
T-Cell Engager Therapy in Prostate Cancer: Molecular Insights into a New Frontier in Immunotherapy



Simple Summary
Abstract
1. Introduction
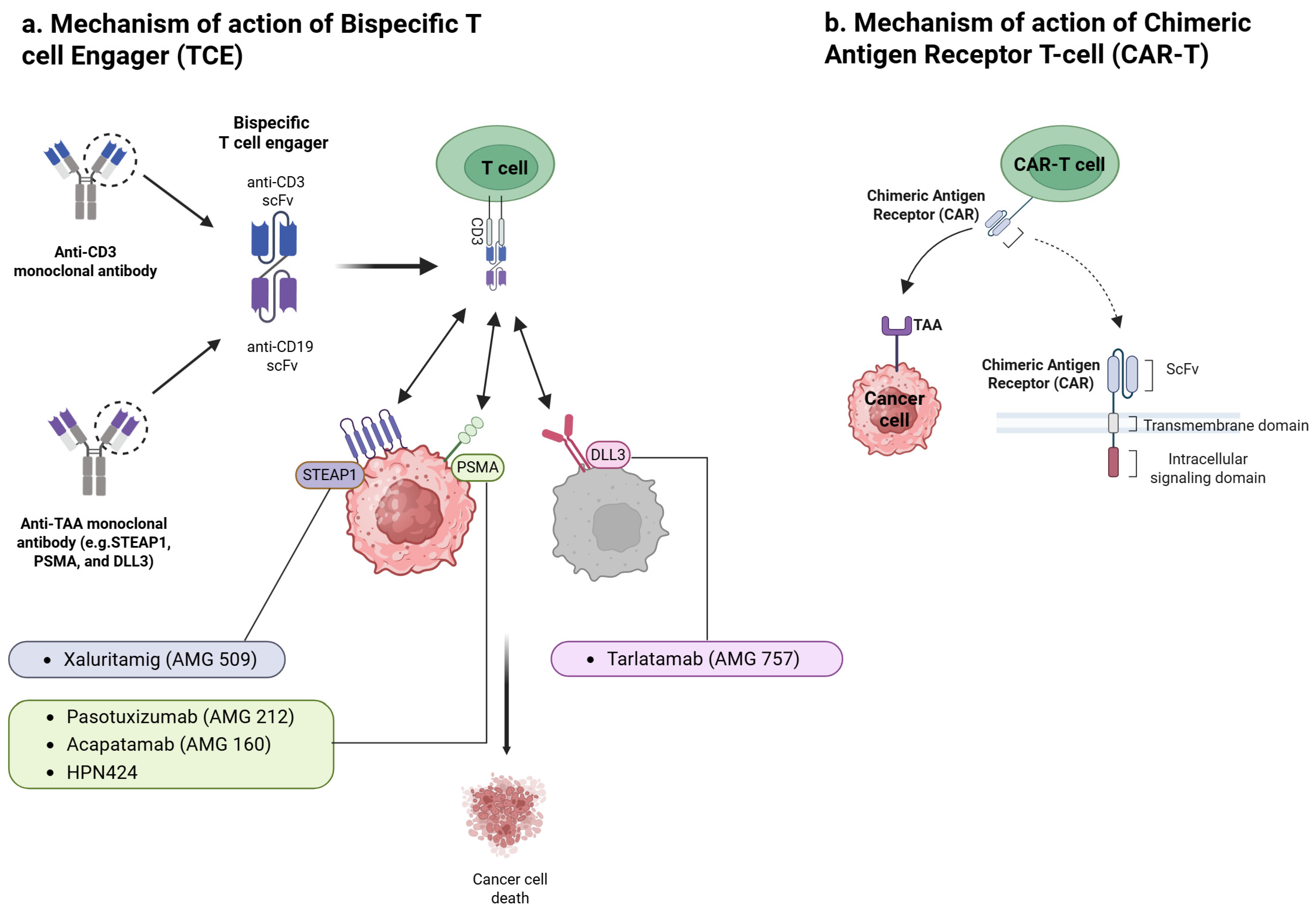
2. Mechanism of Action and Novel Engineering Approaches of T-Cell Engagers
2.1. Mechanism of Action
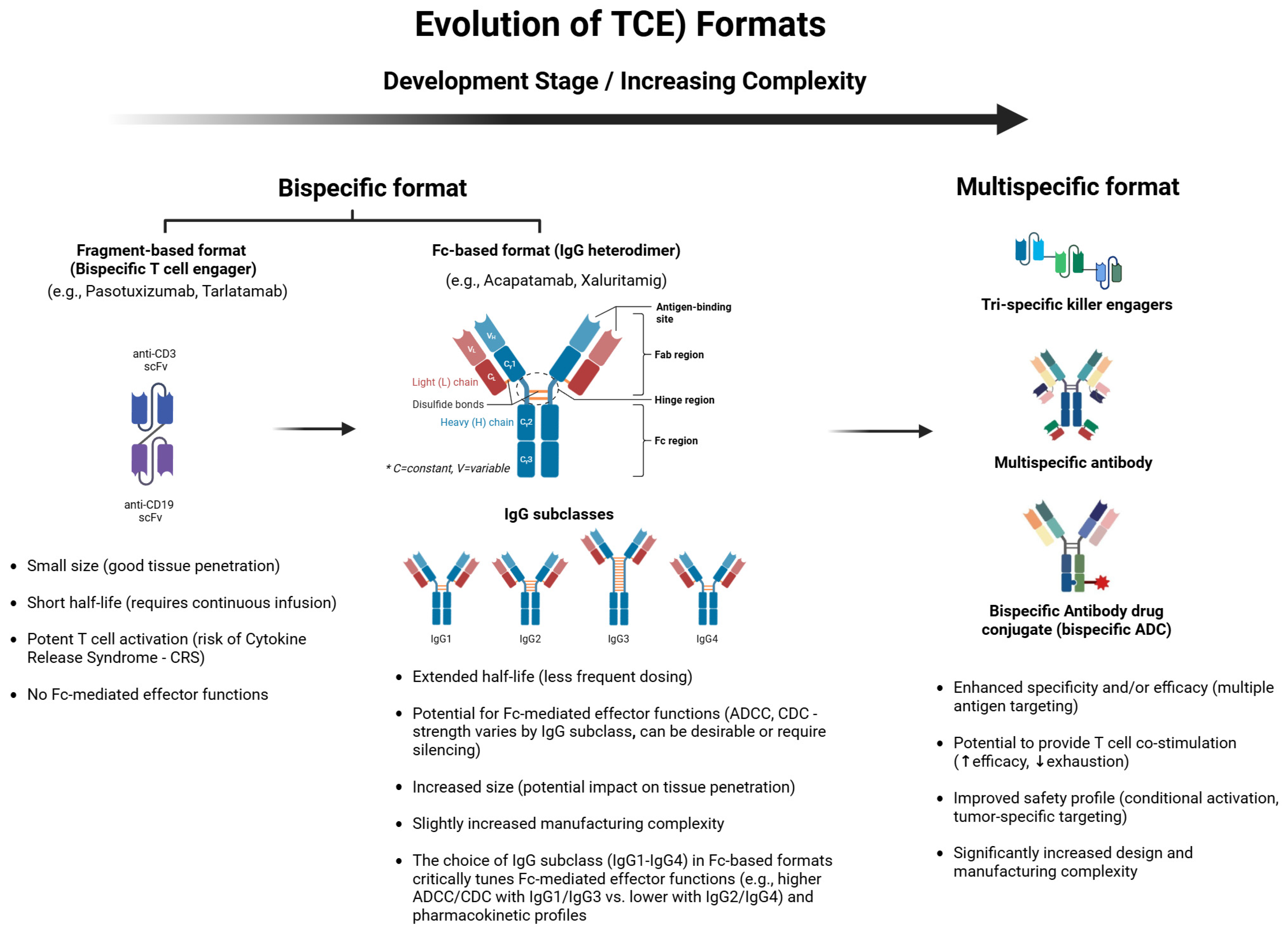
2.2. Novel Engineering Approaches
3. Clinical Development and Therapeutic Value of TCEs for Prostate Cancer
3.1. Key Insights from Approved TCEs in Hematology
3.2. PSMA-Targeting TCEs
3.3. STEAP1-Targeting TCEs
3.4. Delta-like Ligand 3 (DLL3)-Targeting TCEs in Neuroendocrine Prostate Cancer
3.5. Emerging Efficacy Landscape and Intrinsic Therapeutic Value of TCEs in mCRPC
4. Safety and Toxicity Mitigation Strategies
5. Comparison with Existing Therapies
5.1. Comparison with Androgen Receptor (AR)-Targeted Agents
5.2. Comparison with Chemotherapy (Taxanes)
5.3. Comparison with Radioligand Therapy (177Lu-PSMA-617)
5.4. Comparison with Immune Checkpoint Inhibitors (ICIs)
5.5. Comparison with Sipuleucel-T
6. Future Directions and Research Priorities
6.1. Broadening Target Antigens and TCE Platforms
6.2. Beyond CD3—NK Engagers and Multispecific Constructs
6.3. OR-Gated vs. AND-Gated Targeting Strategies
6.4. Biomarker-Driven Patient Selection
6.5. Enhancing Safety and Mitigating Toxicity
6.6. Combination Strategies
6.7. Advancing Clinical Development and Approval
6.8. Earlier Use of TCEs in the Disease Course
6.9. Understanding and Overcoming TCE Resistance
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADA | Anti-Drug Antibody |
| ADCC | Antibody-Dependent Cell-Mediated Cytotoxicity |
| ADC | Antibody-Drug Conjugate |
| ADT | Androgen Deprivation Therapy |
| AE | Adverse Event |
| AMG | Amgen (Company Code for Drug Development) |
| APC | Antigen-Presenting Cell |
| AR | Androgen Receptor |
| ASCO | American Society of Clinical Oncology |
| BiKE | Bispecific Killer Cell Engager |
| BiTE® | Bispecific T-Cell Engager |
| CAR | Chimeric Antigen Receptor |
| CAR-T cell | Chimeric Antigen Receptor T-Cell |
| CD | Cluster of Differentiation |
| CDC | Complement-Dependent Cytotoxicity |
| CDK12mut | Cyclin-Dependent Kinase 12 mutated/mutation |
| CRS | Cytokine Release Syndrome |
| CSF-1R | Colony-Stimulating Factor 1 Receptor |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| DLL3 | Delta-Like Ligand 3 |
| dMMR | Deficient Mismatch Repair |
| Fc | Fragment Crystallizable Region |
| FcRn | Neonatal Fc Receptor |
| G | Grade (for Toxicity Grading) |
| hK2 | Human Kallikrein-Related Peptidase 2 |
| HLA | Human Leukocyte Antigen |
| HLE | Half-Life Extended |
| HPN | Harpoon Therapeutics (Company Code for Drug Development) |
| ICANS | Immune Effector Cell-Associated Neurotoxicity Syndrome |
| ICI | Immune Checkpoint Inhibitor |
| IgG | Immunoglobulin G |
| IHC | Immunohistochemistry |
| IL | Interleukin |
| IV | Intravenous |
| KLK2 | Kallikrein-Related Peptidase 2 |
| LAG-3 | Lymphocyte-Activation Gene 3 |
| MHC | Major Histocompatibility Complex |
| mCRPC | Metastatic Castration-Resistant Prostate Cancer |
| MOA | Mechanism of Action |
| MSI-H | Microsatellite Instability-High |
| NEPC | Neuroendocrine Prostate Cancer |
| NK | Natural Killer |
| ORR | Objective Response Rate |
| OS | Overall Survival |
| PCa | Prostate Cancer |
| PD-1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| PET | Positron Emission Tomography |
| PSA | Prostate-Specific Antigen |
| PSA50 | ≥50% Decline in Prostate-Specific Antigen |
| PSMA | Prostate-Specific Membrane Antigen |
| PTEN | Phosphatase and Tensin Homolog |
| Q2weeks | Every 2 Weeks |
| RB1 | RB Transcriptional Corepressor 1 |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| REGN | Regeneron (Company Code for Drug Development; REGN4336) |
| RLT | Radioligand Therapy |
| SCLC | Small Cell Lung Cancer |
| scFv | Single-Chain Variable Fragment |
| SD | Stable Disease |
| STEAP1 | Six Transmembrane Epithelial Antigen of the Prostate 1 |
| TAA | Tumor-Associated Antigen |
| TCE | T-cell Engager |
| TGF-β | Transforming Growth Factor Beta |
| Th1 | T Helper 1 |
| TIM-3 | T-Cell Immunoglobulin and Mucin-Domain Containing-3 |
| TME | Tumor Microenvironment |
| TNFα | Tumor Necrosis Factor Alpha |
| TP53 | Tumor Protein P53 |
| TriKE | Tandem Trimeric Engager |
| TriTE | Trispecific T-cell Engager |
References
- Francini, E.; Agarwal, N.; Castro, E.; Cheng, H.H.; Chi, K.N.; Clarke, N.; Mateo, J.; Rathkopf, D.; Saad, F.; Tombal, B. Intensification approaches and treatment sequencing in metastatic castration-resistant prostate cancer: A systematic review. Eur. Urol. 2025, 87, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Kwon, W.-A.; Joung, J.Y. Immunotherapy in Prostate Cancer: From a “Cold” Tumor to a “Hot” Prospect. Cancers 2025, 17, 1064. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.N.; Hoimes, C.J.; Gerritsen, W.R.; Vaishampayan, U.N.; Elliott, T.; Hwang, C.; Ten Tije, A.J.; Omlin, A.; McDermott, R.S.; Fradet, Y. Pembrolizumab plus enzalutamide for metastatic castration-resistant prostate cancer progressing on enzalutamide: Cohorts 4 and 5 of the phase 2 KEYNOTE-199 study. Prostate Cancer Prostatic Dis. 2025, 28, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Fucà, G.; Spagnoletti, A.; Ambrosini, M.; De Braud, F.; Di Nicola, M. Immune cell engagers in solid tumors: Promises and challenges of the next generation immunotherapy. ESMO Open 2021, 6, 100046. [Google Scholar] [CrossRef]
- Chehade, C.H.; Ozay, Z.I.; Ostrowski, M.; Mercinelli, C.; Gebrael, G.; Sayegh, N.; Swami, U.; Azad, A.A.; Antonarakis, E.S.; Agarwal, N. T-Cell engagers in prostate cancer. Eur. Urol. 2025, 87, 553–558. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer immune evasion through loss of MHC class I antigen presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef]
- Penny, H.L.; Hainline, K.; Theoharis, N.; Wu, B.; Brandl, C.; Webhofer, C.; McComb, M.; Wittemer-Rump, S.; Koca, G.; Stienen, S. Characterization and root cause analysis of immunogenicity to pasotuxizumab (AMG 212), a prostate-specific membrane antigen-targeting bispecific T-cell engager therapy. Front. Immunol. 2023, 14, 1261070. [Google Scholar] [CrossRef]
- Xu, M.; Evans, L.; Bizzaro, C.L.; Quaglia, F.; Verrillo, C.E.; Li, L.; Stieglmaier, J.; Schiewer, M.J.; Languino, L.R.; Kelly, W.K. STEAP1–4 (six-transmembrane epithelial antigen of the prostate 1–4) and their clinical implications for prostate cancer. Cancers 2022, 14, 4034. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Champiat, S.; Lai, W.V.; Izumi, H.; Govindan, R.; Boyer, M.; Hummel, H.-D.; Borghaei, H.; Johnson, M.L.; Steeghs, N. Tarlatamab, a first-in-class DLL3-targeted bispecific T-cell engager, in recurrent small-cell lung cancer: An open-label, phase I study. J. Clin. Oncol. 2023, 41, 2893–2903. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, J.; Kudlacek, S.; Qi, T.; Dunlap, T.; Cao, Y. Population dynamics of immunological synapse formation induced by bispecific T cell engagers predict clinical pharmacodynamics and treatment resistance. Elife 2023, 12, e83659. [Google Scholar] [CrossRef] [PubMed]
- Dorff, T.; Horvath, L.G.; Autio, K.; Bernard-Tessier, A.; Rettig, M.B.; Machiels, J.-P.; Bilen, M.A.; Lolkema, M.P.; Adra, N.; Rottey, S. A phase I study of acapatamab, a half-life extended, PSMA-targeting bispecific T-cell engager for metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2024, 30, 1488–1500. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, M.; Ren, F.; Meng, X.; Yu, J. The landscape of bispecific T cell engager in cancer treatment. Biomark. Res. 2021, 9, 38. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef]
- Bhatia, V.; Kamat, N.V.; Pariva, T.E.; Wu, L.-T.; Tsao, A.; Sasaki, K.; Sun, H.; Javier, G.; Nutt, S.; Coleman, I. Targeting advanced prostate cancer with STEAP1 chimeric antigen receptor T cell and tumor-localized IL-12 immunotherapy. Nat. Commun. 2023, 14, 2041. [Google Scholar] [CrossRef]
- Aggarwal, R.R.; Rottey, S.; Bernard-Tessier, A.; Mellado-Gonzalez, B.; Kosaka, T.; Stadler, W.M.; Sandhu, S.; Yu, B.; Shaw, C.; Ju, C.-H. Phase 1b study of tarlatamab in de novo or treatment-emergent neuroendocrine prostate cancer (NEPC). In Proceedings of the 2024 ASCO Annual Meeting, Chicago, IL, USA, 31 May–4 June 2024. [Google Scholar]
- Géraud, A.; Hueso, T.; Laparra, A.; Bige, N.; Ouali, K.; Cauquil, C.; Stoclin, A.; Danlos, F.-X.; Hollebecque, A.; Ribrag, V. Reactions and adverse events induced by T-cell engagers as anti-cancer immunotherapies, a comprehensive review. Eur. J. Cancer 2024, 205, 114075. [Google Scholar] [CrossRef]
- Ball, K.; Dovedi, S.J.; Vajjah, P.; Phipps, A. Strategies for clinical dose optimization of T cell-engaging therapies in oncology. In Proceedings of the MABS 2023—The 24th International Workshop on Multi-Agent-Based Simulation, London, UK, 29 May–2 June 2023; p. 2181016. [Google Scholar]
- Kelly, W.K.; Danila, D.C.; Lin, C.-C.; Lee, J.-L.; Matsubara, N.; Ward, P.J.; Armstrong, A.J.; Pook, D.; Kim, M.; Dorff, T.B.; et al. Xaluritamig, a STEAP1 × CD3 XmAb 2+1 Immune Therapy for Metastatic Castration-Resistant Prostate Cancer: Results from Dose Exploration in a First-in-Human Study. Cancer Discov. 2024, 14, 76–89. [Google Scholar] [CrossRef]
- Surowka, M.; Schaefer, W.; Klein, C. Ten years in the making: Application of CrossMab technology for the development of therapeutic bispecific antibodies and antibody fusion proteins. In Proceedings of the 22nd International Workshop on Multi-Agent-Based Simulation, London, UK, 3–7 May 2021; p. 1967714. [Google Scholar]
- De Bono, J.S.; Fong, L.; Beer, T.M.; Gao, X.; Geynisman, D.M.; Burris, H.A., III; Strauss, J.F.; Courtney, K.D.; Quinn, D.I.; VanderWeele, D.J. Results of an ongoing phase 1/2a dose escalation study of HPN424, a tri-specific half-life extended PSMA-targeting T-cell engager, in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2021, 39, 5013. [Google Scholar] [CrossRef]
- Haber, L.; Olson, K.; Kelly, M.P.; Crawford, A.; DiLillo, D.J.; Tavaré, R.; Ullman, E.; Mao, S.; Canova, L.; Sineshchekova, O.; et al. Generation of T-cell-redirecting bispecific antibodies with differentiated profiles of cytokine release and biodistribution by CD3 affinity tuning. Sci. Rep. 2021, 11, 14397. [Google Scholar] [CrossRef]
- Meng, L.; Yang, Y.; Mortazavi, A.; Zhang, J. Emerging immunotherapy approaches for treating prostate cancer. Int. J. Mol. Sci. 2023, 24, 14347. [Google Scholar] [CrossRef]
- Warmuth, S.; Gunde, T.; Snell, D.; Brock, M.; Weinert, C.; Simonin, A.; Hess, C.; Tietz, J.; Johansson, M.; Spiga, F.M. Engineering of a trispecific tumor-targeted immunotherapy incorporating 4-1BB co-stimulation and PD-L1 blockade. Oncoimmunology 2021, 10, 2004661. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Ptacek, J.; Havlinova, B.; Nedvedova, J.; Barinka, C.; Novakova, Z. Targeting prostate Cancer using bispecific T-Cell engagers against prostate-specific membrane Antigen. ACS Pharmacol. Transl. Sci. 2023, 6, 1703–1714. [Google Scholar] [CrossRef]
- Simão, D.C.; Zarrabi, K.K.; Mendes, J.L.; Luz, R.; Garcia, J.A.; Kelly, W.K.; Barata, P.C. Bispecific T-cell engagers therapies in solid tumors: Focusing on prostate cancer. Cancers 2023, 15, 1412. [Google Scholar] [CrossRef] [PubMed]
- Hassel, J.C.; Berking, C.; Forschner, A.; Gebhardt, C.; Heinzerling, L.; Meier, F.; Ochsenreither, S.; Siveke, J.; Hauschild, A.; Schadendorf, D. Practical guidelines for the management of adverse events of the T cell engager bispecific tebentafusp. Eur. J. Cancer 2023, 191, 112986. [Google Scholar] [CrossRef]
- Heitmann, J.S.; Hackenbruch, C.; Walz, J.S.; Jung, S.; Pflügler, M.; Schlenk, R.F.; Ochsenreither, S.; Hadaschik, B.A.; Darr, C.; Jung, G.; et al. Updated results on the bispecific PSMAxCD3 antibody CC-1 for treatment of prostate cancer. J. Clin. Oncol. 2024, 42, 2536. [Google Scholar] [CrossRef]
- Stein, M.; Graham, L.; Baldini, C.; Vinceneux, A.; Kessler, E.; Runcie, K.; Wei, A.; Papadopoulos, K.; Bernard-Tessier, A.; Laurent, M. 1659TiP A phase I, first-in-human, open-label, multicenter, trial-in-progress of the safety, tolerability, and preliminary efficacy of JNJ-87189401 (PSMA-CD28 bispecific antibody) combined with JNJ-78278343 (KLK2-CD3 bispecific antibody) for advanced prostate cancer. Ann. Oncol. 2024, 35, S1000. [Google Scholar]
- Mehra, N.; Robbrecht, D.; Voortman, J.; Parren, P.W.; Macia, S.; Veeneman, J.; Umarale, S.; Winograd, B.; van der Vliet, H.J.; Wise, D.R. Early dose escalation of LAVA-1207, a novel bispecific gamma-delta T-cell engager (Gammabody), in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2023, 41. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A. Effect of blinatumomab vs chemotherapy on event-free survival among children with high-risk first-relapse B-cell acute lymphoblastic leukemia: A randomized clinical trial. JAMA 2021, 325, 843–854. [Google Scholar] [CrossRef]
- Dickinson, M.J.; Carlo-Stella, C.; Morschhauser, F.; Bachy, E.; Corradini, P.; Iacoboni, G.; Khan, C.; Wróbel, T.; Offner, F.; Trněný, M. Glofitamab for relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2022, 387, 2220–2231. [Google Scholar] [CrossRef]
- Moreau, P.; Garfall, A.L.; van de Donk, N.W.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A. Teclistamab in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef]
- Banerjee, R.; Fakhri, B.; Shah, N. Toci or not toci: Innovations in the diagnosis, prevention, and early management of cytokine release syndrome. Leuk. Lymphoma 2021, 62, 2600–2611. [Google Scholar] [CrossRef] [PubMed]
- Belmontes, B.; Sawant, D.V.; Zhong, W.; Tan, H.; Kaul, A.; Aeffner, F.; O’Brien, S.A.; Chun, M.; Noubade, R.; Eng, J. Immunotherapy combinations overcome resistance to bispecific T cell engager treatment in T cell–cold solid tumors. Sci. Transl. Med. 2021, 13, eabd1524. [Google Scholar] [CrossRef] [PubMed]
- Anbari, S.; Wang, H.; Zhang, Y.; Wang, J.; Pilvankar, M.; Nickaeen, M.; Hansel, S.; Popel, A.S. Using quantitative systems pharmacology modeling to optimize combination therapy of anti-PD-L1 checkpoint inhibitor and T cell engager. Front. Pharmacol. 2023, 14, 1163432. [Google Scholar] [CrossRef]
- Wang, F.; Li, Z.; Feng, X.; Yang, D.; Lin, M. Advances in PSMA-targeted therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2022, 25, 11–26. [Google Scholar] [CrossRef]
- Zhang, Y.; Campbell, B.K.; Stylli, S.S.; Corcoran, N.M.; Hovens, C.M. The prostate cancer immune microenvironment, biomarkers and therapeutic intervention. URO 2022, 2, 74–92. [Google Scholar] [CrossRef]
- Kelly, W.K.; Thanigaimani, P.; Sun, F.; Seebach, F.A.; Lowy, I.; Sandigursky, S.; Miller, E. A phase 1/2 study of REGN4336, a PSMAxCD3 bispecific antibody, alone and in combination with cemiplimab in patients with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2022, 40, TPS5105. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, X.; An, R.; Song, H.; Tian, Y.; Wei, X.; Shi, M.; Wang, Z. The Role of STEAP1 in Prostate Cancer: Implications for Diagnosis and Therapeutic Strategies. Biomedicines 2025, 13, 794. [Google Scholar] [CrossRef]
- Tapia-Galisteo, A.; Compte, M.; Álvarez-Vallina, L.; Sanz, L. When three is not a crowd: Trispecific antibodies for enhanced cancer immunotherapy. Theranostics 2023, 13, 1028. [Google Scholar] [CrossRef]
- Raychaudhuri, R.; Schweizer, M.T.; Hawley, J.E.; Fong, L.; Yu, E.Y. Xaluritamig: A first step towards a new target, new mechanism for metastatic prostate cancer. AME Clin. Trials Rev. 2024, 2, 44. [Google Scholar] [CrossRef]
- Zhu, X.; Ding, C.-K.C.; Aggarwal, R.R. Emerging Therapeutic Targets of Neuroendocrine Prostate Cancer. Curr. Oncol. Rep. 2025, 27, 362–374. [Google Scholar] [CrossRef]
- Deb, A.; Patel, N.; Patel, P.A.; Kilinc, E.; Hachem, S.; Elajami, M.; Mansour, E. Immunotherapeutic strategies and immunotherapy resistance in prostate cancer. In Therapy Resistance in Prostate Cancer; Elsevier: Amsterdam, The Netherlands, 2024; pp. 235–253. [Google Scholar]
- Goebeler, M.-E.; Stuhler, G.; Bargou, R. Bispecific and multispecific antibodies in oncology: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2024, 21, 539–560. [Google Scholar] [CrossRef] [PubMed]
- Philipp, N.; Kazerani, M.; Nicholls, A.; Vick, B.; Wulf, J.; Straub, T.; Scheurer, M.; Muth, A.; Hänel, G.; Nixdorf, D. T-cell exhaustion induced by continuous bispecific molecule exposure is ameliorated by treatment-free intervals. Blood J. Am. Soc. Hematol. 2022, 140, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Deluce, J.E.; Cardenas, L.; Lalani, A.-K.; Maleki Vareki, S.; Fernandes, R. Emerging biomarker-guided therapies in prostate cancer. Curr. Oncol. 2022, 29, 5054–5076. [Google Scholar] [CrossRef]
- Beltran, H.; Dowlati, A.; Jain, P.; Johnson, M.L.; Sanborn, R.E.; Thompson, J.R.; Mamdani, H.; Schenk, E.L.; Aggarwal, R.R.; Sankar, K. Interim results from a phase 1/2 study of HPN328, a tri-specific, half-life (T1/2) extended DLL3-targeting T-cell engager, in patients (pts) with neuroendocrine prostate cancer (NEPC) and other neuroendocrine neoplasms (NEN). J. Clin. Oncol. 2024, 42, 121. [Google Scholar] [CrossRef]
- Radtke, K.K.; Bender, B.C.; Li, Z.; Turner, D.C.; Roy, S.; Belousov, A.; Li, C.-C. Clinical Pharmacology of Cytokine Release Syndrome with T-Cell–Engaging Bispecific Antibodies: Current Insights and Drug Development Strategies. Clin. Cancer Res. 2025, 31, 245–257. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Cosenza, M.; Sacchi, S.; Pozzi, S. Cytokine release syndrome associated with T-cell-based therapies for hematological malignancies: Pathophysiology, clinical presentation, and treatment. Int. J. Mol. Sci. 2021, 22, 7652. [Google Scholar] [CrossRef]
- Palecki, J.; Bhasin, A.; Bernstein, A.; Mille, P.J.; Tester, W.J.; Kelly, W.K.; Zarrabi, K.K. T-Cell redirecting bispecific antibodies: A review of a novel class of immuno-oncology for advanced prostate cancer. Cancer Biol. Ther. 2024, 25, 2356820. [Google Scholar] [CrossRef]
- Lepik, K.V.; Markelov, V.V. The Role of the Tumor Microenvironment in T-Cell Redirecting Therapies of Large B-Cell Lymphoma: Lessons Learned from CAR-T to Bispecific Antibodies. Cancers 2025, 17, 317. [Google Scholar] [CrossRef]
- Csizmarik, A.; Hadaschik, B.; Kramer, G.; Nyirady, P.; Szarvas, T. Mechanisms and markers of resistance to androgen signaling inhibitors in patients with metastatic castration-resistant prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2021, 39, 728.e13–728.e24. [Google Scholar] [CrossRef]
- Sommer, U.; Siciliano, T.; Ebersbach, C.; Beier, A.-M.K.; Stope, M.B.; Jöhrens, K.; Baretton, G.B.; Borkowetz, A.; Thomas, C.; Erb, H.H. Impact of androgen receptor activity on prostate-specific membrane antigen expression in prostate cancer cells. Int. J. Mol. Sci. 2022, 23, 1046. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; Bono, J.d.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, S.I.; Ju, X.; Horvath, L.; Clark, G.J. Moving on from sipuleucel-T: New dendritic cell vaccine strategies for prostate cancer. Front. Immunol. 2021, 12, 641307. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Lin, P.; Tombal, B.; Saad, F.; Higano, C.S.; Joshua, A.M.; Parli, T.; Rosbrook, B.; van Os, S.; Beer, T.M. Five-year survival prediction and safety outcomes with enzalutamide in men with chemotherapy-naïve metastatic castration-resistant prostate cancer from the PREVAIL trial. Eur. Urol. 2020, 78, 347–357. [Google Scholar] [CrossRef]
- Cella, D.; Ivanescu, C.; Holmstrom, S.; Bui, C.; Spalding, J.; Fizazi, K. Impact of enzalutamide on quality of life in men with metastatic castration-resistant prostate cancer after chemotherapy: Additional analyses from the AFFIRM randomized clinical trial. Ann. Oncol. 2015, 26, 179–185. [Google Scholar] [CrossRef]
- Miller, K.; Carles, J.; Gschwend, J.E.; Van Poppel, H.; Diels, J.; Brookman-May, S.D. The phase 3 COU-AA-302 study of abiraterone acetate plus prednisone in men with chemotherapy-naïve metastatic castration-resistant prostate cancer: Stratified analysis based on pain, prostate-specific antigen, and Gleason score. Eur. Urol. 2018, 74, 17–23. [Google Scholar] [CrossRef]
- Lee, H.Y.; Chen, H.-L.; Teoh, J.Y.-C.; Chen, T.-C.; Hao, S.-Y.; Tsai, H.-Y.; Huang, W.-H.; Juan, Y.-S.; Cheng, H.-M.; Chang, H.-M. Abiraterone and enzalutamide had different adverse effects on the cardiovascular system: A systematic review with pairwise and network meta-analyses. Prostate Cancer Prostatic Dis. 2021, 24, 244–252. [Google Scholar] [CrossRef]
- Leclercq-Cohen, G.; Bacac, M.; Klein, C. Rationale for combining tyrosine kinase inhibitors and T cell redirecting antibodies to mitigate cytokine release syndrome (CRS). Expert Opin. Biol. Ther. 2023, 23, 223–225. [Google Scholar] [CrossRef]
- Oudard, S. TROPIC: Phase III trial of cabazitaxel for the treatment of metastatic castration-resistant prostate cancer. Future Oncol. 2011, 7, 497–506. [Google Scholar] [CrossRef]
- Liguori, L.; Polcaro, G.; Nigro, A.; Conti, V.; Sellitto, C.; Perri, F.; Ottaiano, A.; Cascella, M.; Zeppa, P.; Caputo, A. Bispecific antibodies: A novel approach for the treatment of solid tumors. Pharmaceutics 2022, 14, 2442. [Google Scholar] [CrossRef]
- Arvedson, T.; Bailis, J.M.; Britten, C.D.; Klinger, M.; Nagorsen, D.; Coxon, A.; Egen, J.G.; Martin, F. Targeting solid tumors with bispecific T cell engager immune therapy. Annu. Rev. Cancer Biol. 2022, 6, 17–34. [Google Scholar] [CrossRef]
- Wang, I.; Song, L.; Wang, B.Y.; Kalebasty, A.R.; Uchio, E.; Zi, X. Prostate cancer immunotherapy: A review of recent advancements with novel treatment methods and efficacy. Am. J. Clin. Exp. Urol. 2022, 10, 210. [Google Scholar] [PubMed]
- Leclercq, G.; Steinhoff, N.; Haegel, H.; De Marco, D.; Bacac, M.; Klein, C. Novel strategies for the mitigation of cytokine release syndrome induced by T cell engaging therapies with a focus on the use of kinase inhibitors. Oncoimmunology 2022, 11, 2083479. [Google Scholar] [CrossRef]
- Singh, A.; Dees, S.; Grewal, I.S. Overcoming the challenges associated with CD3+ T-cell redirection in cancer. Br. J. Cancer 2021, 124, 1037–1048. [Google Scholar] [CrossRef]
- Jairajpuri, D.S.; Hussain, A.; Alajmi, M.F.; Mohammad, T.; Shamsi, A.; Hassan, M.I. Structure-based identification of bioactive phytochemicals targeting kallikrein-related peptidase 2 for prostate cancer therapy. Front. Chem. 2025, 13, 1553987. [Google Scholar] [CrossRef]
- Zhu, A.; Bai, Y.; Nan, Y.; Ju, D. Natural killer cell engagers: From bi-specific to tri-specific and tetra-specific engagers for enhanced cancer immunotherapy. Clin. Transl. Med. 2024, 14, e70046. [Google Scholar] [CrossRef]
- Yang, L.; Gong, L.; Wang, P.; Zhao, X.; Zhao, F.; Zhang, Z.; Li, Y.; Huang, W. Recent Advances in Lipid Nanoparticles for Delivery of mRNA. Pharmaceutics 2022, 14, 2682. [Google Scholar] [CrossRef]
- Arvedson, T.; Bailis, J.M.; Urbig, T.; Stevens, J.L. Considerations for design, manufacture, and delivery for effective and safe T-cell engager therapies. Curr. Opin. Biotechnol. 2022, 78, 102799. [Google Scholar] [CrossRef]
- Qi, T.; Liao, X.; Cao, Y. Development of bispecific T cell engagers: Harnessing quantitative systems pharmacology. Trends Pharmacol. Sci. 2023, 44, 880–890. [Google Scholar] [CrossRef]
- Nikkhoi, S.K.; Yang, G.; Owji, H.; Grizotte-Lake, M.; Cohen, R.I.; Gonzalez, L.G.; Massumi, M.; Hatefi, A. Bispecific immune cell engager enhances the anticancer activity of CD16+ NK cells and macrophages in vitro, and eliminates cancer metastasis in NK humanized NOG mice. J. Immunother. Cancer 2024, 12, e008295. [Google Scholar] [CrossRef]
- Bergom, H.E.; Sena, L.A.; Day, A.; Miller, B.; Miller, C.D.; Lozada, J.R.; Zorko, N.; Wang, J.; Shenderov, E.; Lobo, F.P. Divergent immune microenvironments in two tumor nodules from a patient with mismatch repair-deficient prostate cancer. NPJ Genom. Med. 2024, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhou, L.; Gu, X.; Zhao, J.; Bi, J.; Pan, L. Leveraging T cell co-stimulation for enhanced therapeutic efficacy of trispecific antibodies targeting prostate cancer. J. Immunother. Cancer 2025, 13, e010140. [Google Scholar] [CrossRef] [PubMed]
- Fenis, A.; Demaria, O.; Gauthier, L.; Vivier, E.; Narni-Mancinelli, E. New immune cell engagers for cancer immunotherapy. Nat. Rev. Immunol. 2024, 24, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Smith, R. Logic-gated and contextual control of immunotherapy for solid tumors: Contrasting multi-specific T cell engagers and CAR-T cell therapies. Front. Immunol. 2024, 15, 1490911. [Google Scholar] [CrossRef]
- Frankel, N.W.; Deng, H.; Yucel, G.; Gainer, M.; Leemans, N.; Lam, A.; Li, Y.; Hung, M.; Lee, D.; Lee, C.-T. Precision off-the-shelf natural killer cell therapies for oncology with logic-gated gene circuits. Cell Rep. 2024, 43, 114145. [Google Scholar] [CrossRef]
- Sridaran, D.; Bradshaw, E.; DeSelm, C.; Pachynski, R.; Mahajan, K.; Mahajan, N.P. Prostate cancer immunotherapy: Improving clinical outcomes with a multi-pronged approach. Cell Rep. Med. 2023, 4, 101199. [Google Scholar] [CrossRef]
- Falchook, G.S.; McKean, M.; Kelly, W.K.; Patel, M.R.; Bupathi, M.; Liaw, B.C.-H.; Garmezy, B.; Vuu, I.; Smith, K.; Gokani, P. Phase 1 clinical trial of AMG 340, a prostate-specific membrane antigen (PSMA)-targeted T-cell engager with a novel low-affinity CD3 binding domain designed to mitigate toxicity for the treatment of metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2024, 42, e14587. [Google Scholar] [CrossRef]
- Banerjee, S.; Booth, C.M.; Bruera, E.; Büchler, M.W.; Drilon, A.; Fry, T.J.; Ghobrial, I.M.; Gianni, L.; Jain, R.K.; Kroemer, G. Two decades of advances in clinical oncology—Lessons learned and future directions. Nat. Rev. Clin. Oncol. 2024, 21, 771–780. [Google Scholar] [CrossRef]
- McCue, A.C.; Demarest, S.J.; Froning, K.J.; Hickey, M.J.; Antonysamy, S.; Kuhlman, B. Engineering a tumor-selective prodrug T-cell engager bispecific antibody for safer immunotherapy. In Proceedings of the 25th International Workshop on Multi-Agent-Based Simulation XXV, MABS 2024, Auckland, New Zealand, 6 May 2024; p. 2373325. [Google Scholar]
- Harris, C.T.; Cohen, S. Reducing Immunogenicity by Design: Approaches to Minimize Immunogenicity of Monoclonal Antibodies. BioDrugs 2024, 38, 205–226. [Google Scholar] [CrossRef]
- Crombie, J.L.; Graff, T.; Falchi, L.; Karimi, Y.H.; Bannerji, R.; Nastoupil, L.; Thieblemont, C.; Ursu, R.; Bartlett, N.; Nachar, V. Consensus recommendations on the management of toxicity associated with CD3× CD20 bispecific antibody therapy. Blood 2024, 143, 1565–1575. [Google Scholar] [CrossRef]
- Moore, G.L.; Zeng, V.G.; Diaz, J.E.; Bonzon, C.; Avery, K.N.; Rashid, R.; Qi, J.; Nam, D.H.; Jacinto, J.; Dragovich, M.A. A B7-H3–Targeted CD28 Bispecific Antibody Enhances the Activity of Anti–PD-1 and CD3 T-cell Engager Immunotherapies. Mol. Cancer Ther. 2025, 24, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.; Brailey, P.M.; Song, M.; Bartlett, P.D.; Figueiredo, I.; Gurel, B.; Guo, C.; Brucklacher-Waldert, V.; Thompson, H.L.; Akinwale, J.; et al. CB307: A Dual Targeting Costimulatory Humabody VH Therapeutic for Treating PSMA-Positive Tumors. Clin. Cancer Res. 2024, 30, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Clark, R.; Slaga, D.; Avery, K.; Liu, K.; Schubbert, S.; Varma, R.; Chiang, E.; Totpal, K.; Bernett, M.J. IL-15/IL-15Rα-Fc-Fusion Protein XmAb24306 Potentiates Activity of CD3 Bispecific Antibodies through Enhancing T-Cell Expansion. Mol. Cancer Ther. 2024, 23, 1305–1316. [Google Scholar] [CrossRef]
- Cao, L.; Leclercq-Cohen, G.; Klein, C.; Sorrentino, A.; Bacac, M. Mechanistic insights into resistance mechanisms to T cell engagers. Front. Immunol. 2025, 16, 1583044. [Google Scholar] [CrossRef]
- Mirzaei, S.; Paskeh, M.D.A.; Saghari, Y.; Zarrabi, A.; Hamblin, M.R.; Entezari, M.; Hashemi, M.; Aref, A.R.; Hushmandi, K.; Kumar, A.P. Transforming growth factor-beta (TGF-β) in prostate cancer: A dual function mediator? Int. J. Biol. Macromol. 2022, 206, 435–452. [Google Scholar] [CrossRef]
- Xu, T.; Feng, L.; Zhang, W.; Li, H.; Ma, H.; Abulimiti, M.; Tan, Y.; Deng, F.; Huang, W.; Zou, S. The efficacy and safety of short-course radiotherapy followed by sequential chemotherapy and Cadonilimab for locally advanced rectal cancer: A protocol of a phase II study. BMC Cancer 2024, 24, 501. [Google Scholar] [CrossRef]
- Bakht, M.K.; Beltran, H. Biological determinants of PSMA expression, regulation and heterogeneity in prostate cancer. Nat. Rev. Urol. 2025, 22, 26–45. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Chen, D.; Yu, J. Radiotherapy combined with immunotherapy: The dawn of cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 258. [Google Scholar] [CrossRef]
- Khorasanchi, A.; Hong, F.; Yang, Y.; Singer, E.A.; Wang, P.; Li, M.; Zheng, L.; Monk, P.; Mortazavi, A.; Meng, L. Overcoming drug resistance in castrate-resistant prostate cancer: Current mechanisms and emerging therapeutic approaches. Cancer Drug Resist. 2025, 8, 9. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Li, C.; Liang, L.; Zhan, J.; Estrada, J.; Osgood, T.; Li, F.; Zhang, H.; Case, R.; Murawsky, C.M. AMG 509 (xaluritamig), an anti-STEAP1 XmAb 2+ 1 T-cell redirecting immune therapy with avidity-dependent activity against prostate cancer. Cancer Discov. 2024, 14, 90–103. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, J. Progresses of T-cell-engaging bispecific antibodies in treatment of solid tumors. Int. Immunopharmacol. 2024, 138, 112609. [Google Scholar] [CrossRef] [PubMed]
- Lampe, H.; Tam, L.; Hansen, A.R. Bi-specific T-cell engagers (BiTEs) in prostate cancer and strategies to enhance development: Hope for a BiTE-r future. Front Pharmacol 2024, 15, 1399802. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.C.; Dorff, T.B.; Tran, B. The new era of prostate-specific membrane antigen-directed immunotherapies and beyond in advanced prostate cancer: A review. Ther Adv Med Oncol 2023, 15, 17588359231170474. [Google Scholar] [CrossRef]
- Hackenbruch, C.; Heitmann, J.S.; Walz, J.S.; Federmann, B.; Pflügler, M.; Hadaschik, B.A.; Jung, G.; Salih, H.R. ProSperA: Phase I study to evaluate safety, tolerability and preliminary efficacy of a bispecific PSMAxCD3 antibody in men with biochemical recurrence of prostate cancer. J. Clin. Oncol. 2023, 41, TPS5114. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C.-x. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 156. [Google Scholar] [CrossRef]
- Klein, C.; Brinkmann, U.; Reichert, J.M.; Kontermann, R.E. The present and future of bispecific antibodies for cancer therapy. Nat. Rev. Drug Discov. 2024, 23, 301–319. [Google Scholar] [CrossRef]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef]
- Tomaszewski, B.; Curtis, M.; Foo, C.; Zhang, G.; Chang, P.; Clark, R.; Nguyen, T.; Webster, J.; Rost, S.; Jesudason, R. Abstract PR002: T cell engager therapy affects the spatial distribution and phenotype of T cells in the tumor microenvironment. Cancer Immunol. Res. 2025, 13, PR002. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| TCE Name, Developer | Target Antigen | Format/Key Features | Clinical Stage (Phase) | Key Efficacy Results | Key Toxicities | References |
|---|---|---|---|---|---|---|
| Pasotuximab (AMG 212), Amgen | PSMA | BiTE (small Fc-free) | Phase I (completed) | >50% PSA decline in some pts; Limited ORR and durability | Frequent CRS (various grades), immunogenicity | [8] |
| Acapatamab (AMG 160), Amgen | PSMA | IgG-like, extended half-life | Phase I (Development Discontinued) | PSA50 ~63% (evaluable pts, higher doses); modest ORR reported | CRS common (~high %), mostly G1-2 w/mitigation; Low rate G ≥ 3 | [12] |
| HPN424, Harpoon Therapeutics | PSMA | Trispecific (PSMA × CD3 × albumin) | Phase I (Development Discontinued) | ~20% had any PSA decline; ~5–6% PSA50; SD in ~50% | Mostly G1-2 CRS with step-up dosing | [21] |
| CC-1 (Fully Human) | PSMA | Fully human bispecific (CD3 × PSMA) | Phase I (Reported) | PSA decline up to 60% in all 14 pts (early data) | CRS common (~79%), mostly G1-2; No severe events reported | [28] |
| Xaluritamig (AMG 509), Amgen | STEAP1 | IgG-based TCE | Phase I (Reported; Ph III Planned) | PSA50 ~49% (overall), 59% (high dose); ORR 24% (overall), 41% (high dose) | CRS ~72% (mostly G1-2, 2 G3 cases); Fatigue, myalgia; Manageable immunogenicity | [19] |
| Tarlatamab (AMG 757), Amgen | DLL3 | BiTE-like (CD3 × DLL3) | Phase I (NEPC Cohort Reported) | NEPC cohort: ORR 10.5% (overall), 22.2% (DLL3+ subset); one durable response >2 yrs | CRS 75% (mostly G1-2, one G ≥ 3); Neurotoxicity ~12.5% (one G3) | [10] |
| NJ-78278343, Janssen | KLK2 (Kallikrein-2) | Bispecific antibody (CD3 × KLK2) | Phase I/II (ongoing) | Early data pending | Data pending reporting | [29] |
| LAVA-1207, LAVA Therapeutics | PSMA | Vγ9Vδ2 T-cell engager | Phase I/II (Ongoing) | Early signals of stable disease reported; data maturing | Generally mild AEs reported; Low CRS incidence | [30] |
| Therapy | Mechanism | Typical Efficacy in Late-Line mCRPC | Key Toxicities | Potential Synergy or Considerations | References |
|---|---|---|---|---|---|
| AR-Targeted (e.g., enzalutamide, abiraterone) | Block androgen receptor signaling; efficacy loss in AR-independent disease | PSA50 ~30–40% (earlier lines); less effective later | Fatigue, metabolic disturbances (e.g., hypertension w/abiraterone) | May upregulate PSMA expression, potentially enhancing TCE activity | [54,55] |
| Chemotherapy (e.g., docetaxel, cabazitaxel) | Direct cytotoxicity to dividing cells (non-tumor-specific) | ~35% PSA50, ORR ~10–20% (heavily pretreated); OS benefit | Myelosuppression, neuropathy, alopecia | Cytoreduction prior to TCE might lower tumor burden and reduce CRS risk | [1] |
| Radioligand (177Lu-PSMA-617) | Delivers targeted radiation to PSMA-expressing cells | PSA50 ~46%, ORR ~30% (VISION trial); survival benefit | Hematologic toxicity, xerostomia, potential renal impact | Requires PSMA+; sequential/combination w/TCE under investigation | [6,34,47,56] |
| Immune Checkpoint Inhibitors (e.g., pembrolizumab) | Blocks PD-1/PD-L1 or CTLA-4 to restore T-cell function | Low response (<5–10% ORR) in unselected mCRPC; higher in MSI-H/dMMR/CDK12mut | Immune-related AEs (pneumonitis, hepatitis, colitis) | May reduce T-cell exhaustion, thus enhancing TCE efficacy | [1,14] |
| Sipuleucel-T (autologous vaccine) | Autologous APC vaccine targeting prostatic acid phosphatase | Modest OS gain (~4 mo); low PSA50 rate | Infusion reactions, flu-like symptoms | Could be combined or sequenced with TCE for potential additive immune activation | [4,57] |
| T-cell Engagers (e.g., PSMAxCD3, STEAP1xCD3) | Redirect T-cells to tumor antigens (CD3 × TAA), MHC-independent | Phase I data: PSA50 ~20–60%, ORR ~10–40%+ (agent/target dependent) | Cytokine release syndrome (CRS), neurotoxicity (ICANS), potential off-target effects | Novel MOA bypassing resistance; combinations under study | [5,6,12,19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, W.-A.; Joung, J.Y. T-Cell Engager Therapy in Prostate Cancer: Molecular Insights into a New Frontier in Immunotherapy. Cancers 2025, 17, 1820. https://doi.org/10.3390/cancers17111820
Kwon W-A, Joung JY. T-Cell Engager Therapy in Prostate Cancer: Molecular Insights into a New Frontier in Immunotherapy. Cancers. 2025; 17(11):1820. https://doi.org/10.3390/cancers17111820
Chicago/Turabian StyleKwon, Whi-An, and Jae Young Joung. 2025. "T-Cell Engager Therapy in Prostate Cancer: Molecular Insights into a New Frontier in Immunotherapy" Cancers 17, no. 11: 1820. https://doi.org/10.3390/cancers17111820
APA StyleKwon, W.-A., & Joung, J. Y. (2025). T-Cell Engager Therapy in Prostate Cancer: Molecular Insights into a New Frontier in Immunotherapy. Cancers, 17(11), 1820. https://doi.org/10.3390/cancers17111820