

Future Perspectives in Senescence-Based Therapies for Head and Neck Cancer
 , , and
, , and
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
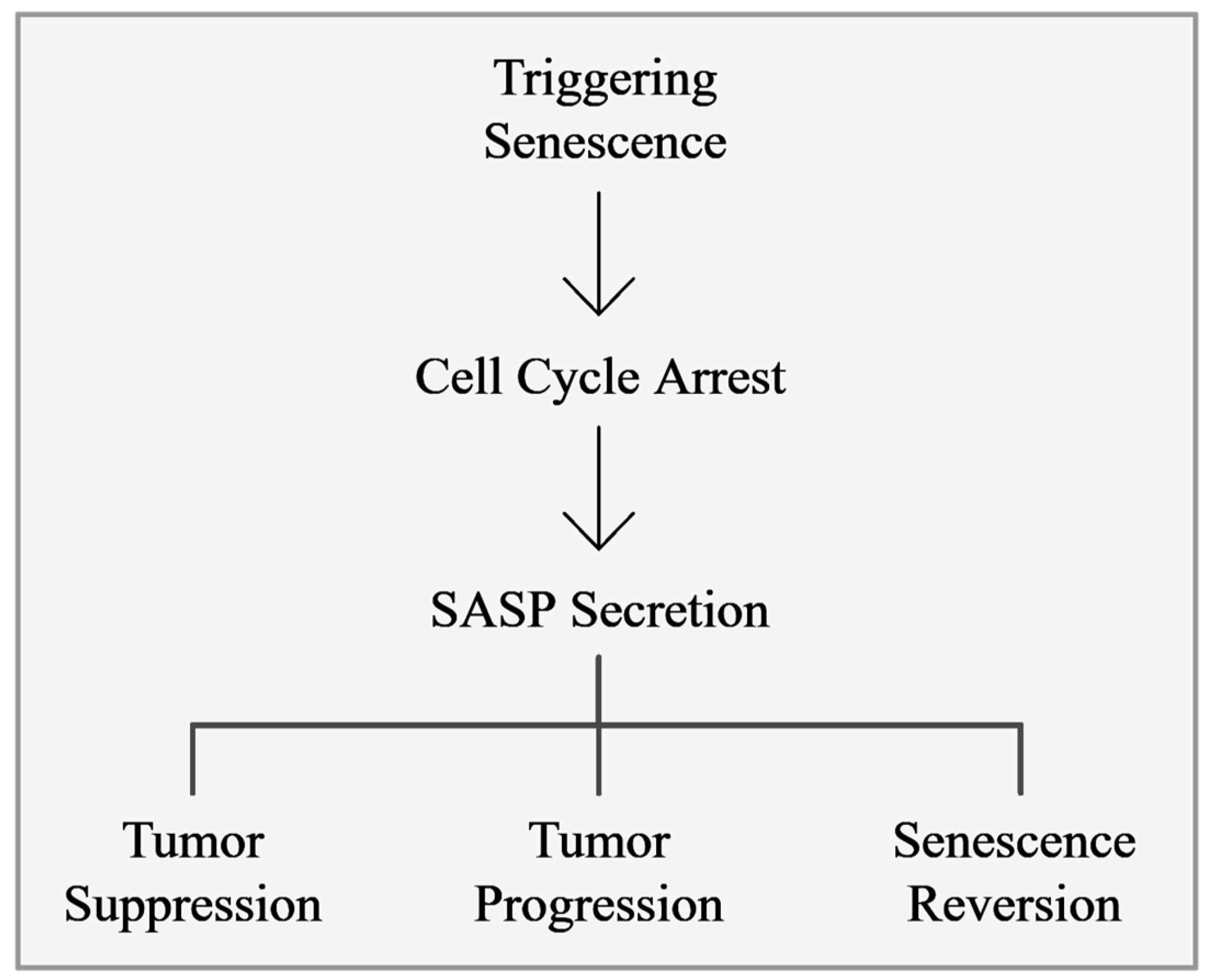
2. Senescence
2.1. Dual Nature
2.2. Characteristics and Biomarkers
3. Senescence Activity on Tumor Cells
3.1. Senescence-Involved Signaling Pathways
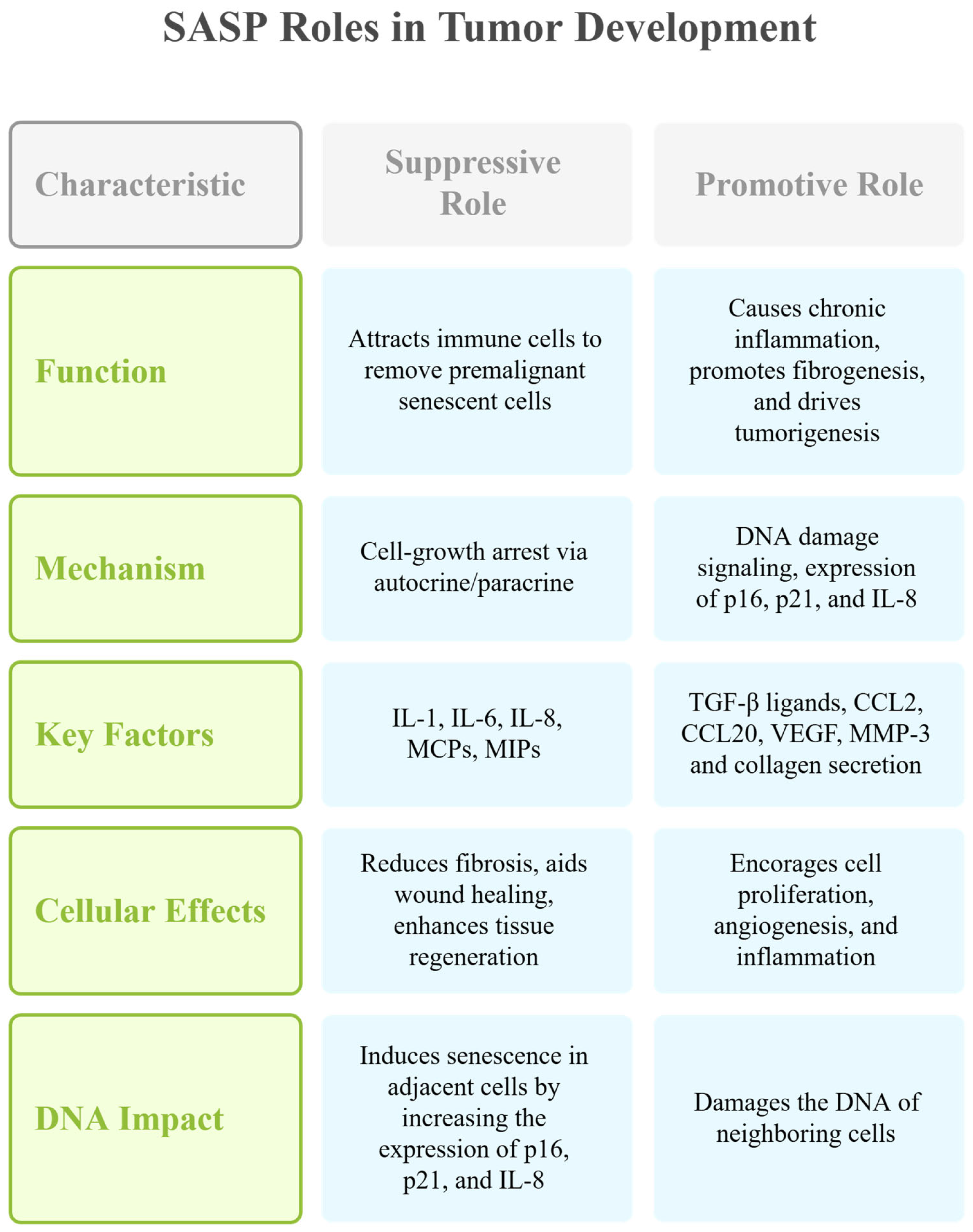
3.2. Senescence-Associated Secretory Phenotype
3.3. Senescence Escape and Tumor Aggressiveness
4. Senescence-Targeted Cancer Therapies
4.1. Oncogene-Induced Senescence and Emerging Therapies Targeting Senescence
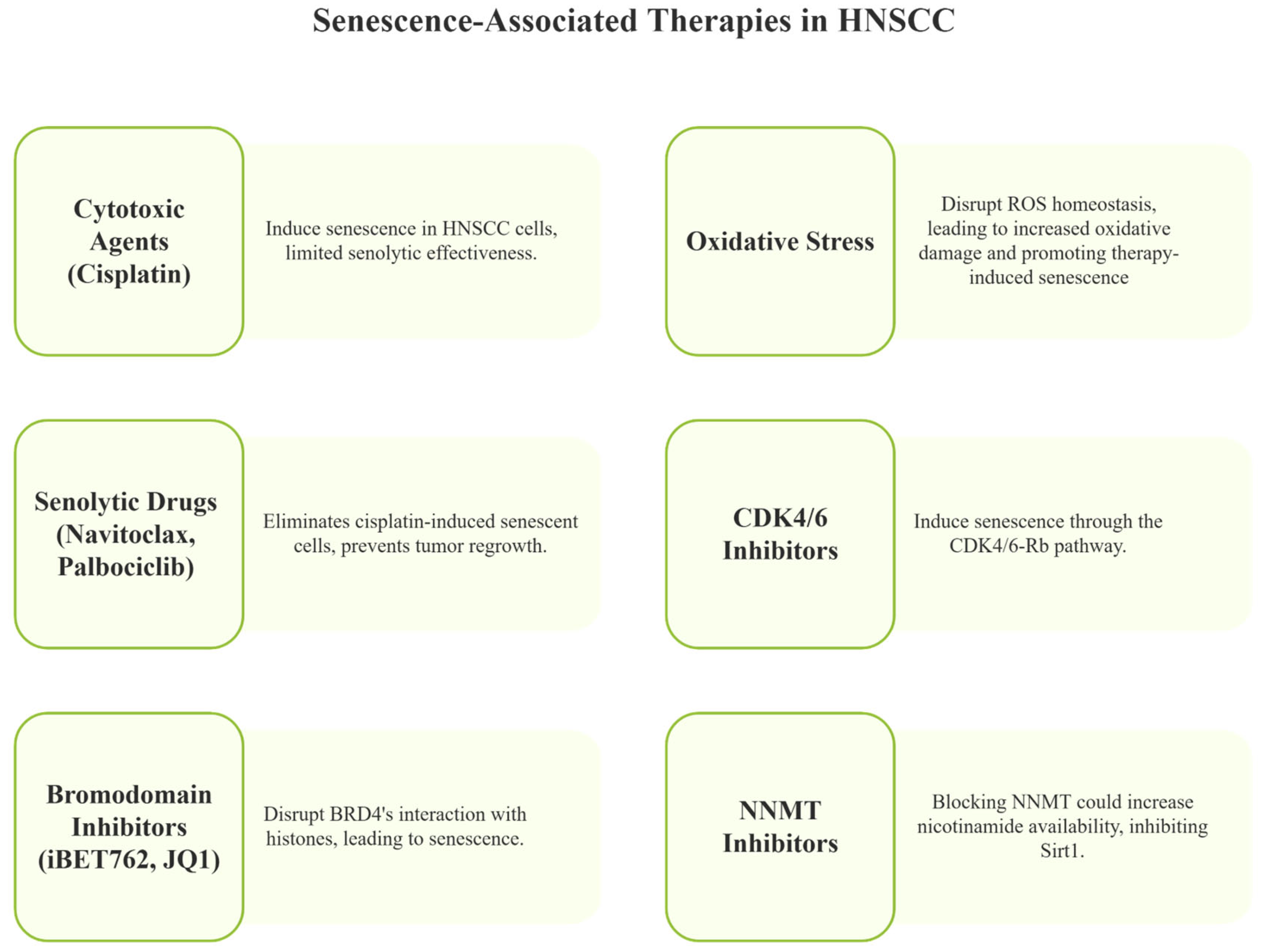
4.2. Targeting Senescence in Cancer—Senotherapies
5. Senescence and Head and Neck Cancers
Emerging Opportunities for Senescence-Induced Therapies
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Declaration of use of Generative AI
References
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Castilho, R.M.; Squarize, C.H.; Almeida, L.O. Epigenetic Modifications and Head and Neck Cancer: Implications for Tumor Progression and Resistance to Therapy. Int. J. Mol. Sci. 2017, 18, 1506. [Google Scholar] [CrossRef] [PubMed]
- Ghantous, Y.; Schussel, J.L.; Brait, M. Tobacco and alcohol-induced epigenetic changes in oral carcinoma. Curr. Opin. Oncol. 2018, 30, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Kirschner, K.; Rattanavirotkul, N.; Quince, M.F.; Chandra, T. Functional heterogeneity in senescence. Biochem. Soc. Trans. 2020, 48, 765–773. [Google Scholar] [CrossRef]
- Tripathi, U.; Misra, A.; Tchkonia, T.; Kirkland, J.L. Impact of Senescent Cell Subtypes on Tissue Dysfunction and Repair: Importance and Research Questions. Mech. Ageing Dev. 2021, 198, 111548. [Google Scholar] [CrossRef]
- Tufail, M.; Huang, Y.Q.; Hu, J.J.; Liang, J.; He, C.Y.; Wan, W.D.; Jiang, C.H.; Wu, H.; Li, N. Cellular Aging and Senescence in Cancer: A Holistic Review of Cellular Fate Determinants. Aging Dis. 2024, 16, 1483–1512. [Google Scholar] [CrossRef]
- Ozdemir, A.; Simay Demir, Y.D.; Yesilyurt, Z.E.; Ark, M. Senescent cells and SASP in cancer microenvironment: New approaches in cancer therapy. Adv. Protein Chem. Struct. Biol. 2023, 133, 115–158. [Google Scholar] [CrossRef]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. Senolytics for Cancer Therapy: Is All That Glitters Really Gold? Cancers 2021, 13, 723. [Google Scholar] [CrossRef]
- Liu, X.; Hoft, D.F.; Peng, G. Senescent T cells within suppressive tumor microenvironments: Emerging target for tumor immunotherapy. J. Clin. Investig. 2020, 130, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Hainer, S.J.; Boskovic, A.; McCannell, K.N.; Rando, O.J.; Fazzio, T.G. Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 2019, 177, 1319–1329.e11. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Xiao, S.; Qin, D.; Hou, X.; Tian, L.; Yu, Y.; Zhang, R.; Lyu, H.; Guo, D.; Chen, X.Z.; Zhou, C.; et al. Cellular senescence: A double-edged sword in cancer therapy. Front. Oncol. 2023, 13, 1189015. [Google Scholar] [CrossRef]
- Lucas, V.; Cavadas, C.; Aveleira, C.A. Cellular Senescence: From Mechanisms to Current Biomarkers and Senotherapies. Pharmacol. Rev. 2023, 75, 675–713. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef]
- Moiseeva, O.; Guillon, J.; Ferbeyre, G. Senescence: A program in the road to cell elimination and cancer. Semin. Cancer Biol. 2022, 81, 48–53. [Google Scholar] [CrossRef]
- Wiley, C.D. Bubble Bubble, Senescent Cells Are a Cauldron of Tumor Trouble. Cancer Res. 2020, 80, 3193–3194. [Google Scholar] [CrossRef] [PubMed]
- Rajaraman, R.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, S.R. Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Int. 2006, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Das, P.K.; Rambukkana, A.; Baas, J.G.; Groothuis, D.G.; Halperin, M. Enzyme-linked immunosorbent assay for distinguishing serological responses of lepromatous and tuberculoid leprosies to the 29/33-kilodalton doublet and 64-kilodalton antigens of Mycobacterium tuberculosis. J. Clin. Microbiol. 1990, 28, 379–382. [Google Scholar] [CrossRef]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Parekh, A.; Das, S.; Parida, S.; Das, C.K.; Dutta, D.; Mallick, S.K.; Wu, P.H.; Kumar, B.N.P.; Bharti, R.; Dey, G.; et al. Multi-nucleated cells use ROS to induce breast cancer chemo-resistance in vitro and in vivo. Oncogene 2018, 37, 4546–4561. [Google Scholar] [CrossRef]
- Guerrero, A.; Guiho, R.; Herranz, N.; Uren, A.; Withers, D.J.; Martinez-Barbera, J.P.; Tietze, L.F.; Gil, J. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 2020, 19, e13133. [Google Scholar] [CrossRef]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Del Toro, N.; Fernandez-Ruiz, A.; Mignacca, L.; Kalegari, P.; Rowell, M.C.; Igelmann, S.; Saint-Germain, E.; Benfdil, M.; Lopes-Paciencia, S.; Brakier-Gingras, L.; et al. Ribosomal protein RPL22/eL22 regulates the cell cycle by acting as an inhibitor of the CDK4-cyclin D complex. Cell Cycle 2019, 18, 759–770. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef]
- Ribeiro, J.D.; Morey, L.; Mas, A.; Gutierrez, A.; Luis, N.M.; Mejetta, S.; Richly, H.; Benitah, S.A.; Keyes, W.M.; Di Croce, L. ZRF1 controls oncogene-induced senescence through the INK4-ARF locus. Oncogene 2013, 32, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Gong, Y.; Yan, G.; Wang, D.; Wang, Q.; Qiao, Y.; Hou, J.; Liu, B.; Tang, C. Atorvastatin treatment modulates p16 promoter methylation to regulate p16 expression. FEBS J. 2017, 284, 1868–1881. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, I. Epigenetic Regulation of Cellular Senescence and Aging. Front. Genet. 2017, 8, 138. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef]
- Calcinotto, A.; Alimonti, A. Aging tumour cells to cure cancer: “pro-senescence” therapy for cancer. Swiss Med. Wkly. 2017, 147, w14367. [Google Scholar] [CrossRef]
- Mikula-Pietrasik, J.; Niklas, A.; Uruski, P.; Tykarski, A.; Ksiazek, K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell Mol. Life Sci. 2020, 77, 213–229. [Google Scholar] [CrossRef]
- Liao, Z.; Yeo, H.L.; Wong, S.W.; Zhao, Y. Cellular Senescence: Mechanisms and Therapeutic Potential. Biomedicines 2021, 9, 1769. [Google Scholar] [CrossRef]
- Sabath, N.; Levy-Adam, F.; Younis, A.; Rozales, K.; Meller, A.; Hadar, S.; Soueid-Baumgarten, S.; Shalgi, R. Cellular proteostasis decline in human senescence. Proc. Natl. Acad. Sci. USA 2020, 117, 31902–31913. [Google Scholar] [CrossRef]
- Abbadie, C.; Pluquet, O. Unfolded Protein Response (UPR) Controls Major Senescence Hallmarks. Trends Biochem. Sci. 2020, 45, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, K.M.; Sehgal, P. A Novel Role of Lamins from Genetic Disease to Cancer Biomarkers. Oncol. Rev. 2016, 10, 309. [Google Scholar] [CrossRef]
- Liu, W.; Huang, X.; Luo, W.; Liu, X.; Chen, W. Progerin Inhibits the Proliferation and Migration of Melanoma Cells by Regulating the Expression of Paxillin. OncoTargets Ther. 2024, 17, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.O.; Guimaraes, D.M.; Martins, M.D.; Martins, M.A.T.; Warner, K.A.; Nor, J.E.; Castilho, R.M.; Squarize, C.H. Unlocking the chromatin of adenoid cystic carcinomas using HDAC inhibitors sensitize cancer stem cells to cisplatin and induces tumor senescence. Stem Cell Res. 2017, 21, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Guo, Q.; Chen, M.; Chen, Q.; Xiao, G.; Chen, Z.; Wang, X.; Huang, Y. Silencing p53 inhibits interleukin 10-induced activated hepatic stellate cell senescence and fibrotic degradation in vivo. Exp. Biol. Med. 2021, 246, 447–458. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Yun, M.H.; Davaapil, H.; Brockes, J.P. Recurrent turnover of senescent cells during regeneration of a complex structure. eLife 2015, 4, e05505. [Google Scholar] [CrossRef]
- Gross, O.; Yazdi, A.S.; Thomas, C.J.; Masin, M.; Heinz, L.X.; Guarda, G.; Quadroni, M.; Drexler, S.K.; Tschopp, J. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 2012, 36, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef]
- Su, Y.; Wang, P.; Shen, H.; Sun, Z.; Xu, C.; Li, G.; Tong, T.; Chen, J. The protein kinase D1-mediated classical protein secretory pathway regulates the Ras oncogene-induced senescence response. J. Cell Sci. 2018, 131, jcs207217. [Google Scholar] [CrossRef]
- Kim, K.M.; Noh, J.H.; Bodogai, M.; Martindale, J.L.; Pandey, P.R.; Yang, X.; Biragyn, A.; Abdelmohsen, K.; Gorospe, M. SCAMP4 enhances the senescent cell secretome. Genes. Dev. 2018, 32, 909–914. [Google Scholar] [CrossRef]
- Georgilis, A.; Klotz, S.; Hanley, C.J.; Herranz, N.; Weirich, B.; Morancho, B.; Leote, A.C.; D’Artista, L.; Gallage, S.; Seehawer, M.; et al. PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells. Cancer Cell 2018, 34, 85–102.e9. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.A.; Kim, S.H.; Campisi, J. Reversible manipulation of telomerase expression and telomere length. Implications for the ionizing radiation response and replicative senescence of human cells. J. Biol. Chem. 2002, 277, 28609–28617. [Google Scholar] [CrossRef]
- Shaheen, F.; Grammatopoulos, D.K.; Muller, J.; Zammit, V.A.; Lehnert, H. Extra-nuclear telomerase reverse transcriptase (TERT) regulates glucose transport in skeletal muscle cells. Biochim. Biophys. Acta 2014, 1842, 1762–1769. [Google Scholar] [CrossRef]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Takasugi, M.; Yoshida, Y.; Hara, E.; Ohtani, N. The role of cellular senescence and SASP in tumour microenvironment. FEBS J. 2023, 290, 1348–1361. [Google Scholar] [CrossRef]
- Yu, Y.; Schleich, K.; Yue, B.; Ji, S.; Lohneis, P.; Kemper, K.; Silvis, M.R.; Qutob, N.; van Rooijen, E.; Werner-Klein, M.; et al. Targeting the Senescence-Overriding Cooperative Activity of Structurally Unrelated H3K9 Demethylases in Melanoma. Cancer Cell 2018, 33, 785. [Google Scholar] [CrossRef]
- Zhu, H.; Blake, S.; Kusuma, F.K.; Pearson, R.B.; Kang, J.; Chan, K.T. Oncogene-induced senescence: From biology to therapy. Mech. Ageing Dev. 2020, 187, 111229. [Google Scholar] [CrossRef]
- Zampetidis, C.P.; Papantonis, A.; Gorgoulis, V.G. Escape from senescence: Revisiting cancer therapeutic strategies. Mol. Cell Oncol. 2022, 9, 2030158. [Google Scholar] [CrossRef]
- Chambers, C.R.; Ritchie, S.; Pereira, B.A.; Timpson, P. Overcoming the senescence-associated secretory phenotype (SASP): A complex mechanism of resistance in the treatment of cancer. Mol. Oncol. 2021, 15, 3242–3255. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Ding, J.; Meng, L.H. Oncogene-induced senescence: A double edged sword in cancer. Acta Pharmacol. Sin. 2018, 39, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef] [PubMed]
- Jochems, F.; Thijssen, B.; De Conti, G.; Jansen, R.; Pogacar, Z.; Groot, K.; Wang, L.; Schepers, A.; Wang, C.; Jin, H.; et al. The Cancer SENESCopedia: A delineation of cancer cell senescence. Cell Rep. 2021, 36, 109441. [Google Scholar] [CrossRef]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef]
- Markman, R.L.; Webber, L.P.; Nascimento Filho, C.H.V.; Reis, L.A.; Vargas, P.A.; Lopes, M.A.; Zanella, V.; Martins, M.D.; Squarize, C.H.; Castilho, R.M. Interfering with bromodomain epigenome readers as therapeutic option in mucoepidermoid carcinoma. Cell. Oncol. 2019, 42, 143–155. [Google Scholar] [CrossRef]
- Webber, L.P.; Yujra, V.Q.; Vargas, P.A.; Martins, M.D.; Squarize, C.H.; Castilho, R.M. Interference with the bromodomain epigenome readers drives p21 expression and tumor senescence. Cancer Lett. 2019, 461, 10–20. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2019, 139, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Guan, X.; LaPak, K.M.; Hennessey, R.C.; Yu, C.Y.; Shakya, R.; Zhang, J.; Burd, C.E. Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol. Cancer Res. 2017, 15, 237–249. [Google Scholar] [CrossRef]
- Bollard, J.; Miguela, V.; Ruiz de Galarreta, M.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sanchez, P.; Nguyen, C.B.; et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017, 66, 1286–1296. [Google Scholar] [CrossRef]
- Muhleder, S.; Fernandez-Chacon, M.; Garcia-Gonzalez, I.; Benedito, R. Endothelial sprouting, proliferation, or senescence: Tipping the balance from physiology to pathology. Cell Mol. Life Sci. 2021, 78, 1329–1354. [Google Scholar] [CrossRef]
- Jin, P.; Duan, X.; Li, L.; Zhou, P.; Zou, C.G.; Xie, K. Cellular senescence in cancer: Molecular mechanisms and therapeutic targets. MedComm 2024, 5, e542. [Google Scholar] [CrossRef]
- Ge, M.; Hu, L.; Ao, H.; Zi, M.; Kong, Q.; He, Y. Senolytic targets and new strategies for clearing senescent cells. Mech. Ageing Dev. 2021, 195, 111468. [Google Scholar] [CrossRef]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. eBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [PubMed]
- Chrienova, Z.; Rysanek, D.; Oleksak, P.; Stary, D.; Bajda, M.; Reinis, M.; Mikyskova, R.; Novotny, O.; Andrys, R.; Skarka, A.; et al. Discovery of small molecule mechanistic target of rapamycin inhibitors as anti-aging and anti-cancer therapeutics. Front. Aging Neurosci. 2022, 14, 1048260. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, L.; Qu, C.; Chen, L.; Geng, Y.; Cheng, C.; Yu, S.; Wang, D.; Yang, L.; Meng, Z.; et al. Kaempferol induces ROS-dependent apoptosis in pancreatic cancer cells via TGM2-mediated Akt/mTOR signaling. BMC Cancer 2021, 21, 396. [Google Scholar] [CrossRef]
- Ahmadinejad, F.; Bos, T.; Hu, B.; Britt, E.; Koblinski, J.; Souers, A.J.; Leverson, J.D.; Faber, A.C.; Gewirtz, D.A.; Harada, H. Senolytic-Mediated Elimination of Head and Neck Tumor Cells Induced Into Senescence by Cisplatin. Mol. Pharmacol. 2022, 101, 168–180. [Google Scholar] [CrossRef]
- Witte, I.; Altenhofer, S.; Wilgenbus, P.; Amort, J.; Clement, A.M.; Pautz, A.; Li, H.; Forstermann, U.; Horke, S. Beyond reduction of atherosclerosis: PON2 provides apoptosis resistance and stabilizes tumor cells. Cell Death Dis. 2011, 2, e112. [Google Scholar] [CrossRef]
- Kruger, M.; Pabst, A.M.; Al-Nawas, B.; Horke, S.; Moergel, M. Paraoxonase-2 (PON2) protects oral squamous cell cancer cells against irradiation-induced apoptosis. J. Cancer Res. Clin. Oncol. 2015, 141, 1757–1766. [Google Scholar] [CrossRef]
- Kruger, M.; Amort, J.; Wilgenbus, P.; Helmstadter, J.P.; Grechowa, I.; Ebert, J.; Tenzer, S.; Moergel, M.; Witte, I.; Horke, S. The anti-apoptotic PON2 protein is Wnt/beta-catenin-regulated and correlates with radiotherapy resistance in OSCC patients. Oncotarget 2016, 7, 51082–51095. [Google Scholar] [CrossRef]
- Kamal, M.V.; Prabhu, K.; Sharan, K.; Pai, A.; Chakrabarty, S.; Damerla, R.R.; Shetty, P.S.; Belle, V.S.; Rao, M.; Kumar, N.A.N. Investigation of the Molecular Mechanisms of Paraoxonase-2 Mediated Radiotherapy and Chemotherapy Resistance in Oral Squamous Cell Carcinoma. Clin. Transl. Sci. 2025, 18, e70201. [Google Scholar] [CrossRef]
- Campagna, R.; Belloni, A.; Pozzi, V.; Salvucci, A.; Notarstefano, V.; Togni, L.; Mascitti, M.; Sartini, D.; Giorgini, E.; Salvolini, E.; et al. Role Played by Paraoxonase-2 Enzyme in Cell Viability, Proliferation and Sensitivity to Chemotherapy of Oral Squamous Cell Carcinoma Cell Lines. Int. J. Mol. Sci. 2022, 24, 338. [Google Scholar] [CrossRef]
- Belloni, A.; Campagna, R.; Notarstefano, V.; Pozzi, V.; Orilisi, G.; Pompei, V.; Togni, L.; Mascitti, M.; Sartini, D.; Giorgini, E.; et al. Deepening Cisplatin sensitivity on Oral Squamous cell Carcinoma cell lines after PON2 knockdown: A FTIRM investigation. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2025, 330, 125726. [Google Scholar] [CrossRef]
- Woo, S.H.; Yang, L.P.; Chuang, H.C.; Fitzgerald, A.; Lee, H.Y.; Pickering, C.; Myers, J.N.; Skinner, H.D. Down-regulation of malic enzyme 1 and 2: Sensitizing head and neck squamous cell carcinoma cells to therapy-induced senescence. Head Neck 2016, 38 (Suppl. 1), E934–E940. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Mi, J.; Ruan, X.; Zhang, G.; Pan, Y.; Wang, R. Identification and Analysis of Senescence-Related Genes in Head and Neck Squamous Cell Carcinoma by a Comprehensive Bioinformatics Approach. Mediators Inflamm. 2022, 2022, 4007469. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, C.C.; Sun, H.C.; Li, Q.; Hu, J.D.; Jiang, T.; Zhou, S. Identification of several senescence-associated genes signature in head and neck squamous cell carcinoma. J. Clin. Lab. Anal. 2022, 36, e24555. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lin, J.; Wen, Y.; Lan, B.; Xiong, J.; Fu, Y.; Chen, Y.; Chen, C.B. A senescence-related lncRNA signature predicts prognosis and reflects immune landscape in HNSCC. Oral. Oncol. 2024, 149, 106659. [Google Scholar] [CrossRef]
- Morales-Valencia, J.; David, G. The Contribution of Physiological and Accelerated Aging to Cancer Progression Through Senescence-Induced Inflammation. Front. Oncol. 2021, 11, 747822. [Google Scholar] [CrossRef]
- Skwarska, A.; Konopleva, M. BCL-xL Targeting to Induce Apoptosis and to Eliminate Chemotherapy-Induced Senescent Tumor Cells: From Navitoclax to Platelet-Sparing BCL-xL PROTACs. Cancer Res. 2023, 83, 3501–3503. [Google Scholar] [CrossRef]
- Gadsden, N.J.; Fulcher, C.D.; Li, D.; Shrivastava, N.; Thomas, C.; Segall, J.E.; Prystowsky, M.B.; Schlecht, N.F.; Gavathiotis, E.; Ow, T.J. Palbociclib Renders Human Papilloma Virus-Negative Head and Neck Squamous Cell Carcinoma Vulnerable to the Senolytic Agent Navitoclax. Mol. Cancer Res. 2021, 19, 862–873. [Google Scholar] [CrossRef]
- Gu, Z.; Shi, C.; Li, J.; Han, Y.; Sun, B.; Zhang, W.; Wu, J.; Zhou, G.; Ye, W.; Li, J.; et al. Palbociclib-based high-throughput combination drug screening identifies synergistic therapeutic options in HPV-negative head and neck squamous cell carcinoma. BMC Med. 2022, 20, 175. [Google Scholar] [CrossRef]
- Togni, L.; Mascitti, M.; Sartini, D.; Campagna, R.; Pozzi, V.; Salvolini, E.; Offidani, A.; Santarelli, A.; Emanuelli, M. Nicotinamide N-Methyltransferase in Head and Neck Tumors: A Comprehensive Review. Biomolecules 2021, 11, 1594. [Google Scholar] [CrossRef]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef]
- Ostrowska, K.; Niewinski, P.; Piotrowski, I.; Ostapowicz, J.; Koczot, S.; Suchorska, W.M.; Golusinski, P.; Masternak, M.M.; Golusinski, W. Senescence in head and neck squamous cell carcinoma: Relationship between senescence-associated secretory phenotype (SASP) mRNA expression level and clinicopathological features. Clin. Transl. Oncol. 2024, 26, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Brauning, A.; Rae, M.; Zhu, G.; Fulton, E.; Admasu, T.D.; Stolzing, A.; Sharma, A. Aging of the Immune System: Focus on Natural Killer Cells Phenotype and Functions. Cells 2022, 11, 1017. [Google Scholar] [CrossRef] [PubMed]
- Solana, R.; Campos, C.; Pera, A.; Tarazona, R. Shaping of NK cell subsets by aging. Curr. Opin. Immunol. 2014, 29, 56–61. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palomares, B.H.; Martins, M.D.; Martins, M.A.T.; Squarize, C.H.; Castilho, R.M. Future Perspectives in Senescence-Based Therapies for Head and Neck Cancer. Cancers 2025, 17, 1965. https://doi.org/10.3390/cancers17121965
Palomares BH, Martins MD, Martins MAT, Squarize CH, Castilho RM. Future Perspectives in Senescence-Based Therapies for Head and Neck Cancer. Cancers. 2025; 17(12):1965. https://doi.org/10.3390/cancers17121965
Chicago/Turabian StylePalomares, Bruna Haddad, Manoela Domingues Martins, Marco Antonio Trevizani Martins, Cristiane Helena Squarize, and Rogerio Moraes Castilho. 2025. "Future Perspectives in Senescence-Based Therapies for Head and Neck Cancer" Cancers 17, no. 12: 1965. https://doi.org/10.3390/cancers17121965
APA StylePalomares, B. H., Martins, M. D., Martins, M. A. T., Squarize, C. H., & Castilho, R. M. (2025). Future Perspectives in Senescence-Based Therapies for Head and Neck Cancer. Cancers, 17(12), 1965. https://doi.org/10.3390/cancers17121965