acid oxidation drives senescence. Sci. Adv. 2024, 10, eado5887. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.A.; Wang, B.; Demaria, M. Senescence and cancer—role and therapeutic opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. Aging of mice is associated with p16 (Ink4a)-and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 2016, 8, 1294. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Acosta, J.C.; Adams, P.D.; di Fagagna, F.D.A.; Baker, D.J.; Bishop, C.L.; Chandra, T.; Collado, M.; Gil, J.; Gorgoulis, V.; et al. Guidelines for minimal information on cellular senescence experimentation in vivo. Cell 2024, 187, 4150–4175. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the regulation of cellular senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef]
- Takeuchi, S.; Takahashi, A.; Motoi, N.; Yoshimoto, S.; Tajima, T.; Yamakoshi, K.; Hirao, A.; Yanagi, S.; Fukami, K.; Ishikawa, Y.; et al. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo. Cancer Res. 2010, 70, 9381–9390. [Google Scholar] [CrossRef]
- Cho, K.A.; Ryu, S.J.; Oh, Y.S.; Park, J.H.; Lee, J.W.; Kim, H.P.; Kim, K.T.; Jang, I.S.; Park, S.C. Morphological adjustment of senescent cells by modulating caveolin-1 status. J. Biol. Chem. 2004, 279, 42270–42278. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular senescence: Aging, cancer, and injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell. Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef]
- Morisaki, H.; Ando, A.; Nagata, Y.; Pereira-Smith, O.; Smith, J.R.; Ikeda, K.; Nakanishi, M. Complex mechanisms underlying impaired activation of Cdk4 and Cdk2 in replicative senescence: Roles of p16, p21, and cyclin D1. Exp. Cell Res. 1999, 253, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Yoneda-Kato, N.; Kato, J.Y. CSN5 specifically interacts with CDK2 and controls senescence in a cytoplasmic cyclin E-mediated manner. Sci. Rep. 2013, 3, 1054. [Google Scholar] [CrossRef]
- Hydbring, P.; Larsson, L.G. Tipping the balance: Cdk2 enables Myc to suppress senescence. Cancer Res. 2010, 70, 6687–6691. [Google Scholar] [CrossRef] [PubMed]
- Campaner, S.; Doni, M.; Hydbring, P.; Verrecchia, A.; Bianchi, L.; Sardella, D.; Schleker, T.; Perna, D.; Tronnersjö, S.; Murga, M.; et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 2010, 12, 54–59. [Google Scholar] [CrossRef]
- Li, Y.; Tian, W.; Zhang, H.; Zhang, Z.; Zhao, Q.; Chang, L.; Lei, N.; Zhang, W. MSLN correlates with immune infiltration and chemoresistance as a prognostic biomarker in ovarian cancer. Front. Oncol. 2022, 12, 830570. [Google Scholar] [CrossRef]
- Harada, G.; Neng, Q.; Fujiki, T.; Katakura, Y. Molecular mechanisms for the p38-induced cellular senescence in normal human fibroblast. J. Biochem. 2014, 156, 283–290. [Google Scholar] [CrossRef]
- Han, X.; Lei, Q.; Xie, J.; Liu, H.; Li, J.; Zhang, X.; Zhang, T.; Gou, X. Potential regulators of the senescence-associated secretory phenotype during senescence and aging. J. Gerontol. A 2022, 77, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Al Bitar, S.; Gali-Muhtasib, H. The role of the cyclin dependent kinase inhibitor p21cip1/waf1 in targeting cancer: Molecular mechanisms and novel therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Zingoni, A.; Antonangeli, F.; Sozzani, S.; Santoni, A.; Cippitelli, M.; Soriani, A. The senescence journey in cancer immunoediting. Mol. Cancer 2024, 23, 68. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Tanaka, I.; Huang, H.; Okado, S.; Imamura, Y.; Nomata, Y.; Takenaka, H.; Watanabe, H.; Kawasumi, Y.; Nakanishi, K.; et al. Molecular mechanisms of tumor progression and novel therapeutic and diagnostic strategies in mesothelioma. Int. J. Mol. Sci. 2025, 26, 4299. [Google Scholar] [CrossRef]
- Kindler, H.L.; Novello, S.; Bearz, A.; Ceresoli, G.L.; Aerts, J.G.; Spicer, J.; Taylor, P.; Nackaerts, K.; Greystoke, A.; Jennens, R.; et al. Anetumab ravtansine versus vinorelbine in patients with relapsed, mesothelin-positive malignant pleural mesothelioma (ARCS-M): A randomised, open-label phase 2 trial. Lancet Oncol. 2022, 23, 540–552. [Google Scholar] [CrossRef]
- Matsuzawa, F.; Kamachi, H.; Mizukami, T.; Einama, T.; Kawamata, F.; Fujii, Y.; Fukai, M.; Kobayashi, N.; Hatanaka, Y.; Taketomi, A. Mesothelin blockage by Amatuximab suppresses cell invasiveness, enhances gemcitabine sensitivity and regulates cancer cell stemness in mesothelin-positive pancreatic cancer cells. BMC Cancer 2021, 21, 200. [Google Scholar] [CrossRef]
- Hassan, R.; Blumenschein, G.R., Jr.; Moore, K.N.; Santin, A.D.; Kindler, H.L.; Nemunaitis, J.J.; Seward, S.M.; Thomas, A.; Kim, S.K.; Rajagopalan, P.; et al. First-in-human, multicenter, phase I dose-escalation and expansion study of anti-mesothelin antibody–drug conjugate anetumab ravtansine in advanced or metastatic solid tumors. J. Clin. Oncol. 2020, 38, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chan, A.; Tai, C.H.; Andresson, T.; Pastan, I. Multiple proteases are involved in mesothelin shedding by cancer cells. Commun. Biol. 2020, 3, 728. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, J.A.; Belisle, J.; Onda, M.; Rancourt, C.; Migneault, M.; Ho, M.; Bera, T.K.; Connor, J.; Sathyanarayana, B.K.; Lee, B.; et al. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol. Cancer 2006, 5, 50. [Google Scholar] [CrossRef]
- Kaneko, O.; Gong, L.; Zhang, J.; Hansen, J.K.; Hassan, R.; Lee, B.; Ho, M. A binding domain on mesothelin for CA125/MUC16. J. Biol. Chem. 2009, 284, 3739–3749. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for targeting senescent cells in human disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.U.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- González-Gualda, E.; Pàez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R., III; González-López, C.; Ou, H.L.; Mirón-Barroso, S.; Zhang, Z.; Lérida-Viso, A.; et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. eBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Hickson, L.J.; Prata, L.G.L.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. eBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Lu, J.; Chen, C.; Yao, Q. Mesothelin-Associated Anti-Senescence Through P53 in Pancreatic Ductal Adenocarcinoma. Cancers 2025, 17, 2058. https://doi.org/10.3390/cancers17122058
Liu D, Lu J, Chen C, Yao Q. Mesothelin-Associated Anti-Senescence Through P53 in Pancreatic Ductal Adenocarcinoma. Cancers. 2025; 17(12):2058. https://doi.org/10.3390/cancers17122058
Chicago/Turabian StyleLiu, Dongliang, Jianming Lu, Changyi Chen, and Qizhi Yao. 2025. "Mesothelin-Associated Anti-Senescence Through P53 in Pancreatic Ductal Adenocarcinoma" Cancers 17, no. 12: 2058. https://doi.org/10.3390/cancers17122058
APA StyleLiu, D., Lu, J., Chen, C., & Yao, Q. (2025). Mesothelin-Associated Anti-Senescence Through P53 in Pancreatic Ductal Adenocarcinoma. Cancers, 17(12), 2058. https://doi.org/10.3390/cancers17122058