
RNA Sequencing Identifies WT1 Overexpression as a Predictor of Poor Outcomes in Acute Myeloid Leukemia

 ,
,  , , , , , add
Show full author list
, , , , , add
Show full author list
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
- Patients aged 18 years or older of either gender.
- De novo AML patients who had not received prior treatment.
- Patients with any concurrent solid or hematologic malignancy.
- Individuals with a history of chemotherapy or radiotherapy.
- Individuals who declined informed consent or refused participation.
- Patients with specific hematologic conditions such as APL, Down syndrome-associated myeloid neoplasms, MDS, t-AML, or myeloid sarcoma.
2.2. Bone Marrow Mononuclear Cells (BMMCs) Isolation
2.3. RNA Extraction and Quantification
2.4. Discovery Phase: Comprehensive Analysis of WT1 Expression via RNA Sequencing
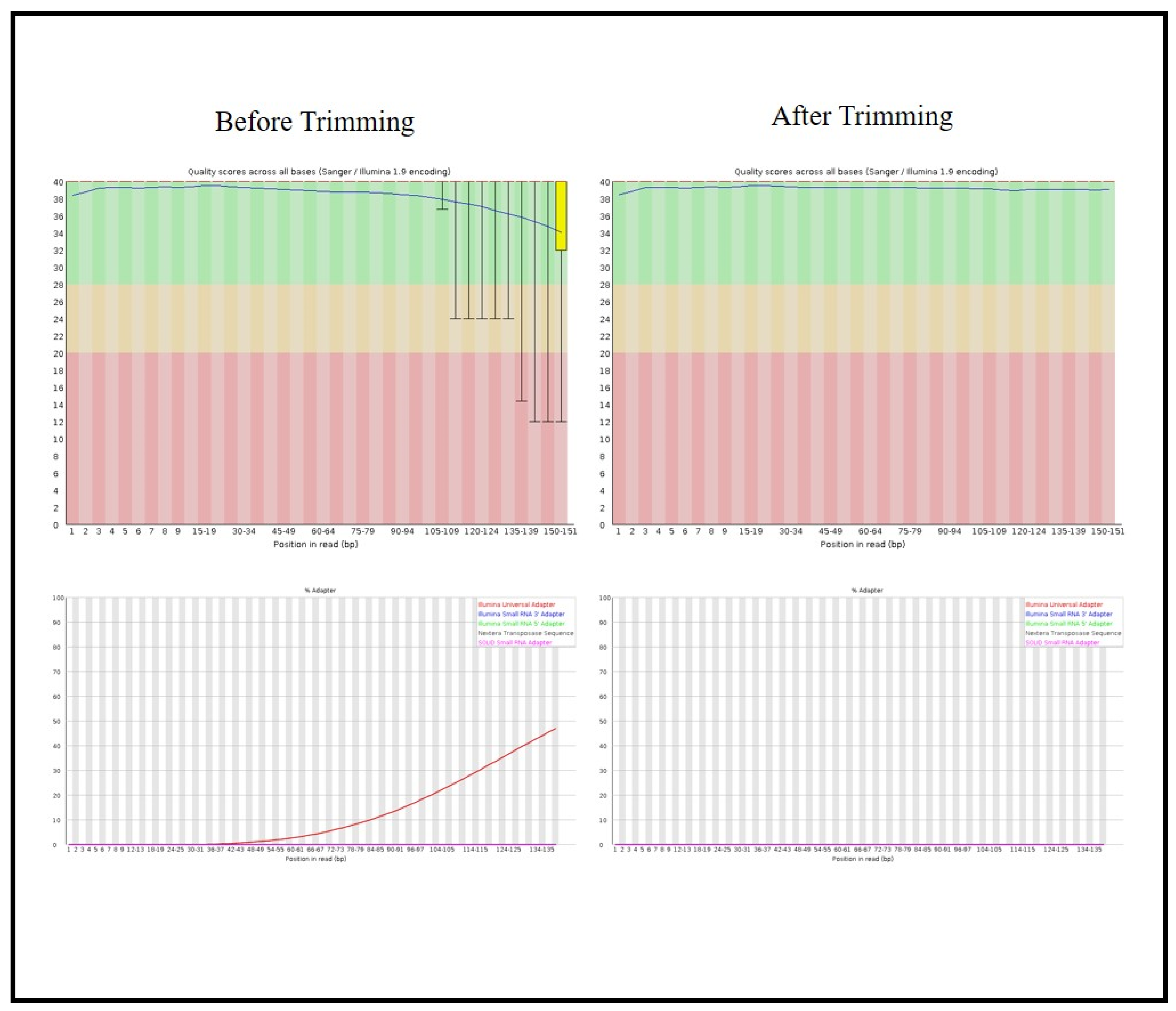
2.5. Library Preparation and Sequencing
2.6. Bioinformatics Analysis
2.7. Validation of WT1 Gene Expression Profiling by Quantitative RT-PCR
2.8. Statistical Analysis
3. Results
3.1. Patient Characterstics
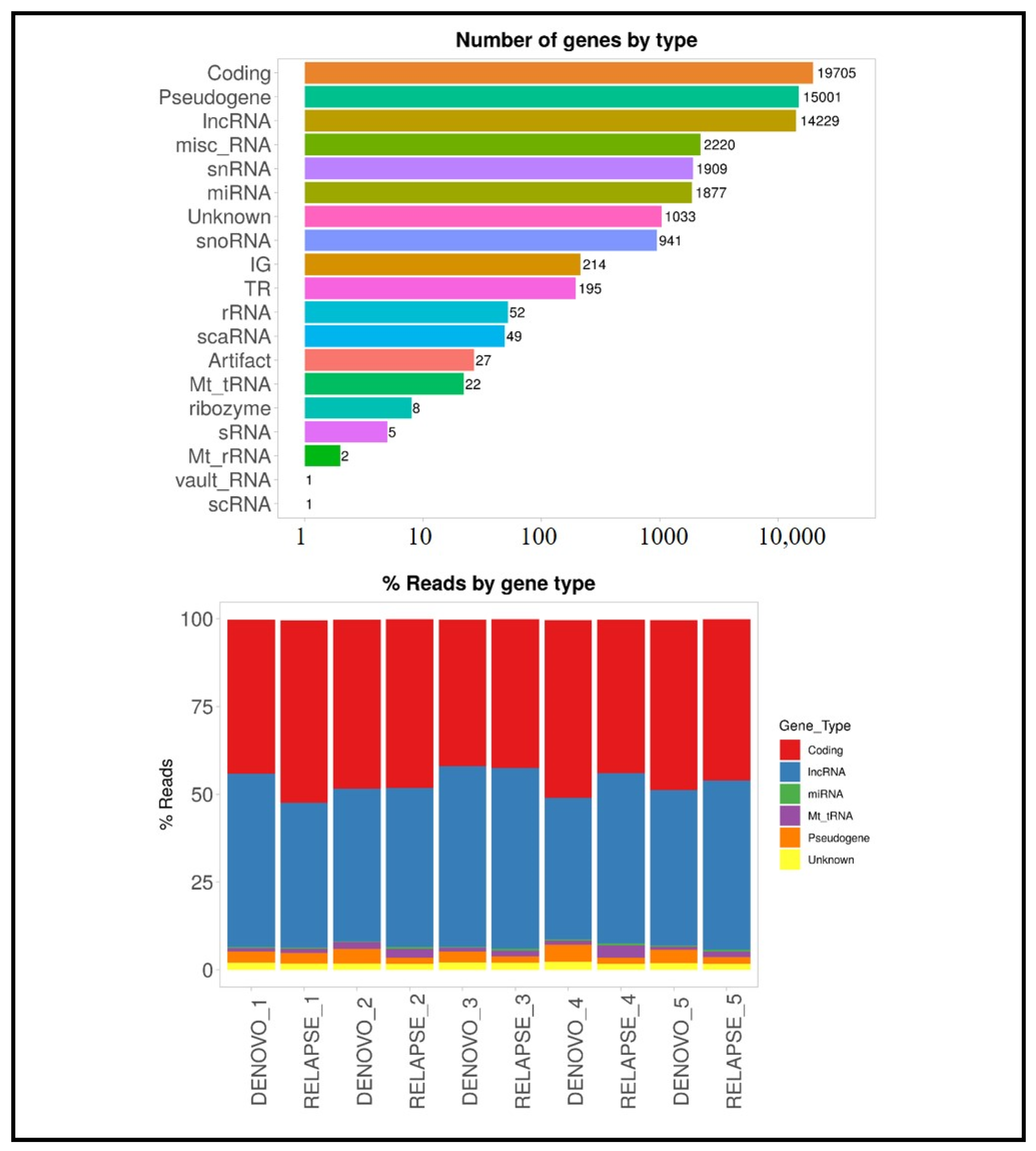
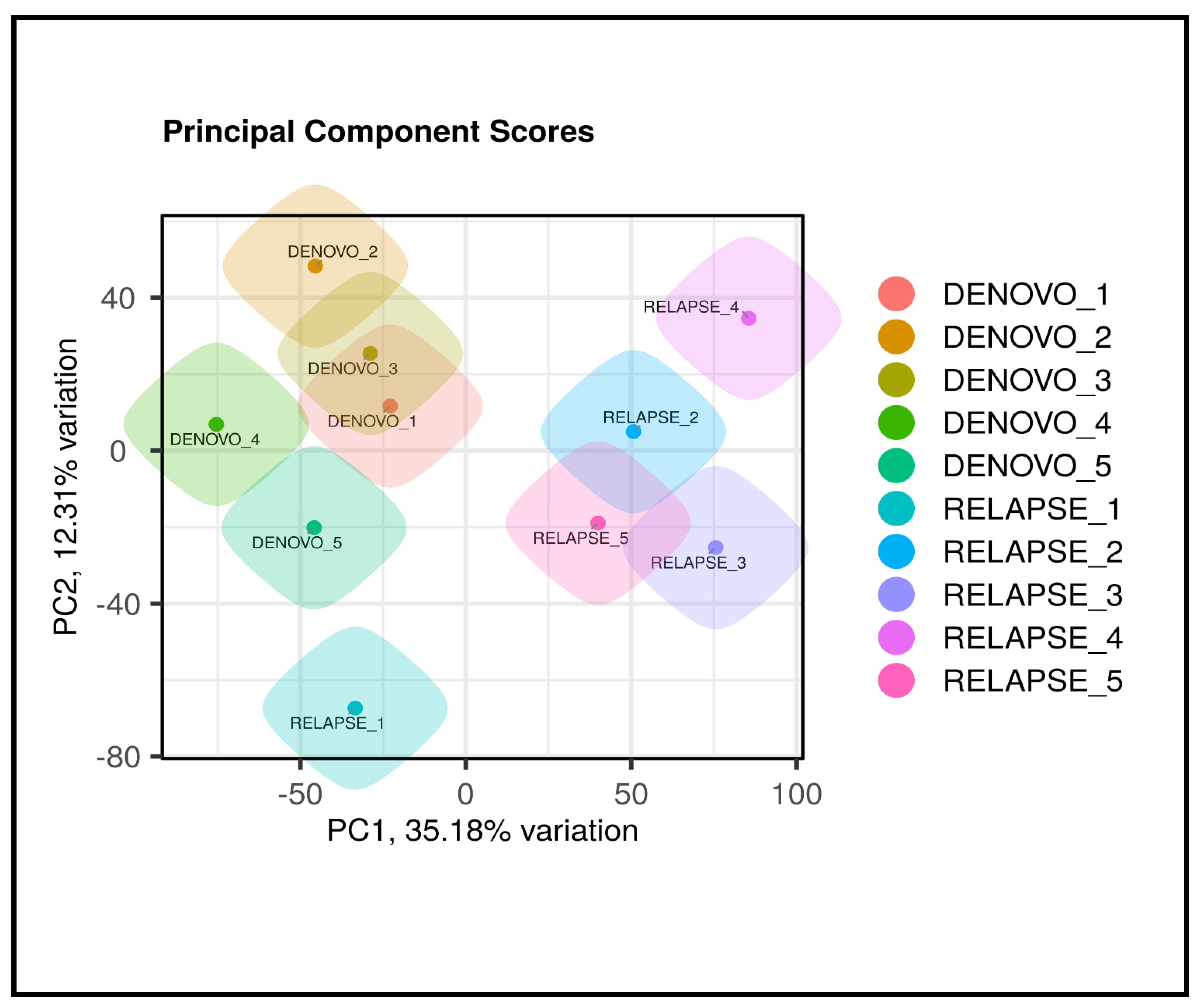
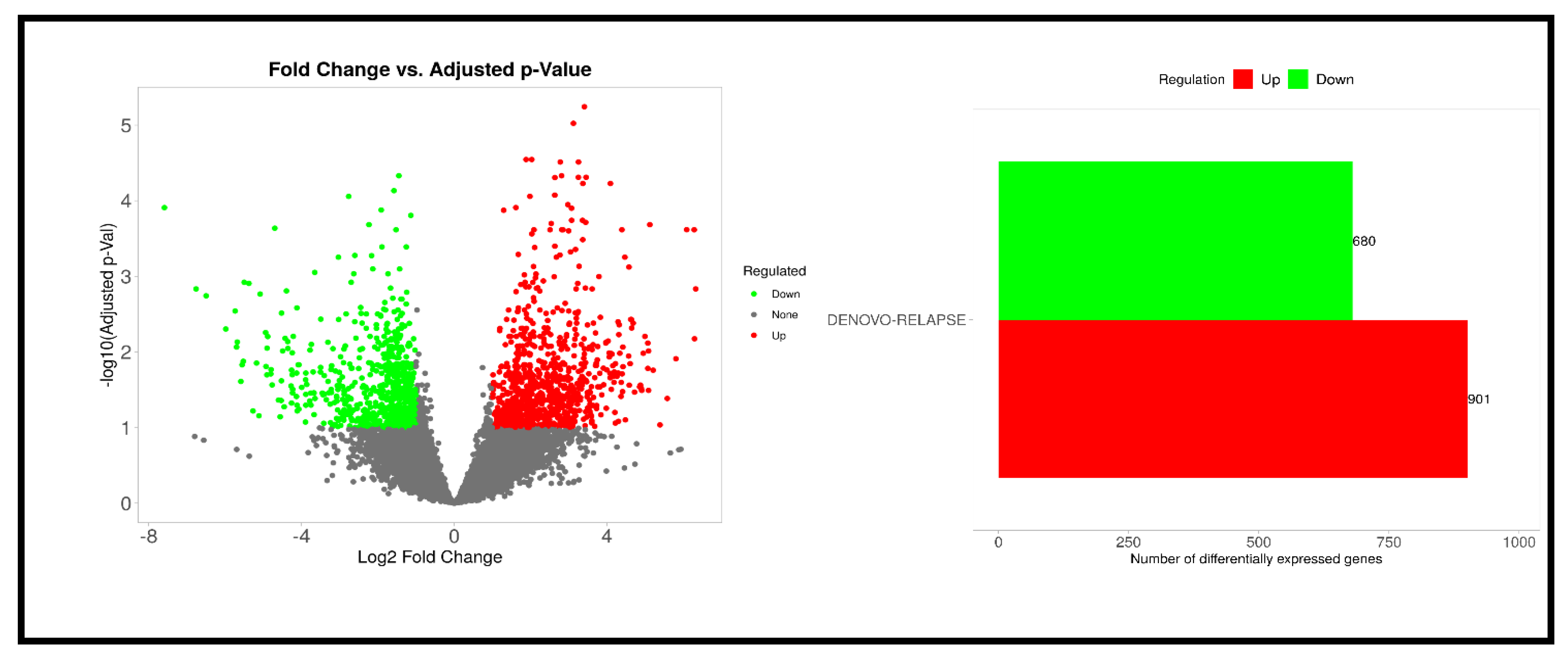
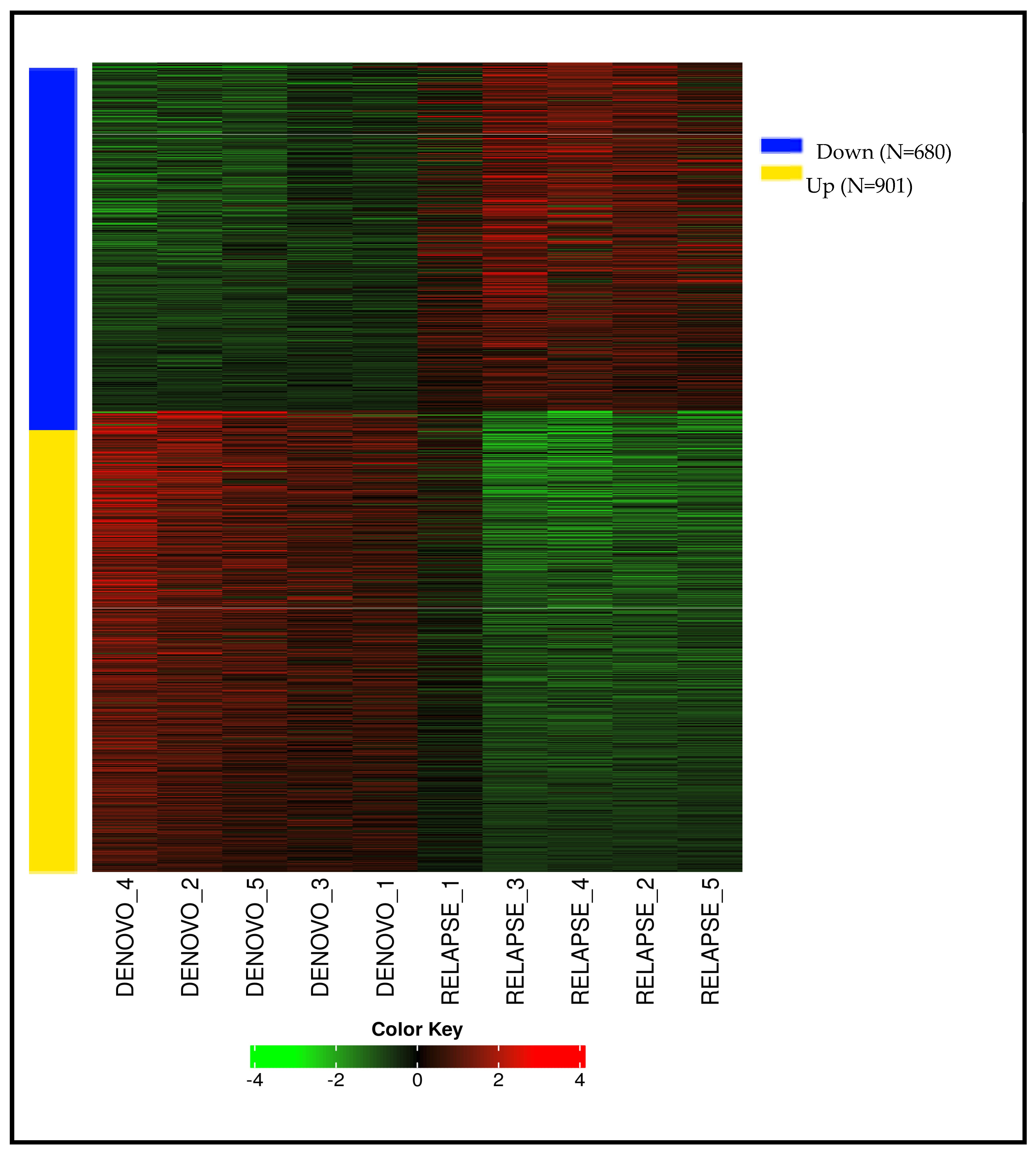
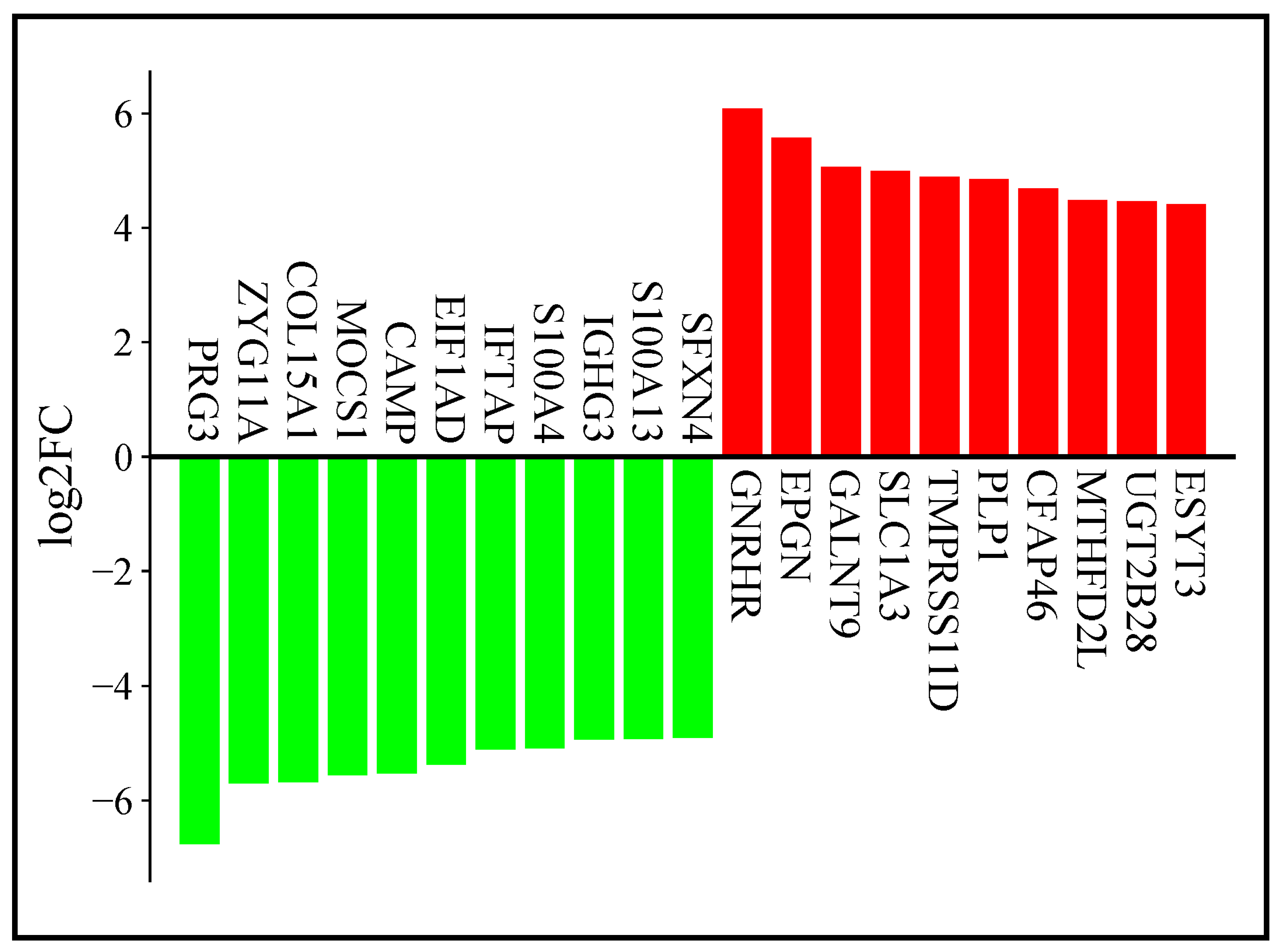
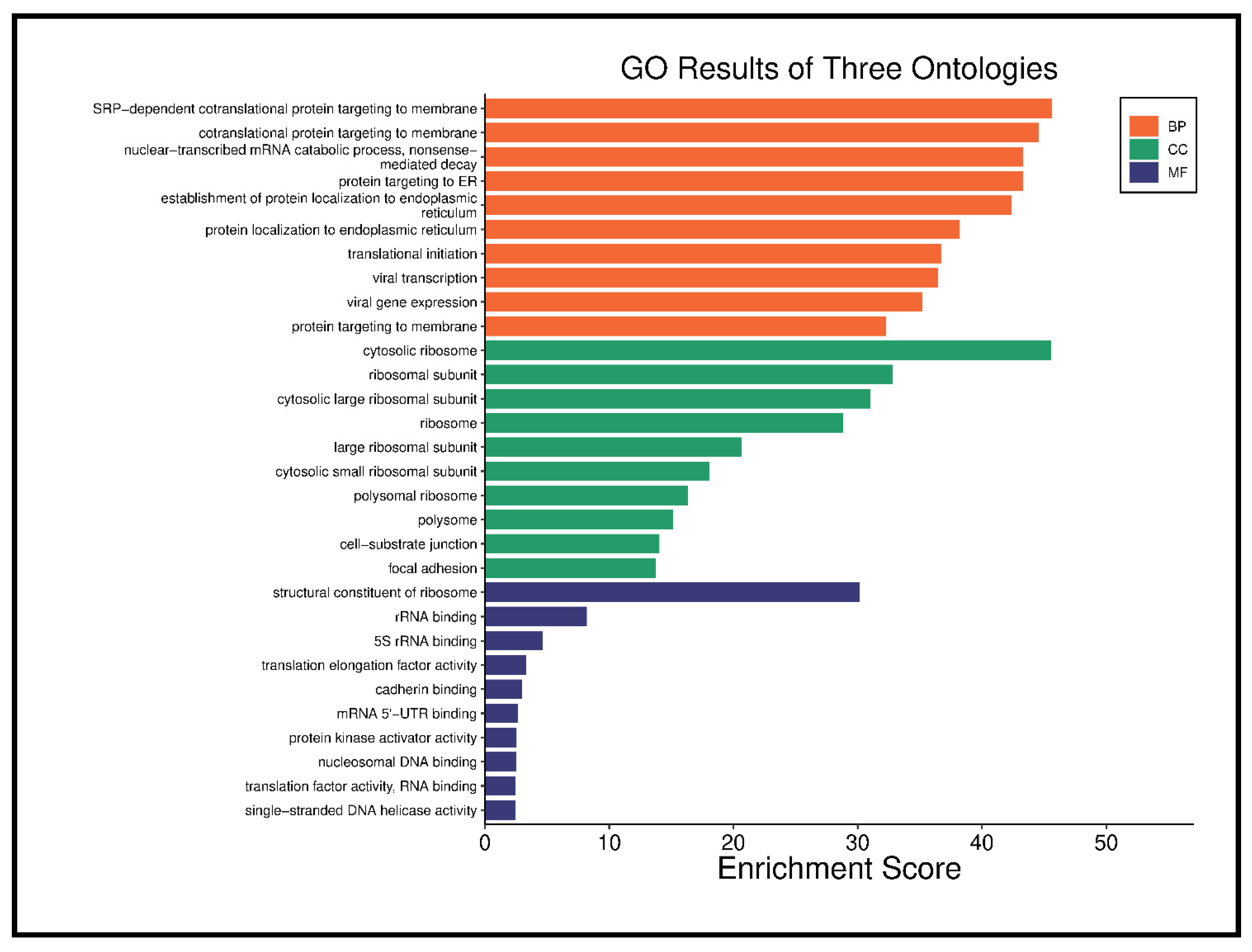
3.2. RNA Sequencing Results
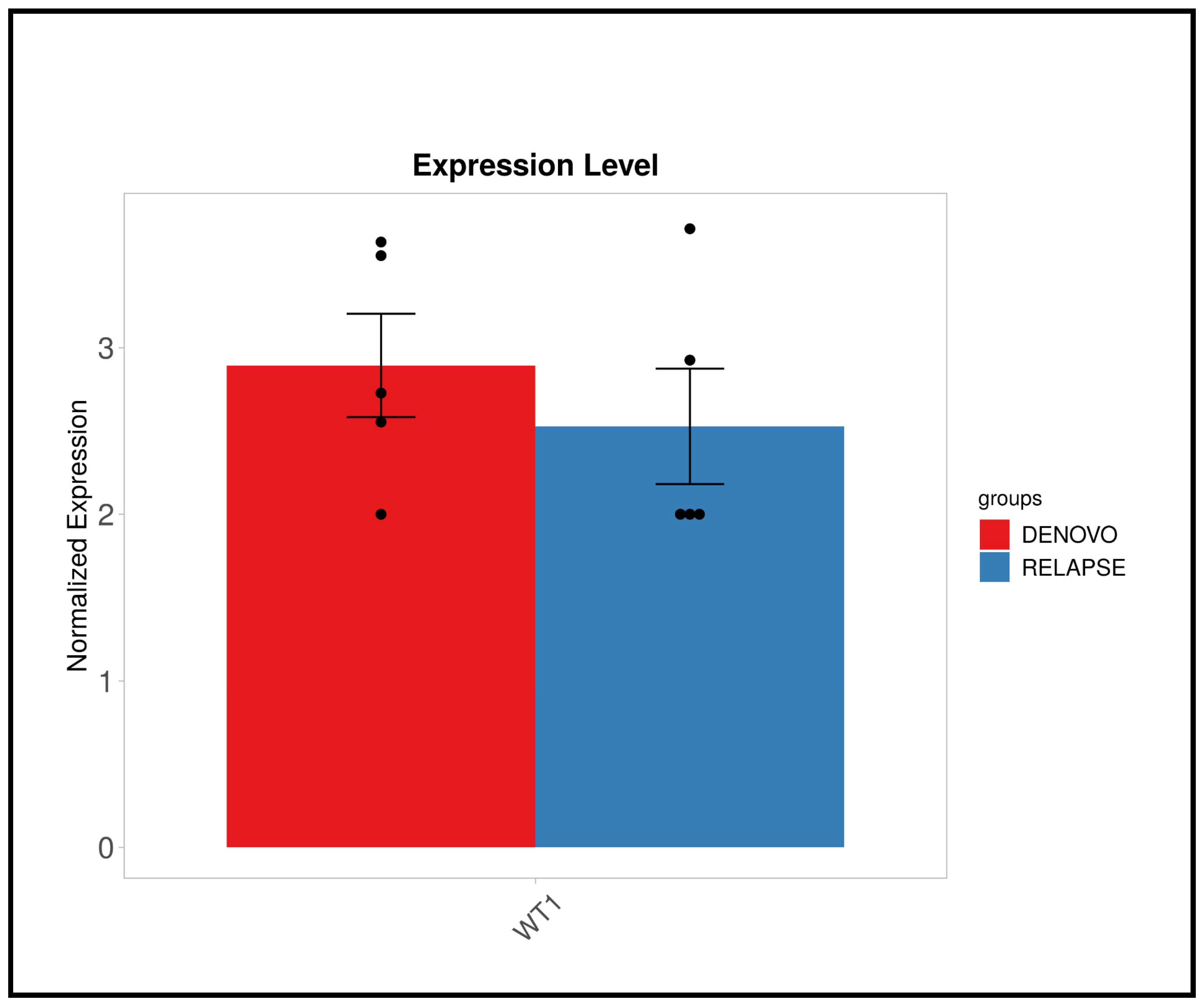
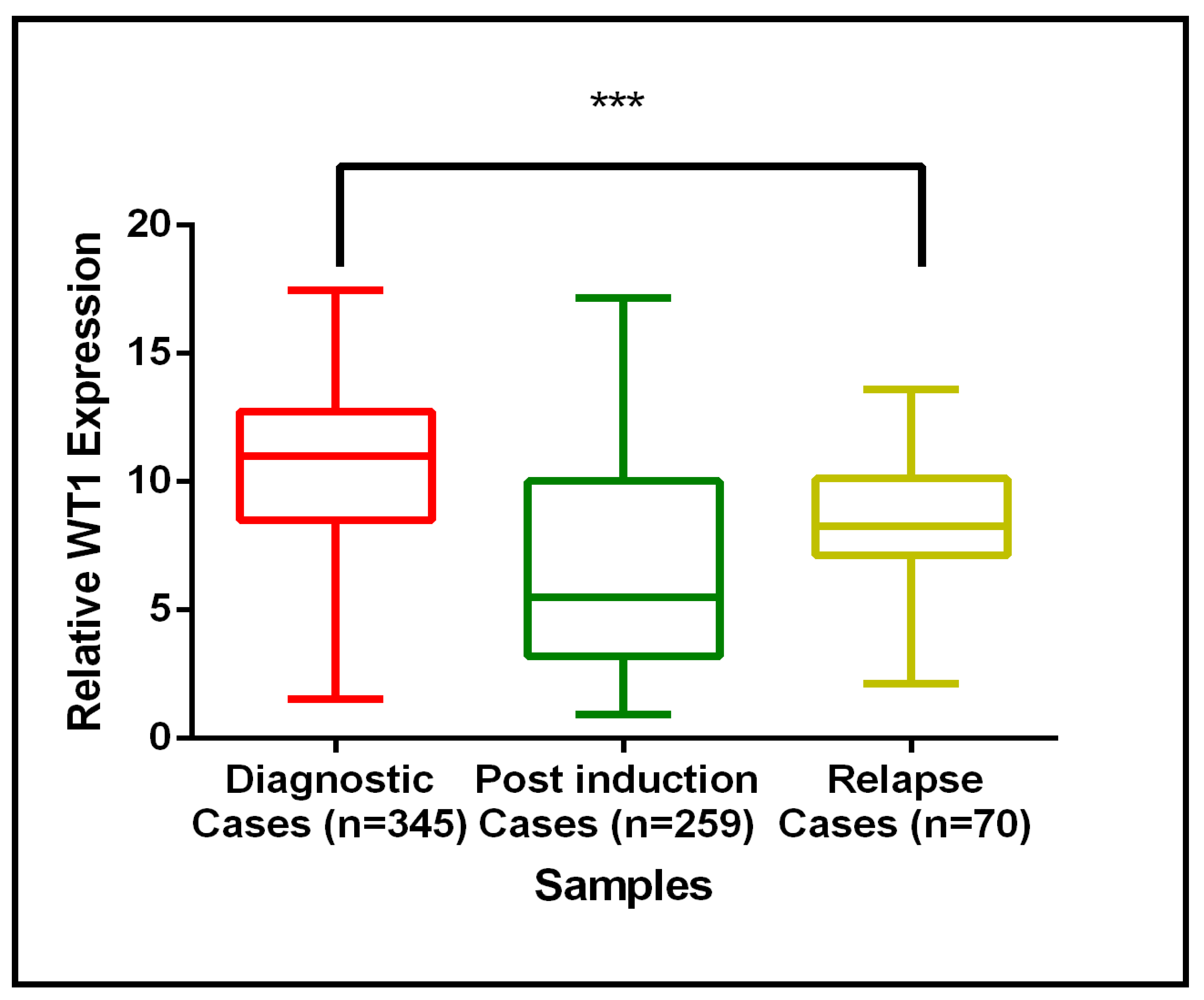
3.3. Validation Result of WT1 Gene Expression Using qRT-PCR
3.4. Association of WT1 mRNA Expression with Clinical and Hematological Parameters in AML Patients
3.5. Prognostic Significance of WT1 Gene Expression in AML: Univariate and Multivariate Analysis
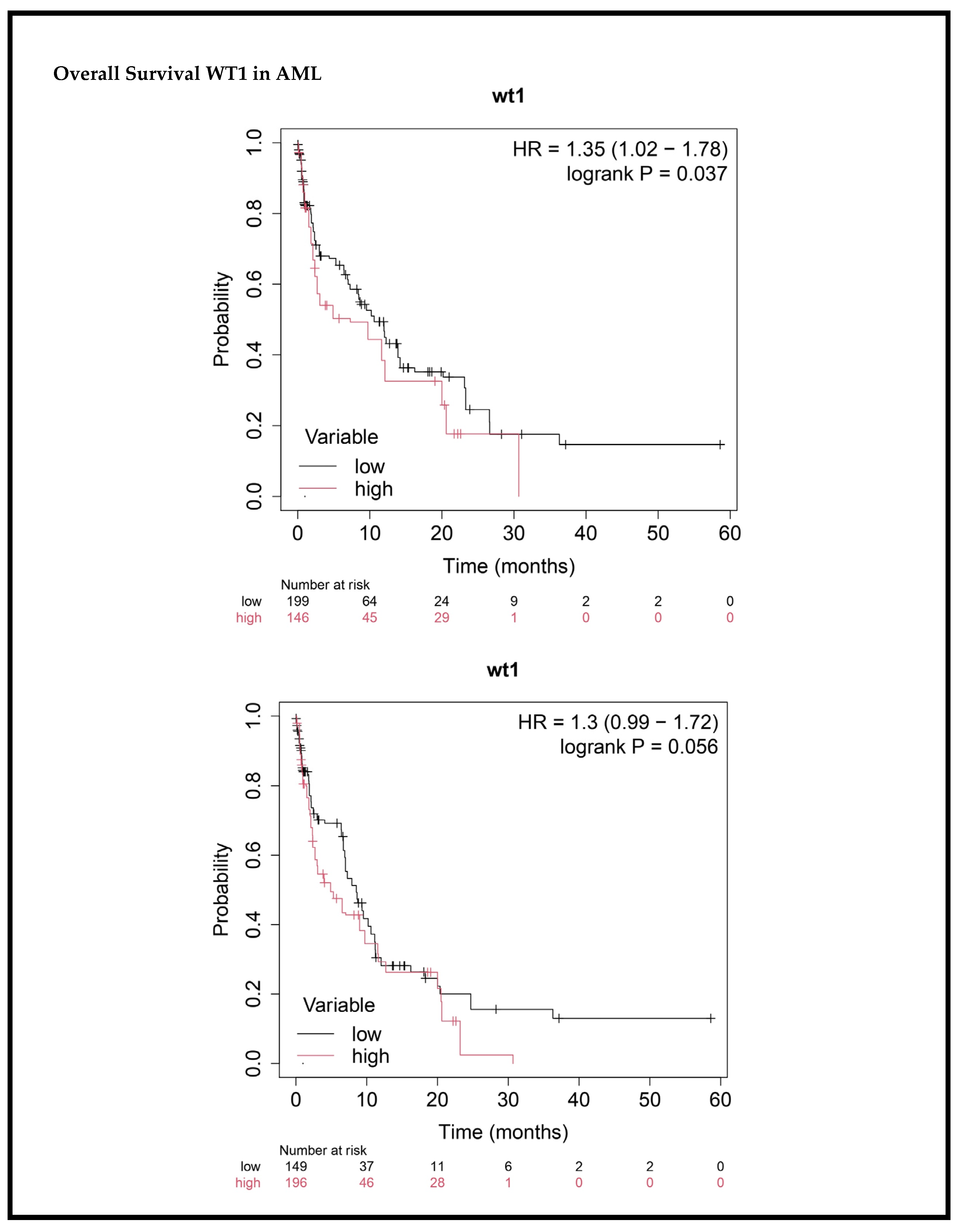
3.6. Impact of WT1 Gene Expression on Survival Outcomes in AML Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute Myeloid Leukemia |
| WT1 | Wilms’ Tumor 1 |
| BM | Bone Marrow |
| PB | Peripheral Blood |
| OS | Overall Survival |
| EFS | Event-Free Survival |
| FAB | French-American-British |
| WHO | World Health Organization |
| CR | Complete Remission |
| PR | Partial Remission |
| FAB | French-American-British |
| WHO | World Health Organization |
| MDS | Myelodysplastic Syndromes |
| MPN | Myeloproliferative Neoplasms |
| t-AML | Therapy-Related AML |
| EMT | Epithelial-Mesenchymal Transitions |
| ELN | European Leukemia Net |
| APL | Acute Promyelocytic Leukemia |
| MRD | Minimal Residual Disease |
| AHSCT | Allogeneic Hematopoietic Stem Cell Transplantation |
| ROC | Receiver Operating Characteristic |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No | Sample | Read Orientation | Mean Read Quality (Phred Score) | Number of Reads | % GC | % Q > 30 | Number of Bases (MB) | Mean Read Length (bp) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1I | R1 | 38.74 | 30,189,372 | 49.49 | 94.04 | 4558.6 | 151.0 |
| R2 | 38.43 | 30,189,372 | 49.49 | 92.66 | 4558.6 | 151.0 | ||
| 2 | 1III | R1 | 38.21 | 47,726,631 | 46.96 | 91.73 | 7206.72 | 151.0 |
| R2 | 38.14 | 47,726,631 | 46.92 | 91.32 | 7206.72 | 151.0 | ||
| 3 | 2I | R1 | 38.83 | 23,990,789 | 49.75 | 94.42 | 3622.61 | 151.0 |
| R2 | 38.55 | 23,990,789 | 49.4 | 93.17 | 3622.61 | 151.0 | ||
| 4 | 2III | R1 | 38.59 | 29,223,493 | 52.74 | 93.37 | 4412.75 | 151.0 |
| R2 | 38.33 | 29,223,493 | 52.91 | 92.22 | 4412.75 | 151.0 | ||
| 5 | 3I | R1 | 38.7 | 25,487,420 | 50.16 | 93.88 | 3848.6 | 151.0 |
| R2 | 38.15 | 25,487,420 | 50.35 | 91.62 | 3848.6 | 151.0 | ||
| 6 | 3III | R1 | 38.79 | 27,077,096 | 53.5 | 94.26 | 4088.64 | 151.0 |
| R2 | 38.47 | 27,077,096 | 53.28 | 92.82 | 4088.64 | 151.0 | ||
| 7 | 4I | R1 | 38.58 | 29,404,952 | 47.95 | 93.41 | 4440.15 | 151.0 |
| R2 | 38.36 | 29,404,952 | 47.86 | 92.39 | 4440.15 | 151.0 | ||
| 8 | 4III | R1 | 38.74 | 28,882,135 | 53.22 | 94.02 | 4361.2 | 151.0 |
| R2 | 38.34 | 28,882,135 | 53.12 | 92.27 | 4361.2 | 151.0 | ||
| 9 | 5I | R1 | 38.78 | 28,208,542 | 48.73 | 94.22 | 4259.49 | 151.0 |
| R2 | 38.42 | 28,208,542 | 48.89 | 92.66 | 4259.49 | 151.0 | ||
| 10 | 5III | R1 | 38.75 | 26,060,227 | 52.05 | 94.09 | 3935.09 | 151.0 |
| R2 | 38.34 | 26,060,227 | 51.95 | 92.23 | 3935.09 | 151.0 |
Appendix B


References
- Deschler, B.; Lübbert, M. Acute myeloid leukemia: Epidemiology and etiology. Cancer 2006, 107, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.; Rahul, E.; Gupta, A.K.; Meena, J.P.; Chopra, A.; Ranjan, A.; Hussain, S.; Rath, G.; Tanwar, P. Molecular update on biology of Wilms Tumor 1 gene and its applications in acute myeloid leukemia. Am. J. Blood Res. 2020, 10, 151–160. [Google Scholar] [PubMed]
- Juliusson, G.; Antunovic, P.; Derolf, A.; Lehmann, S.; Möllgård, L.; Stockelberg, D.; Tidefelt, U.; Wahlin, A.; Höglund, M. Age and acute myeloid leukemia: Real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood 2009, 113, 4179–4187. [Google Scholar] [CrossRef] [PubMed]
- McNerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-Related Myeloid Neoplasms: When Genetics and Environment Collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef]
- Goel, H.; Rahul, E.; Gupta, I.; Chopra, A.; Ranjan, A.; Gupta, A.K.; Meena, J.P.; Viswanathan, G.K.; Bakhshi, S.; Misra, A.; et al. Molecular and genomic landscapes in secondary & therapy related acute myeloid leukemia. Am. J. Blood Res. 2021, 11, 472–497. [Google Scholar]
- Leonard, J.P.; Martin, P.; Roboz, G.J. Practical Implications of the 2016 Revision of the World Health Organization Classification of Lymphoid and Myeloid Neoplasms and Acute Leukemia. J. Clin. Oncol. 2017, 35, 2708–2715. [Google Scholar] [CrossRef]
- Adès, L.; Itzykson, R.; Fenaux, P. Myelodysplastic syndromes. Lancet 2014, 383, 2239–2252. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposal for the recognition of minimally differentiated acute myeloid leukaemia (AML-MO). Br. J. Haematol. 1991, 78, 325–329. [Google Scholar] [CrossRef]
- Peters, J.M.; Ansari, M.Q. Multiparameter flow cytometry in the diagnosis and management of acute leukemia. Arch. Pathol. Lab. Med. 2011, 135, 44–54. [Google Scholar] [CrossRef]
- Betz, B.L.; Hess, J.L. Acute myeloid leukemia diagnosis in the 21st century. Arch. Pathol. Lab. Med. 2010, 134, 1427–1433. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Plass, C.; Oakes, C.; Blum, W.; Marcucci, G. Epigenetics in acute myeloid leukemia. Semin. Oncol. 2008, 35, 378–387. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Yang, H.; Kuang, S.-Q.; O’Brien, S.; Thomas, D.; Kantarjian, H. Epigenetics of acute lymphocytic leukemia. Semin. Hematol. 2009, 46, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.W.; Gilliland, D.G. Leukemia stem cells. Semin. Cancer Biol. 2010, 20, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Jordan, C.T. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017, 129, 1627–1635. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Sasaki, K.; Ravandi, F.; Kadia, T.M.; DiNardo, C.D.; Short, N.J.; Borthakur, G.; Jabbour, E.; Kantarjian, H.M. De novo acute myeloid leukemia: A population-based study of outcome in the United States based on the Surveillance, Epidemiology, and End Results (SEER) database, 1980 to 2017. Cancer 2021, 127, 2049–2061. [Google Scholar] [CrossRef]
- Dinner, S.N.; Giles, F.J.; Altman, J.K. New strategies for relapsed acute myeloid leukemia: Fertile ground for translational research. Curr. Opin. Hematol. 2014, 21, 79–86. [Google Scholar] [CrossRef]
- Jimenez-Chillon, C.; Dillon, R.; Russell, N. Optimal Post-Remission Consolidation Therapy in Patients with AML. Acta Haematol. 2023, 147, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.G.E. Transcriptional regulation by WT1 in development. Curr. Opin. Genet. Dev. 2005, 15, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Essafi, A.; Hastie, N.D. WT1 the oncogene: A tale of death and HtrA. Mol. Cell 2010, 37, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Belluti, S.; Rigillo, G.; Imbriano, C. Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells 2020, 9, 760. [Google Scholar] [CrossRef]
- Hosen, N.; Sonoda, Y.; Oji, Y.; Kimura, T.; Minamiguchi, H.; Tamaki, H.; Kawakami, M.; Asada, M.; Kanato, K.; Motomura, M.; et al. Very low frequencies of human normal CD34+ haematopoietic progenitor cells express the Wilms’ tumour gene WT1 at levels similar to those in leukaemia cells. Br. J. Haematol. 2002, 116, 409–420. [Google Scholar] [CrossRef]
- Wang, D.; Horton, J.R.; Zheng, Y.; Blumenthal, R.M.; Zhang, X.; Cheng, X. Role for first zinc finger of WT1 in DNA sequence specificity: Denys–Drash syndrome-associated WT1 mutant in ZF1 enhances affinity for a subset of WT1 binding sites. Nucleic Acids Res. 2018, 46, 3864–3877. [Google Scholar] [CrossRef]
- Hastie, N.D. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development 2017, 144, 2862–2872. [Google Scholar] [CrossRef]
- Madden, S.L.; Cook, D.M.; Morris, J.F.; Gashler, A.; Sukhatme, V.P.; Rauscher, F.J. Transcriptional repression mediated by the WT1 Wilms tumor gene product. Science 1991, 253, 1550–1553. [Google Scholar] [CrossRef]
- Artibani, M.; Sims, A.H.; Slight, J.; Aitken, S.; Thornburn, A.; Muir, M.; Brunton, V.G.; Del-Pozo, J.; Morrison, L.R.; Katz, E.; et al. WT1 expression in breast cancer disrupts the epithelial/mesenchymal balance of tumour cells and correlates with the metabolic response to docetaxel. Sci. Rep. 2017, 7, 45255. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, F.; Wu, H.; Liu, J.; Zong, B.; Xu, C.; Jiang, J. Wilms’ tumor 1 (WT1) expression and prognosis in solid cancer patients: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 8924. [Google Scholar] [CrossRef]
- Pronier, E.; Bowman, R.L.; Ahn, J.; Glass, J.; Kandoth, C.; Merlinsky, T.R.; Whitfield, J.T.; Durham, B.H.; Gruet, A.; Hanasoge Somasundara, A.V.; et al. Genetic and epigenetic evolution as a contributor to WT1-mutant leukemogenesis. Blood 2018, 132, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Haber, D.A.; Sohn, R.L.; Buckler, A.J.; Pelletier, J.; Call, K.M.; Housman, D.E. Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc. Natl. Acad. Sci. USA 1991, 88, 9618–9622. [Google Scholar] [CrossRef]
- Reddy, J.C.; Hosono, S.; Licht, J.D. The transcriptional effect of WT1 is modulated by choice of expression vector. J. Biol. Chem. 1995, 270, 29976–29982. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Qiu, Q.Q.; Huang, J.; Gurrieri, M.; Deuel, T.F. Products of alternatively spliced transcripts of the Wilms’ tumor suppressor gene, wt1, have altered DNA binding specificity and regulate transcription in different ways. Oncogene 1995, 10, 415–422. [Google Scholar]
- Miwa, H.; Beran, M.; Saunders, G.F. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia 1992, 6, 405–409. [Google Scholar]
- Chau, Y.-Y.; Hastie, N.D. The role of Wt1 in regulating mesenchyme in cancer, development, and tissue homeostasis. Trends Genet. 2012, 28, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Sugiyama, H.; Ogawa, H.; Nakagawa, M.; Yamagami, T.; Miwa, H.; Kita, K.; Hiraoka, A.; Masaoka, T.; Nasu, K. WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood 1994, 84, 3071–3079. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Ghio, F.; Guerrini, F.; Ciabatti, E.; Grassi, S.; Ferreri, M.I.; Petrini, M. WT1 expression levels at diagnosis could predict long-term time-to-progression in adult patients affected by acute myeloid leukaemia and myelodysplastic syndromes. Br. J. Haematol. 2010, 149, 451–454. [Google Scholar] [CrossRef]
- Nomdedéu, J.F.; Hoyos, M.; Carricondo, M.; Bussaglia, E.; Estivill, C.; Esteve, J.; Tormo, M.; Duarte, R.; Salamero, O.; de Llano, M.P.Q.; et al. Bone marrow WT1 levels at diagnosis, post-induction and post-intensification in adult de novo AML. Leukemia 2013, 27, 2157–2164. [Google Scholar] [CrossRef]
- Brieger, J.; Weidmann, E.; Fenchel, K.; Mitrou, P.S.; Hoelzer, D.; Bergmann, L. The expression of the Wilms’ tumor gene in acute myelocytic leukemias as a possible marker for leukemic blast cells. Leukemia 1994, 8, 2138–2143. [Google Scholar]
- Ahmad, E.I.; El-Akad, G.M.; Ismail, W.I.; Al Nagar, A.A.R. Study of Wilms’ tumor 1 gene expression in patients with acute myeloid leukemia. Egypt. J. Haematol. 2019, 44, 195–203. [Google Scholar] [CrossRef]
- Xu, N.; Chen, W.-M.; Li, L.-D.; Long, L.-Y.; Wang, X.; Jiang, Q.; Jiang, H.; Huang, X.-J.; Qin, Y.-Z. High WT1 expression predicted induction chemotherapy failure in acute myeloid leukemia patients with non-favorable cytogenetic risk. Clin. Exp. Med. 2023, 23, 2629–2638. [Google Scholar] [CrossRef]
- Bergmann, L.; Miething, C.; Maurer, U.; Brieger, J.; Karakas, T.; Weidmann, E.; Hoelzer, D. High levels of Wilms’ tumor gene (wt1) mRNA in acute myeloid leukemias are associated with a worse long-term outcome. Blood 1997, 90, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Lapillonne, H.; Renneville, A.; Auvrignon, A.; Flamant, C.; Blaise, A.; Perot, C.; Lai, J.-L.; Ballerini, P.; Mazingue, F.; Fasola, S.; et al. High WT1 expression after induction therapy predicts high risk of relapse and death in pediatric acute myeloid leukemia. J. Clin. Oncol. 2006, 24, 1507–1515. [Google Scholar] [CrossRef]
- Woehlecke, C.; Wittig, S.; Arndt, C.; Gruhn, B. Prognostic impact of WT1 expression prior to hematopoietic stem cell transplantation in children with malignant hematological diseases. J. Cancer Res. Clin. Oncol. 2015, 141, 523–529. [Google Scholar] [CrossRef]
- Weisser, M.; Kern, W.; Rauhut, S.; Schoch, C.; Hiddemann, W.; Haferlach, T.; Schnittger, S. Prognostic impact of RT-PCR-based quantification of WT1 gene expression during MRD monitoring of acute myeloid leukemia. Leukemia 2005, 19, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Renneville, A.; Hermitte, F.; Hills, R.K.; Daly, S.; Jovanovic, J.V.; Gottardi, E.; Fava, M.; Schnittger, S.; Weiss, T.; et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leukemia: A European LeukemiaNet study. J. Clin. Oncol. 2009, 27, 5195–5201. [Google Scholar] [CrossRef] [PubMed]
- Barragán, E.; Cervera, J.; Bolufer, P.; Ballester, S.; Martín, G.; Fernández, P.; Collado, R.; Sayas, M.J.; Sanz, M.A. Prognostic implications of Wilms’ tumor gene (WT1) expression in patients with de novo acute myeloid leukemia. Haematologica 2004, 89, 926–933. [Google Scholar] [PubMed]
- Mehralizadeh, H.; Aliparasti, M.R.; Talebi, M.; Salekzamani, S.; Almasi, S.; Raeisi, M.; Yousefi, M.; Movassaghpour, A. WT-1, BAALC, and ERG Expressions in Iranian Patients with Acute Myeloid Leukemia Pre- and Post-chemotherapy. Adv. Pharm. Bull. 2020, 11, 197–203. [Google Scholar] [CrossRef]
- Ahmed, S.; Elsherif, M.; Yassin, D.; Elsharkawy, N.; Mohamed, A.S.; Yasser, N.; Elnashar, A.; Hafez, H.; Kolb, E.A.; Elhaddad, A. Integration of measurable residual disease by WT1 gene expression and flow cytometry identifies pediatric patients with high risk of relapse in acute myeloid leukemia. Front. Oncol. 2024, 14, 1340909. [Google Scholar] [CrossRef]
- Løvvik Juul-Dam, K.; Guldborg Nyvold, C.; Vålerhaugen, H.; Zeller, B.; Lausen, B.; Hasle, H.; Beier Ommen, H. Measurable residual disease monitoring using Wilms tumor gene 1 expression in childhood acute myeloid leukemia based on child-specific reference values. Pediatr. Blood Cancer 2019, 66, e27671. [Google Scholar] [CrossRef]
- Østergaard, M.; Olesen, L.H.; Hasle, H.; Kjeldsen, E.; Hokland, P. WT1 gene expression: An excellent tool for monitoring minimal residual disease in 70% of acute myeloid leukaemia patients—Results from a single-centre study. Br. J. Haematol. 2004, 125, 590–600. [Google Scholar] [CrossRef]
- Pozzi, S.; Geroldi, S.; Tedone, E.; Luchetti, S.; Grasso, R.; Colombo, N.; Di Grazia, C.; Lamparelli, T.; Gualandi, F.; Ibatici, A.; et al. Leukaemia relapse after allogeneic transplants for acute myeloid leukaemia: Predictive role of WT1 expression. Br. J. Haematol. 2013, 160, 503–509. [Google Scholar] [CrossRef]
- Toska, E.; Roberts, S.G.E. Mechanisms of transcriptional regulation by WT1 (Wilms’ tumour 1). Biochem. J. 2014, 461, 15–32. [Google Scholar] [CrossRef]
- Maslak, P.G.; Dao, T.; Bernal, Y.; Chanel, S.M.; Zhang, R.; Frattini, M.; Rosenblat, T.; Jurcic, J.G.; Brentjens, R.J.; Arcila, M.E.; et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv. 2018, 2, 224–234. [Google Scholar] [CrossRef]
- Raguraman, P.; Balachandran, A.A.; Chen, S.; Diermeier, S.D.; Veedu, R.N. Antisense Oligonucleotide-Mediated Splice Switching: Potential Therapeutic Approach for Cancer Mitigation. Cancers 2021, 13, 5555. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Li, Y.; Qiao, J.; Meng, X.; He, Z.; Gao, X.; Yu, L. Antigen-Specific TCR-T Cells for Acute Myeloid Leukemia: State of the Art and Challenges. Front. Oncol. 2022, 12, 787108. [Google Scholar] [CrossRef] [PubMed]
| Gender Distribution | Male 200 (57.97)% | Female 145 (42.02)% | ||
|---|---|---|---|---|
| Median age in years (range) | 40 (19–76) | |||
| Hemogram Details of Enrolled Patients | ||||
| Median Total Leukocytes Count (×103/µL) | 20.73 (0–310.4) | |||
| Median Hemoglobin (g/dL) (range) | 7.34 (2.4–14.8) | |||
| Median Platelets (×103/µL) | 49.0 (6–321) | |||
| Median BM Blast Percentage (%) | 80.7 (22–100) | |||
| cMPO Selection (n = 276) | Positive 161 (58.33)% | Negative 115 (41.66)% | ||
| Fusion Gene Mutation Analysis by RT-PCR | ||||
| NPM1 (n = 180) | FLT3-ITD (n = 192) | FLT3-TKD (n = 182) | CEBPA (n = 161) | |
| Positive | 29 | 45 | 21 | 18 |
| Negative | 151 | 147 | 161 | 143 |
| Karyotype Report | ||||
| AML-ETO (n = 152) | CBFB-MYH (n = 148) | Normal karyotype (n = 116) | Complex Karyotype (n = 116) | |
| Positive | 26 | 10 | 37 | 3 |
| Negative | 125 | 138 | 79 | 113 |
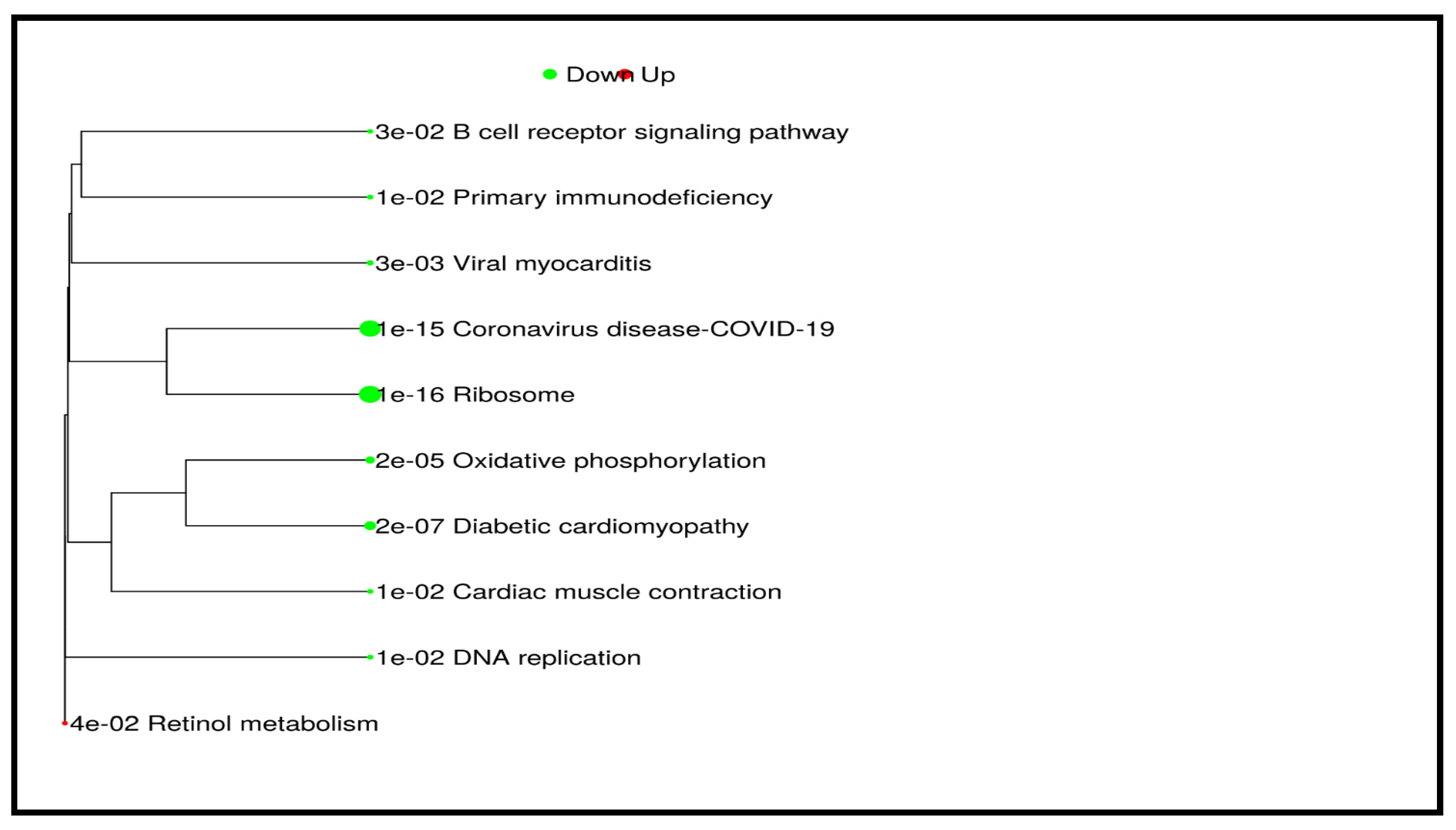
| Direction | GSEA Analysis: DENOVO vs. RELAPSE | NES | Genes | Adj. p-Value |
|---|---|---|---|---|
| Down | Ribosome | −0.6827 | 87 | 1.5 × 10−16 |
| Coronavirus disease-COVID-19 | −0.6165 | 118 | 9.5 × 10−16 | |
| Diabetic cardiomyopathy | −0.5134 | 103 | 2.5 × 10−7 | |
| Oxidative phosphorylation | −0.5593 | 60 | 2.3 × 10−5 | |
| Viral myocarditis | −0.7306 | 15 | 2.8 × 10−3 | |
| DNA replication | −0.6316 | 19 | 1.0 × 10−2 | |
| Primary immunodeficiency | −0.6811 | 15 | 1.3 × 10−2 | |
| Cardiac muscle contraction | −0.4949 | 35 | 1.5 × 10−2 | |
| B cell receptor signaling pathway | −0.4565 | 42 | 2.5 × 10−2 | |
| Up | Retinol metabolism | 0.6895 | 14 | 3.5 × 10−2 |
| Variable | WT1 Low Expression (n = 202) | WT1 High Expression (n = 143) | p-Value |
|---|---|---|---|
| Age (years) | 0.82 | ||
| <40 | 105 (52.0%) | 72 (50.3%) | |
| ≥40 | 97 (48.0%) | 71 (49.7%) | |
| Total Leukocyte Count (×103/µL) | 0.08 | ||
| <50 | 144 (71.3%) | 89 (62.2%) | |
| ≥50 | 58 (28.7%) | 54 (37.8%) | |
| Blast Percentage (%) | 0.05 | ||
| <80 | 96 (47.5%) | 63 (44.1%) | |
| ≥80 | 106 (52.5%) | 80 (55.9%) | |
| Hemoglobin (g/dL) | 0.0001 | ||
| <8 | 115 (56.9%) | 115 (80.4%) | |
| ≥8 | 87 (43.1%) | 28 (19.6%) | |
| Platelets (×103/µL) | 0.23 | ||
| <50 | 104 (51.5%) | 64 (44.8%) | |
| ≥50 | 98 (48.5%) | 79 (55.3%) | |
| Sex | 0.31 | ||
| Male | 122 (61.4%) | 78 (54.6%) | |
| Female | 80 (39.6%) | 65 (45.4%) |
| Variables | Event Free Survival | Overall Survival | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p Value | HR | 95% CI | p Value | |
| Age at Diagnosis | ||||||
| <40 years (n = 117) ≥40 years (n = 168) | 1.057876 | 0.81–1.38 | 0.673 | 1.097428 | 0.83–1.44 | 0.510 |
| Gender | ||||||
| Male (n = 200) Female (n = 145) | 1.455092 | 1.11–1.89 | 0.006 | 1.470668 | 1.11–1.94 | 0.007 |
| Total Leukocyte Count (×109/L) | ||||||
| <50 (n = 233) ≥50 (n = 112) | 1.043832 | 0.79–1.37 | 0.757 | 1.092357 | 0.82–1.45 | 0.542 |
| BM Blast (%) | ||||||
| <80% (n = 159) ≥80% (n = 186) | 0.9498176 | 0.73–1.23 | 0.701 | 1.21451 | 0.92–1.60 | 0.169 |
| Hemoglobin (g/dL) | ||||||
| <8 (n = 230) ≥8 (n = 115) | 0.7187736 | 0.53–0.96 | 0.029 | 0.580122 | 0.41–0.80 | 0.001 |
| Platelets (×103/µL) | ||||||
| <50 (n = 168) ≥50 (n = 175) | 0.5778738 | 0.44–0.75 | 0.0001 | 0.7278208 | 0.55–0.96 | 0.025 |
| WT1 Expression | ||||||
| High (n = 143) Low (n = 202) | 1.204193 | 0.92–1.57 | 0.056 | 1.329035 | 1.00–1.76 | 0.037 |
| Variables | Event Free Survival | Overall Survival | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p Value | HR | 95% CI | p Value | |
| Age at Diagnosis | ||||||
| <40 years (n = 117) ≥40 years (n = 168) | 0.9420834 | 0.62–1.41 | 0.775 | 0.9210855 | 0.57–1.46 | 0.728 |
| Gender | ||||||
| Male (n = 200) Female (n = 145) | 1.620069 | 1.05–2.48 | 0.028 | 1.515567 | 0.93–2.44 | 0.089 |
| Total Leukocyte Count (×109/L) | ||||||
| <50 (n = 233) ≥50 (n = 112) | 1.371956 | 0.83–2.26 | 0.217 | 1.457944 | 0.87–2.42 | 0.148 |
| BM Blast (%) | ||||||
| <80% (n = 159) ≥80% (n = 186) | 0.9102344 | 0.57–1.44 | 0.689 | 1.181538 | 0.75–1.84 | 0.465 |
| Hemoglobin (g/dL) | ||||||
| <8 (n = 230) ≥8 (n = 115) | 0.4168227 | 0.26–0.64 | 0.0001 | 0.2639707 | 0.15–0.45 | 0.001 |
| Platelets (×103/µL) | ||||||
| <50 (n = 168) ≥50 (n = 175) | 0.9836502 | 0.63–1.51 | 0.940 | 0.7934608 | 0.48–1.30 | 0.363 |
| WT1 Expression | ||||||
| High (n = 143) Low (n = 202) | 1.070796 | 0.66–1.71 | 0.777 | 1.202246 | 0.72–1.98 | 0.472 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goel, H.; Pandey, A.K.; Kumar, R.; Kumar, R.; Ningombam, S.S.; Naz, F.; Makkar, H.; Singh, J.; Ali, S.; Chopra, A.; et al. RNA Sequencing Identifies WT1 Overexpression as a Predictor of Poor Outcomes in Acute Myeloid Leukemia. Cancers 2025, 17, 1818. https://doi.org/10.3390/cancers17111818
Goel H, Pandey AK, Kumar R, Kumar R, Ningombam SS, Naz F, Makkar H, Singh J, Ali S, Chopra A, et al. RNA Sequencing Identifies WT1 Overexpression as a Predictor of Poor Outcomes in Acute Myeloid Leukemia. Cancers. 2025; 17(11):1818. https://doi.org/10.3390/cancers17111818
Chicago/Turabian StyleGoel, Harsh, Avanish Kumar Pandey, Rahul Kumar, Rakesh Kumar, Somorjit Singh Ningombam, Farhat Naz, Harshita Makkar, Jay Singh, Shadab Ali, Anita Chopra, and et al. 2025. "RNA Sequencing Identifies WT1 Overexpression as a Predictor of Poor Outcomes in Acute Myeloid Leukemia" Cancers 17, no. 11: 1818. https://doi.org/10.3390/cancers17111818
APA StyleGoel, H., Pandey, A. K., Kumar, R., Kumar, R., Ningombam, S. S., Naz, F., Makkar, H., Singh, J., Ali, S., Chopra, A., Ranjan, A., Gupta, A. K., Meena, J. P., Viswanathan, G. K., Bakhshi, S., Sahoo, R. K., Batra, A., Rath, G. K., Hussain, S., ... Tanwar, P. (2025). RNA Sequencing Identifies WT1 Overexpression as a Predictor of Poor Outcomes in Acute Myeloid Leukemia. Cancers, 17(11), 1818. https://doi.org/10.3390/cancers17111818