
Current Perspectives on Mesenchymal Dendritic Cell Neoplasms of Lymphoid Tissue: Insights into Ontogeny, Updates on Classification, and Clinicopathologic Characteristics
 ,
, 
Simple Summary
Abstract
1. Introduction
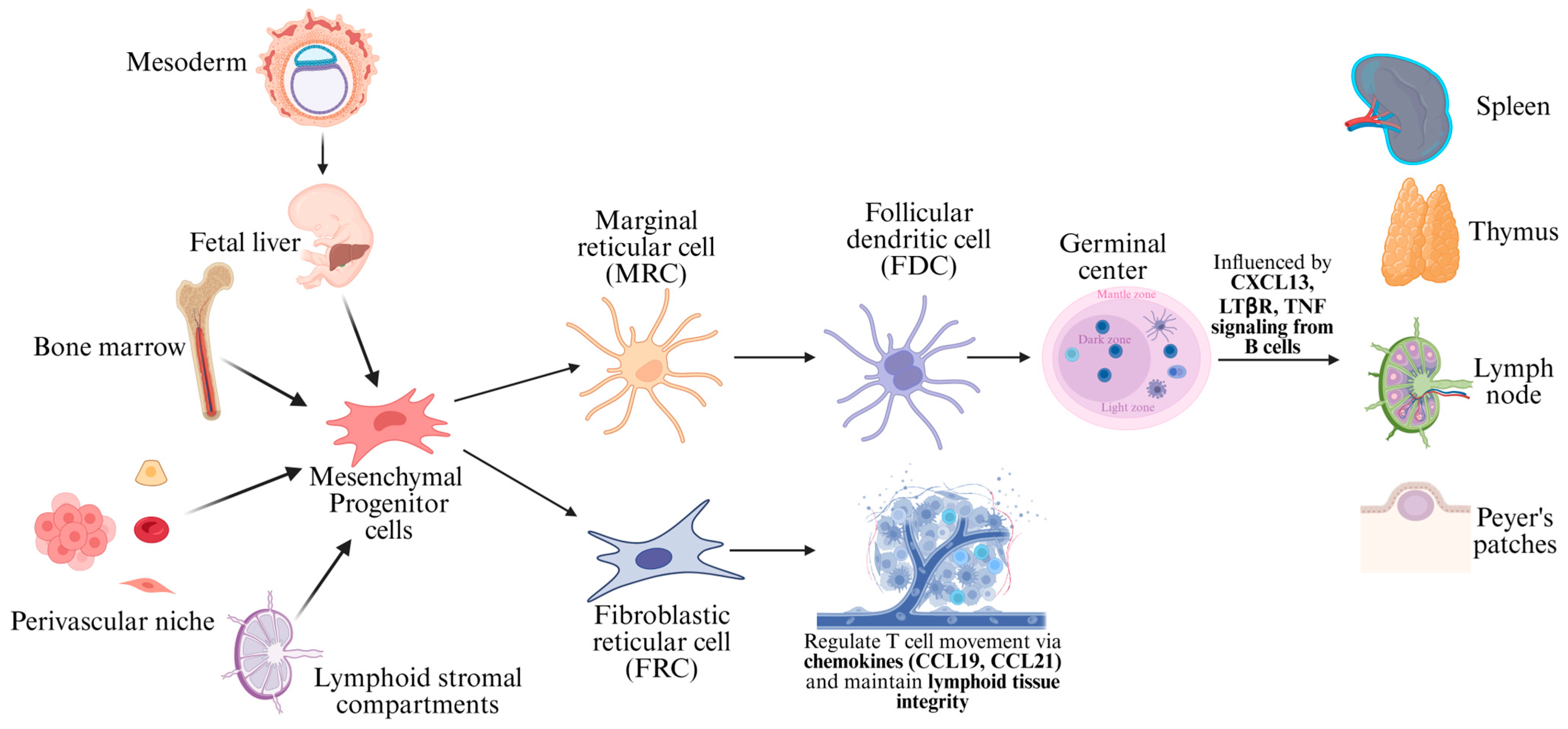
2. Ontogeny of Mesenchymal Dendritic Cells Versus the Mononuclear Phagocyte System
2.1. Ontogeny of Mesenchymal Dendritic Cells
2.2. Ontogeny of the Mononuclear Phagocyte System
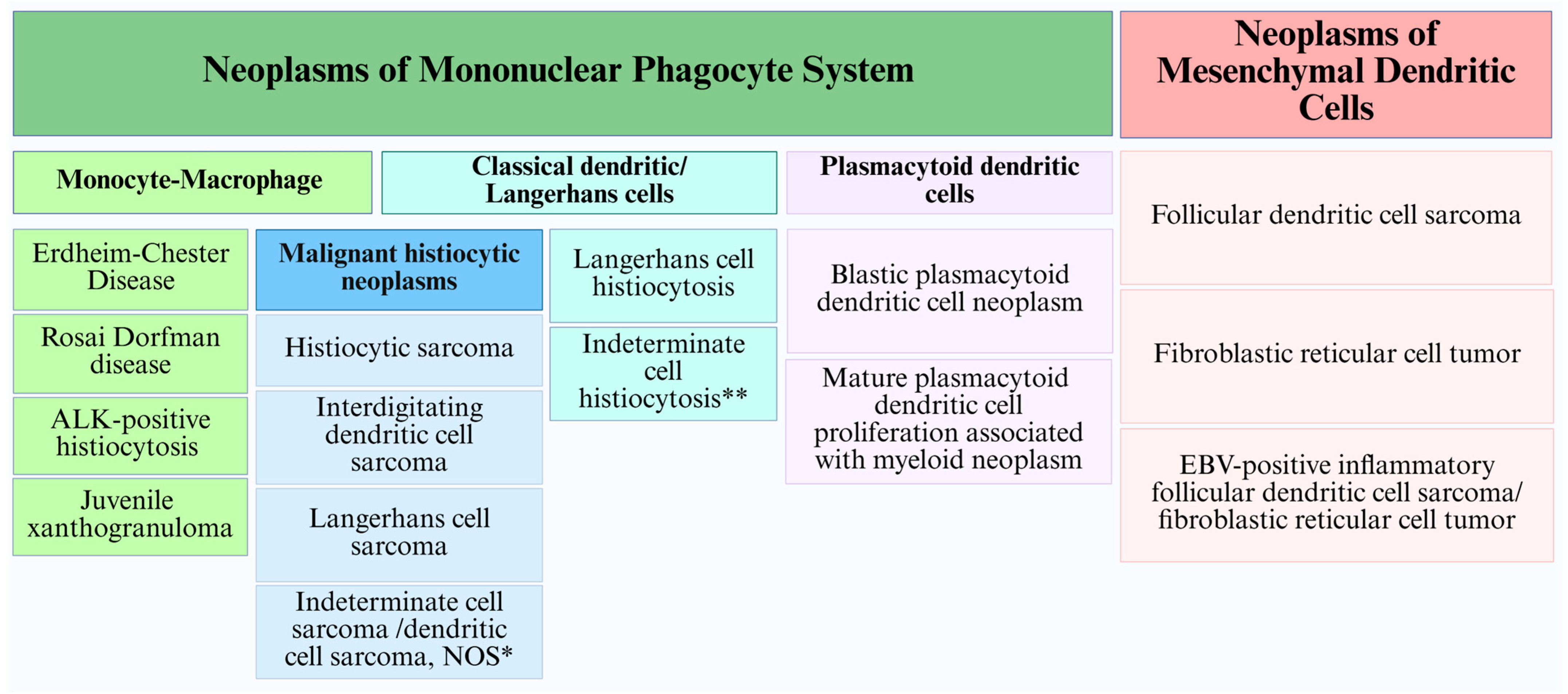
3. Classification: Past, Present and Future
4. Clinical Features and Histopathologic Characterization of Mesenchymal Dendritic Cell Neoplasms
4.1. Follicular Dendritic Cell Sarcoma
4.1.1. Clinical Features
4.1.2. Histopathologic Features
4.1.3. Follicular Dendritic Cell Sarcoma and Castleman Disease: Understanding the Connection
4.2. Fibroblastic Reticular Cell Tumor (FRCT)
4.2.1. Clinical Features
4.2.2. Histopathologic Features
4.3. EBV-Positive Inflammatory FDCS/FRCT
4.4. Overlap of FDCS and FRCT Morphologies
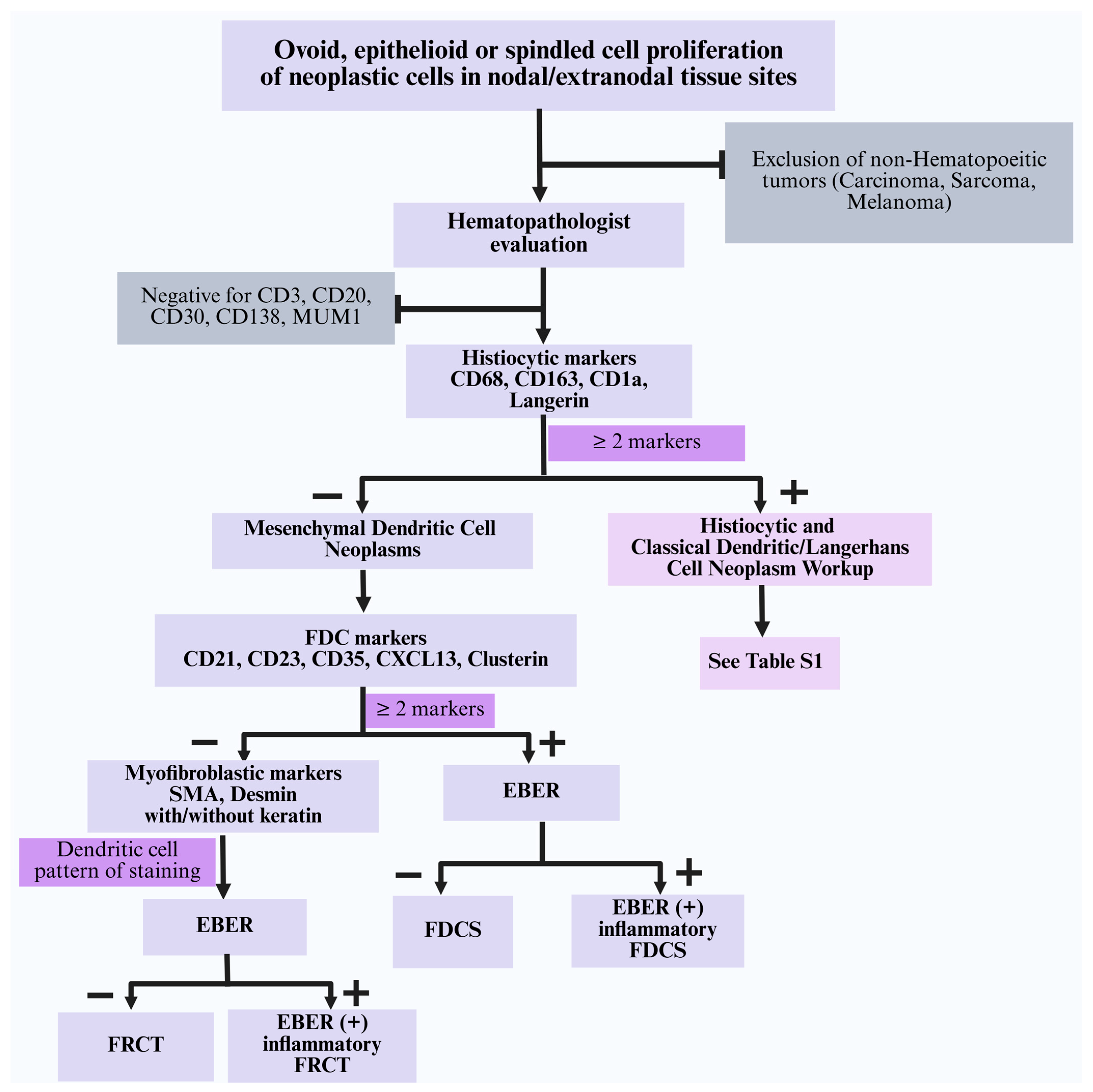
4.5. Differential Diagnosis of Mesenchymal Dendritic Cell Neoplasms
- (i)
- Histiocytic and classical dendritic/Langerhans cell neoplasms: Tumor cells in this category show expression of at least two monocyte/macrophage markers (CD68, CD163, CD4, CD14) or classical dendritic/Langerhans cell markers (CD1a, langerin). They are consistently negative for specific FDC markers (CD21, CD23, CD35, CXCL13), myoid markers (SMA, desmin), and EBER [63]. A summary of the morphologic and immunophenotypic characteristics of various subtypes is described in Table S1.
- (ii)
- Blastic plasmacytoid dendritic cell neoplasm (BPDCN): This aggressive disease is considered an acute leukemia; thus, its clinical presentation should effectively distinguish this entity from the other lesions discussed herein. Histologically, BPDCN shows sheets of small to medium-sized cells with a uniform blast-like morphology, scant cytoplasm, and fine chromatin, often mimicking a myeloid sarcoma. BPDCN is positive for pDC markers (CD123, CD303, CD304, TCL1, TCF4), CD4, and CD56, and is negative for histiocytic and classical dendritic/Langerhans cell markers, FDC markers, SMA, desmin, and EBER [11,28]. Morphologically and clinically, these immature pDC-derived tumors are distinct from other histiocytic and classical dendritic/Langerhans cell neoplasms and are therefore excluded from the IHC algorithm (Figure 4).
- (iii)
- Inflammatory myofibroblastic tumors (IMTs): IMTs show spindled or stellate cells with mixed inflammatory infiltrates. Tumor cells are positive for SMA and variably positive for desmin. Approximately 50–60% of cases exhibit cytoplasmic ALK expression due to ALK gene rearrangement. These cases are negative for FDC markers and EBER [52,64]. ALK-negative IMTs may be more challenging to distinguish from FRCT; in such cases, molecular analysis is helpful as IMTs may harbor ROS1, NTRK, PDGFRβ, and RET gene fusions, amongst others [64].
- (iv)
- Kaposi sarcoma: The morphologic features of these sarcomas are characterized by slit-like vascular spaces with spindled endothelial proliferation that are positive for HHV8, D2-40, and vascular markers (CD31, CD34, ERG, FLI-1). These tumors lack SMA, desmin, FDC markers, and EBER.
- (v)
- Metastatic carcinoma: These cases typically show nests or sheets of epithelioid cells with marked cytologic atypia and demonstrate uniform expression of keratins. The presence of true epithelial differentiation and absence of FDC or myoid markers exclude FDCS/FRCT.
- (vi)
- Metastatic melanoma: These cases have varying morphologies composed of ovoid/epithelioid or spindled cells, often with prominent nucleoli; the cytoplasm may contain brown pigment (melanin). Immunostains are positive for ≥ 2 melanocytic markers (S100, SOX10, Melan A, HMB45). They are negative for FDC markers, SMA, desmin, and EBER.
- (vii)
- Leiomyosarcoma: The histology of these sarcomas shows intersecting fascicles of spindled cells with cigar-shaped nuclei, varying degrees of nuclear pleomorphism, frequent mitoses, and areas of coagulative necrosis. Immunostains show diffuse SMA positivity and variable desmin and caldesmon expression [65]. These tumors are negative for FDC markers, helping to distinguish them from FDCS. However, distinction from FRCT may be challenging due to overlapping phenotypes (SMA+, variably desmin+) and thus require careful correlation with site(s) of involvement, including the absence of lymphoid scaffold characteristics, architectural patterns, and cytologic features (including lack of delicate cytoplasmic extensions on IHC). Hormone receptor positivity, if present, can be useful for distinguishing leiomyosarcoma (estrogen receptor+, progesterone receptor+) from FRCT.
- (viii)
- Rhabdomyosarcoma: These cases show small round or elongated/spindled cells with variable skeletal muscle differentiation. IHC is positive for myogenic markers including myogenin, MyoD1, desmin, and muscle-specific actin. These tumors are negative for FDC markers and EBER. While there is some immunophenotypic overlap with FRCT (variably desmin+), the presence of positive markers of skeletal muscle differentiation (myogenin, myoD1) distinguishes these tumors from FRCT.
- (ix)
- Undifferentiated sarcoma (pleomorphic sarcoma, undifferentiated): These tumors are composed of highly pleomorphic cells, including bizarre multinucleated, spindled, and epithelioid forms. These tumors may show variable and often focal SMA and desmin expression but lack a consistent immunophenotype. They are negative for FDC markers and EBER. Differentiation from FRCT requires correlation with cytomorphologic features, which appear to be more uniform in FRCT compared to the varying degree of nuclear pleomorphism in pleomorphic sarcomas.
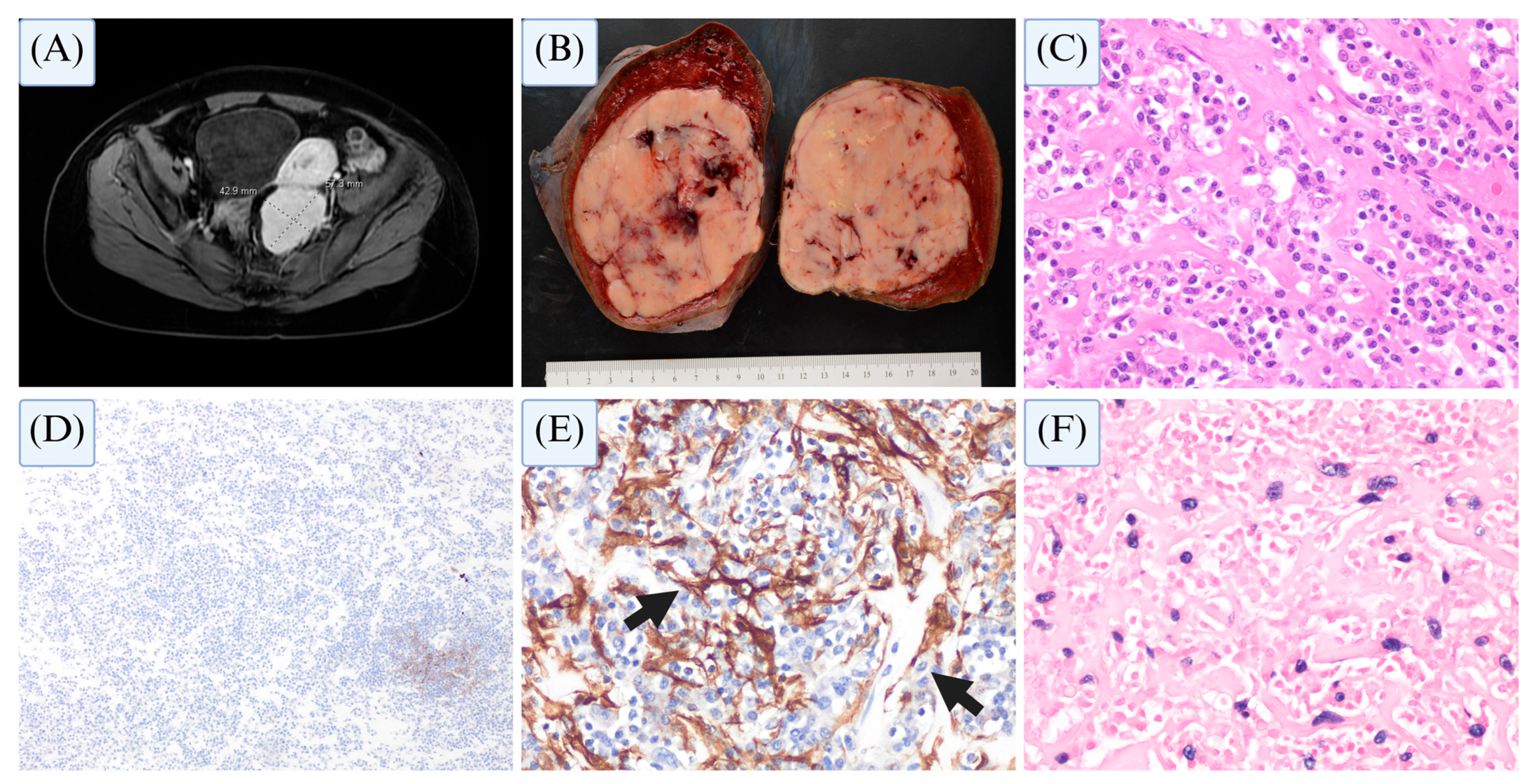
4.6. Mesenchymal Dendritic Cell Neoplasms—Case Studies
4.6.1. Case 1
4.6.2. Case 2
5. Mutational Landscape of Mesenchymal Dendritic Cell Neoplasms
6. Prognosis and Treatment Outcomes
6.1. Prognosis
6.2. Treatment
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Falini, B.; Martino, G.; Lazzi, S. A comparison of the International Consensus and 5th World Health Organization classifications of mature B-cell lymphomas. Leukemia 2023, 37, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Ferry, J.A.; Hill, B.; Hsi, E.D. Mature B, T and NK-cell, plasma cell and histiocytic/dendritic cell neoplasms: Classification according to the World Health Organization and International Consensus Classification. J. Hematol. Oncol. 2024, 17, 51. [Google Scholar] [CrossRef]
- Dalia, S.; Jaglal, M.; Chervenick, P.; Cualing, H.; Sokol, L. Clinicopathologic characteristics and outcomes of histiocytic and dendritic cell neoplasms: The moffitt cancer center experience over the last twenty five years. Cancers 2014, 6, 2275–2295. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, F.; Pileri, S.A.; Lorenzi, L.; Tabanelli, V.; Rimsza, L.; Pittaluga, S.; Dirnhofer, S.; Copie-Bergman, C.; de Leval, L.; Rosenwald, A.; et al. Histiocytic and dendritic cell neoplasms: What have we learnt by studying 67 cases. Virchows Arch. 2017, 471, 467–489. [Google Scholar] [CrossRef]
- El Shikh, M.E.; Pitzalis, C. Follicular dendritic cells in health and disease. Front. Immunol. 2012, 3, 292. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.L.; Acton, S.E.; Knoblich, K. Lymph node fibroblastic reticular cells in health and disease. Nat. Rev. Immunol. 2015, 15, 350–361. [Google Scholar] [CrossRef]
- Durham, B.H. Molecular characterization of the histiocytoses: Neoplasia of dendritic cells and macrophages. Semin. Cell Dev. Biol. 2019, 86, 62–76. [Google Scholar] [CrossRef]
- Durham, B.H. Molecular Pathogenesis of the Histiocytic and Dendritic Cell Neoplasms. Hematol. Oncol. Clin. N. Am. 2025, 39, 471–490. [Google Scholar] [CrossRef]
- Griffin, G.K.; Sholl, L.M.; Lindeman, N.I.; Fletcher, C.D.; Hornick, J.L. Targeted genomic sequencing of follicular dendritic cell sarcoma reveals recurrent alterations in NF-kappaB regulatory genes. Mod. Pathol. 2016, 29, 67–74. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253. [Google Scholar] [CrossRef] [PubMed]
- Rezk, S.A.; Nathwani, B.N.; Zhao, X.; Weiss, L.M. Follicular dendritic cells: Origin, function, and different disease-associated patterns. Hum. Pathol. 2013, 44, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Brown, F.D.; Turley, S.J. Fibroblastic reticular cells: Organization and regulation of the T lymphocyte life cycle. J. Immunol. 2015, 194, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Jarjour, M.; Jorquera, A.; Mondor, I.; Wienert, S.; Narang, P.; Coles, M.C.; Klauschen, F.; Bajenoff, M. Fate mapping reveals origin and dynamics of lymph node follicular dendritic cells. J. Exp. Med. 2014, 211, 1109–1122. [Google Scholar] [CrossRef]
- Allen, C.D.; Cyster, J.G. Follicular dendritic cell networks of primary follicles and germinal centers: Phenotype and function. Semin. Immunol. 2008, 20, 14–25. [Google Scholar] [CrossRef]
- van Nierop, K.; de Groot, C. Human follicular dendritic cells: Function, origin and development. Semin. Immunol. 2002, 14, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Heesters, B.A.; van Megesen, K.; Tomris, I.; de Vries, R.P.; Magri, G.; Spits, H. Characterization of human FDCs reveals regulation of T cells and antigen presentation to B cells. J. Exp. Med. 2021, 218, e20210790. [Google Scholar] [CrossRef]
- Haniffa, M.; Bigley, V.; Collin, M. Human mononuclear phagocyte system reunited. Semin. Cell Dev. Biol. 2015, 41, 59–69. [Google Scholar] [CrossRef]
- Collin, M.; Milne, P. Langerhans cell origin and regulation. Curr. Opin. Hematol. 2016, 23, 28–35. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Sobenin, I.A.; Bobryshev, Y.V. Plasmacytoid dendritic cells: Development, functions, and role in atherosclerotic inflammation. Front. Physiol. 2014, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Gerber-Ferder, Y.; Vaivode, K.; Bourdely, P.; Helft, J. Origin and development of classical dendritic cells. Int. Rev. Cell Mol. Biol. 2019, 349, 1–54. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Segura, E. Human dendritic cell subsets: An updated view of their ontogeny and functional specialization. Eur. J. Immunol. 2022, 52, 1759–1767. [Google Scholar] [CrossRef]
- Cytlak, U.; Resteu, A.; Pagan, S.; Green, K.; Milne, P.; Maisuria, S.; McDonald, D.; Hulme, G.; Filby, A.; Carpenter, B.; et al. Differential IRF8 Transcription Factor Requirement Defines Two Pathways of Dendritic Cell Development in Humans. Immunity 2020, 53, 353–370.e8. [Google Scholar] [CrossRef]
- Emile, J.F.; Abla, O.; Fraitag, S.; Horne, A.; Haroche, J.; Donadieu, J.; Requena-Caballero, L.; Jordan, M.B.; Abdel-Wahab, O.; Allen, C.E.; et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016, 127, 2672–2681. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Perkins, S.M.; Shinohara, E.T. Interdigitating and follicular dendritic cell sarcomas: A SEER analysis. Am. J. Clin. Oncol. 2013, 36, 395–398. [Google Scholar] [CrossRef]
- Surveillance, Epidemiology, and End Results Program. Available online: https://seer.cancer.gov/data/ (accessed on 5 May 2025).
- Friedman, S.; Negoita, S. History of the Surveillance, Epidemiology, and End Results (SEER) Program. J. Natl. Cancer Inst. Monogr. 2024, 2024, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Grogg, K.L.; Lae, M.E.; Kurtin, P.J.; Macon, W.R. Clusterin expression distinguishes follicular dendritic cell tumors from other dendritic cell neoplasms: Report of a novel follicular dendritic cell marker and clinicopathologic data on 12 additional follicular dendritic cell tumors and 6 additional interdigitating dendritic cell tumors. Am. J. Surg. Pathol. 2004, 28, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Uzunaslan, D.; Ozguroglu, M.; Senocak, M.; Tuzuner, N. Dendritic cell sarcoma: A pooled analysis including 462 cases with presentation of our case series. Crit. Rev. Oncol. Hematol. 2013, 88, 253–271. [Google Scholar] [CrossRef]
- Mao, S.; Dong, J.; Wang, Y.; Zhang, C.; Dong, A.; Shen, J. Follicular Dendritic Cell Sarcomas: CT and MRI Findings in 20 Patients. AJR Am. J. Roentgenol. 2021, 216, 835–843. [Google Scholar] [CrossRef]
- Lee, I.J.; Kim, S.C.; Kim, H.S.; Bang, D.; Yang, W.I.; Jung, W.H.; Chi, H.S. Paraneoplastic pemphigus associated with follicular dendritic cell sarcoma arising from Castleman’s tumor. J. Am. Acad. Dermatol. 1999, 40, 294–297. [Google Scholar] [CrossRef]
- Walters, M.; Pittelkow, M.R.; Hasserjian, R.P.; Harris, N.L.; Macon, W.R.; Kurtin, P.J.; Rech, K.L.G. Follicular Dendritic Cell Sarcoma With Indolent T-Lymphoblastic Proliferation Is Associated With Paraneoplastic Autoimmune Multiorgan Syndrome. Am. J. Surg. Pathol. 2018, 42, 1647–1652. [Google Scholar] [CrossRef]
- Vermi, W.; Lonardi, S.; Bosisio, D.; Uguccioni, M.; Danelon, G.; Pileri, S.; Fletcher, C.; Sozzani, S.; Zorzi, F.; Arrigoni, G.; et al. Identification of CXCL13 as a new marker for follicular dendritic cell sarcoma. J. Pathol. 2008, 216, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, L.; Doring, C.; Rausch, T.; Benes, V.; Lonardi, S.; Bugatti, M.; Campo, E.; Cabecadas, J.; Simonitsch-Klupp, I.; Borges, A.; et al. Identification of novel follicular dendritic cell sarcoma markers, FDCSP and SRGN, by whole transcriptome sequencing. Oncotarget 2017, 8, 16463–16472. [Google Scholar] [CrossRef]
- Tao, L.L.; Huang, Y.H.; Chen, Y.L.; Yu, G.Y.; Yin, W.H. SSTR2a Is a Useful Diagnostic Marker for Follicular Dendritic Cells and Their Related Tumors. Am. J. Surg. Pathol. 2019, 43, 374–381. [Google Scholar] [CrossRef]
- Yu, H.; Gibson, J.A.; Pinkus, G.S.; Hornick, J.L. Podoplanin (D2-40) is a novel marker for follicular dendritic cell tumors. Am. J. Clin. Pathol. 2007, 128, 776–782. [Google Scholar] [CrossRef]
- Schelbert, S.; Maurus, K.; Roth, S.; Ott, G.; Kurz, K.S.; Mogler, C.; Wollenberg, B.; Linde, J.; Zamo, A.; Anagnostopoulos, I.; et al. Morphological, immunohistochemical and molecular analysis of follicular dendritic cell sarcomas: L1CAM as a new diagnostic marker. Histopathology 2025, Epub ahead of print. [CrossRef]
- Wang, H.; Su, Z.; Hu, Z.; Wen, J.; Liu, B. Follicular dendritic cell sarcoma: A report of six cases and a review of the Chinese literature. Diagn. Pathol. 2010, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Hisijos, N.; Omman, R.; Pambuccian, S.; Mirza, K. Follicular Dendritic Cell Sarcoma or Not? A Series of 5 Diagnostically Challenging Cases. Clin. Med. Insights Oncol. 2019, 13, 1179554919844531. [Google Scholar] [CrossRef]
- Perez-Ordonez, B.; Erlandson, R.A.; Rosai, J. Follicular dendritic cell tumor: Report of 13 additional cases of a distinctive entity. Am. J. Surg. Pathol. 1996, 20, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; You, W.; Wan, P.; Jiang, X.; Chen, J.; Zheng, Y.; Li, W.; Tan, J.; Zhang, S. Clinicopathological and prognostic significance of PD-L1 expression in sarcoma: A systematic review and meta-analysis. Medicine 2018, 97, e11004. [Google Scholar] [CrossRef]
- Hwang, S.O.; Lee, T.H.; Bae, S.H.; Cho, H.D.; Choi, K.H.; Park, S.H.; Kim, C.H.; Kim, S.J. Transformation of Castleman’s disease into follicular dendritic cell sarcoma, presenting as an asymptomatic intra-abdominal mass. Korean J. Gastroenterol. 2013, 62, 131–134. [Google Scholar] [CrossRef]
- Jimenez-Heffernan, J.A.; Diaz Del Arco, C.; Adrados, M. A Cytological Review of Follicular Dendritic Cell-Derived Tumors with Emphasis on Follicular Dendritic Cell Sarcoma and Unicentric Castleman Disease. Diagnostics 2022, 12, 406. [Google Scholar] [CrossRef]
- Sun, X.; Chang, K.C.; Abruzzo, L.V.; Lai, R.; Younes, A.; Jones, D. Epidermal growth factor receptor expression in follicular dendritic cells: A shared feature of follicular dendritic cell sarcoma and Castleman’s disease. Hum. Pathol. 2003, 34, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Vermi, W.; Giurisato, E.; Lonardi, S.; Balzarini, P.; Rossi, E.; Medicina, D.; Bosisio, D.; Sozzani, S.; Pellegrini, W.; Doglioni, C.; et al. Ligand-dependent activation of EGFR in follicular dendritic cells sarcoma is sustained by local production of cognate ligands. Clin. Cancer Res. 2013, 19, 5027–5038. [Google Scholar] [CrossRef]
- Phulware, R.H.; Ramteke, P.; Yadav, R.; Iyer, V.K.; Mallick, S. Cytology of Castleman’s disease (hyaline-vascular type) masquerading as Hodgkin’s lymphoma. Am. J. Blood Res. 2022, 12, 196–200. [Google Scholar]
- Chan, J.; Naresh, K.N.; Saygin, C.; Kaji, S. Fibroblastic Reticular Cell tumor. In WHO Classification of Tumours Editorial Board. Haematolymphoid Tumours [Internet], 5th ed.; WHO Classification of Tumours Series; International Agency for Research on Cancer: Lyon, France, 2024; Volume 11. [Google Scholar]
- Pescia, C.; Lopez, G.; Gianelli, U.; Croci, G.A. Fibroblastic/cytokeratin-positive interstitial reticular cell tumor of the spleen with indolent behavior: A case report with review of the literature. Virchows Arch. 2023, 482, 1069–1077. [Google Scholar] [CrossRef]
- Chan, A.C.; Serrano-Olmo, J.; Erlandson, R.A.; Rosai, J. Cytokeratin-positive malignant tumors with reticulum cell morphology: A subtype of fibroblastic reticulum cell neoplasm? Am. J. Surg. Pathol. 2000, 24, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Goto, N.; Tsurumi, H.; Takami, T.; Futamura, M.; Morimitsu, K.; Takata, K.; Sato, Y.; Yoshino, T.; Adachi, S.; Saito, K.; et al. Cytokeratin-positive fibroblastic reticular cell tumor with follicular dendritic cell features: A case report and review of the literature. Am. J. Surg. Pathol. 2015, 39, 573–580. [Google Scholar] [CrossRef]
- Martel, M.; Sarli, D.; Colecchia, M.; Coppa, J.; Romito, R.; Schiavo, M.; Mazzaferro, V.; Rosai, J. Fibroblastic reticular cell tumor of the spleen: Report of a case and review of the entity. Hum. Pathol. 2003, 34, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Andriko, J.W.; Kaldjian, E.P.; Tsokos, M.; Abbondanzo, S.L.; Jaffe, E.S. Reticulum cell neoplasms of lymph nodes: A clinicopathologic study of 11 cases with recognition of a new subtype derived from fibroblastic reticular cells. Am. J. Surg. Pathol. 1998, 22, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; You, Z.; Chen, X.; Wang, C. Clinicopathological and molecular genetic insights into EBV-positive inflammatory follicular dendritic cell sarcoma. Hum. Pathol. 2024, 153, 105668. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Kamel, O.W.; van de Rijn, M.; Davis, R.E.; Medeiros, L.J.; Jaffe, E.S.; Weiss, L.M. Frequent presence of the Epstein-Barr virus in inflammatory pseudotumor. Hum. Pathol. 1995, 26, 1093–1098. [Google Scholar] [CrossRef]
- Selves, J.; Meggetto, F.; Brousset, P.; Voigt, J.J.; Pradere, B.; Grasset, D.; Icart, J.; Mariame, B.; Knecht, H.; Delsol, G. Inflammatory pseudotumor of the liver. Evidence for follicular dendritic reticulum cell proliferation associated with clonal Epstein-Barr virus. Am. J. Surg. Pathol. 1996, 20, 747–753. [Google Scholar] [CrossRef]
- Ke, X.; He, H.; Zhang, Q.; Yuan, J.; Ao, Q. Epstein-Barr virus-positive inflammatory follicular dendritic cell sarcoma presenting as a solitary colonic mass: Two rare cases and a literature review. Histopathology 2020, 77, 832–840. [Google Scholar] [CrossRef]
- Morales-Vargas, B.; Deeb, K.; Peker, D. Clinicopathologic and Molecular Analysis of Inflammatory Pseudotumor-Like Follicular/Fibroblastic Dendritic Cell Sarcoma: A Case Report and Review of Literature. Turk. Patoloji Derg. 2021, 37, 266–272. [Google Scholar] [CrossRef]
- Ravindran, A.; Dasari, S.; Ruan, G.J.; Artymiuk, C.J.; He, R.; Viswanatha, D.S.; Abeykoon, J.P.; Zanwar, S.; Young, J.R.; Goyal, G.; et al. Malignant Histiocytosis Comprises a Phenotypic Spectrum That Parallels the Lineage Differentiation of Monocytes, Macrophages, Dendritic Cells, and Langerhans Cells. Mod. Pathol. 2023, 36, 100268. [Google Scholar] [CrossRef]
- Chmiel, P.; Sl, O.A.; Banaszek, L.; Szumera, C.A.; Szostakowski, B.; MJ, S.P.; Switaj, T.; Rutkowski, P.; Czarnecka, A.M. Inflammatory myofibroblastic tumor from molecular diagnostics to current treatment. Oncol. Res. 2024, 32, 1141–1162. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Kusakabe, T.; Hoshi, N.; Saito, A.; Suzuki, T. h-Caldesmon in leiomyosarcoma and tumors with smooth muscle cell-like differentiation: Its specific expression in the smooth muscle cell tumor. Hum. Pathol. 1999, 30, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Bhaduri, A.; Shahmarvand, N.; Shahryari, J.; Zehnder, J.L.; Warnke, R.A.; Mughal, T.; Ali, S.; Ohgami, R.S. Next-generation sequencing of idiopathic multicentric and unicentric Castleman disease and follicular dendritic cell sarcomas. Blood Adv. 2018, 2, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Ye, H.; Luo, S.; Wang, J. Description of two cases of follicular dendritic cell sarcoma, including next-generation sequencing analysis. Diagn. Pathol. 2025, 20, 19. [Google Scholar] [CrossRef]
- Shia, J.; Chen, W.; Tang, L.H.; Carlson, D.L.; Qin, J.; Guillem, J.G.; Nobrega, J.; Wong, W.D.; Klimstra, D.S. Extranodal follicular dendritic cell sarcoma: Clinical, pathologic, and histogenetic characteristics of an underrecognized disease entity. Virchows Arch. 2006, 449, 148–158. [Google Scholar] [CrossRef]
- Shinagare, A.B.; Ramaiya, N.H.; Jagannathan, J.P.; Hornick, J.L.; Swanson, R.S. Primary follicular dendritic cell sarcoma of liver treated with cyclophosphamide, doxorubicin, vincristine, and prednisone regimen and surgery. J. Clin. Oncol. 2011, 29, e849–e851. [Google Scholar] [CrossRef]
- Jain, P.; Milgrom, S.A.; Patel, K.P.; Nastoupil, L.; Fayad, L.; Wang, M.; Pinnix, C.C.; Dabaja, B.S.; Smith, G.L.; Yu, J.; et al. Characteristics, management, and outcomes of patients with follicular dendritic cell sarcoma. Br. J. Haematol. 2017, 178, 403–412. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, D.; Zhang, G. Clinicopathological characteristics of extranodal follicular dendritic cell sarcoma: A report of two cases. Oncol. Lett. 2021, 21, 182. [Google Scholar] [CrossRef]
- Vojjala, N.; Yadav, S.K.; Tan Aldecoa, K.A.; Liu, B.; Hussein, G.; Azar, I.; Yadlapalli, S.; Krishnamoorthy, G.; Goodman, J.R. Clinical profile and response to adjuvant treatments in patients with follicular dendritic cell sarcoma in the United States: Insights from SEER analysis. J. Clin. Oncol. 2024, 42, e23537. [Google Scholar] [CrossRef]
- Soriano, A.O.; Thompson, M.A.; Admirand, J.H.; Fayad, L.E.; Rodriguez, A.M.; Romaguera, J.E.; Hagemeister, F.B.; Pro, B. Follicular dendritic cell sarcoma: A report of 14 cases and a review of the literature. Am. J. Hematol. 2007, 82, 725–728. [Google Scholar] [CrossRef]
- Gounder, M.; Desai, V.; Kuk, D.; Agaram, N.; Arcila, M.; Durham, B.; Keohan, M.L.; Dickson, M.A.; D’Angelo, S.P.; Shukla, N.; et al. Impact of surgery, radiation and systemic therapy on the outcomes of patients with dendritic cell and histiocytic sarcomas. Eur. J. Cancer 2015, 51, 2413–2422. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ren, M.; Bi, F.; Chen, Y.; Li, Z. Favorable response to PD-1 inhibitor plus chemotherapy as first-line treatment for metastatic follicular dendritic cell sarcoma of the spleen: A case report. Front. Immunol. 2023, 14, 1228653. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Du, Z.; Liang, Y.; Zhou, J. Apatinib manifests an unexpectedly favorable outcome in the management of axillary lymph node follicular dendritic cell sarcoma: A case report. Front. Oncol. 2024, 14, 1388982. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhao, S.; Jiang, M. Unexpected Favorable Outcome to PD-1 Antibody Plus Lenvatinib in a Patient With Recurrent Intestinal Follicular Dendritic Cell Sarcoma: A Case Report and Literature Review. Front. Immunol. 2021, 12, 653319. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WHO (Revised Fourth Edition, 2016) | WHO (Fifth Edition, 2022) | ICC (2022) |
|---|---|---|
Follicular dendritic cell sarcoma Fibroblastic reticular cell tumor Inflammatory pseudotumor-like follicular/fibroblastic dendritic cell sarcoma Langerhans cell histiocytosis Langerhans cell sarcoma Indeterminate dendritic cell tumor Interdigitating dendritic cell sarcoma Erdheim–Chester disease Disseminated JXG Histiocytic sarcoma | Mesenchymal Dendritic Cell Neoplasms Follicular dendritic cell sarcoma Fibroblastic reticular cell tumor EBV-positive inflammatory FDCS # Histiocytic/Dendritic Cell Neoplasms Blastic plasmacytoid dendritic cell neoplasm Mature plasmacytoid dendritic cell proliferation associated with myeloid neoplasm * Langerhans cell histiocytosis Langerhans cell sarcoma Indeterminate dendritic cell tumor Interdigitating dendritic cell sarcoma Erdheim–Chester disease JXG Histiocytic sarcoma Rosai–Dorfman disease * ALK-positive histiocytosis * | Follicular dendritic cell sarcoma Fibroblastic reticular cell tumor EBV-positive inflammatory FDCS/FRCT Langerhans cell histiocytosis Langerhans cell sarcoma Indeterminate dendritic cell histiocytosis Interdigitating dendritic cell sarcoma Erdheim-Chester disease Disseminated JXG Histiocytic sarcoma Rosai-Dorfman disease * ALK-positive histiocytosis * |
| Treatment Modality | Description | Relative Efficacy |
|---|---|---|
| Surgery | Mainstay of treatment for localized disease | Complete surgical excision optimizes progression-free survival (PFS) and overall survival (OS) [70,72,73] |
| Adjuvant Radiotherapy | Considered post-surgery | Improves local control, PFS, and OS, especially after gross total resection |
| Systemic Chemotherapy | Used for metastatic or advanced tumors due to the lack of standard protocols | Higher overall response rates (~80%) in gemcitabine-based regimens in FDCS [34,70,74] Inadequate data on lymphoma-based regimens (CHOP, ICE, ABVD) |
| Immune Checkpoint Inhibitors | Pembrolizumab and other PD-1 inhibitors show potential when combined with chemotherapy | Potential for durable responses, especially in tumors with PD-L1 expression [46,75] |
| Targeted Therapies | Rare cases with MAPK pathway alterations may be amenable to treatment with BRAF/MEK-inhibitors | Offers tailored treatment with potential for improved outcomes [9] |
| Small Molecule Inhibitors | Apatinib (anti-angiogenic) and lenvatinib (multi-kinase inhibitor) have shown promise in individual cases | Limited data; additional research is required to evaluate long-term outcomes [76,77] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seth, N.; Abeykoon, J.P.; Goyal, G.; Go, R.S.; Tessier, S.; King, R.L.; Ravindran, A. Current Perspectives on Mesenchymal Dendritic Cell Neoplasms of Lymphoid Tissue: Insights into Ontogeny, Updates on Classification, and Clinicopathologic Characteristics. Cancers 2025, 17, 2055. https://doi.org/10.3390/cancers17122055
Seth N, Abeykoon JP, Goyal G, Go RS, Tessier S, King RL, Ravindran A. Current Perspectives on Mesenchymal Dendritic Cell Neoplasms of Lymphoid Tissue: Insights into Ontogeny, Updates on Classification, and Clinicopathologic Characteristics. Cancers. 2025; 17(12):2055. https://doi.org/10.3390/cancers17122055
Chicago/Turabian StyleSeth, Neha, Jithma P. Abeykoon, Gaurav Goyal, Ronald S. Go, Steven Tessier, Rebecca L. King, and Aishwarya Ravindran. 2025. "Current Perspectives on Mesenchymal Dendritic Cell Neoplasms of Lymphoid Tissue: Insights into Ontogeny, Updates on Classification, and Clinicopathologic Characteristics" Cancers 17, no. 12: 2055. https://doi.org/10.3390/cancers17122055
APA StyleSeth, N., Abeykoon, J. P., Goyal, G., Go, R. S., Tessier, S., King, R. L., & Ravindran, A. (2025). Current Perspectives on Mesenchymal Dendritic Cell Neoplasms of Lymphoid Tissue: Insights into Ontogeny, Updates on Classification, and Clinicopathologic Characteristics. Cancers, 17(12), 2055. https://doi.org/10.3390/cancers17122055