
Classification of Gene Variants in a Danish Population with Suspected Predisposition to Hereditary Breast and/or Ovarian Cancer
 , , , , and
, , , , and
Simple Summary
Abstract
1. Introduction
- To classify gene variants as either pathogenic, likely pathogenic, likely benign, benign, or a variant of unknown significance (VUS) and to determine their respective distributions;
- To reclassify VUSs using two different methods:
- (a)
- Association analysis comparing the prevalence of VUSs in this Danish population to the Swedish population using gnomAD;
- (b)
- Splice analysis using RNA sequencing.
2. Materials and Methods
2.1. Study Population and Gene Panels
2.2. Data Collection
2.3. Gene Panel Analysis
2.4. Variant Classification
2.4.1. Classification of Copy Number Variants (CNV)
2.4.2. Classification of 5′-UTR Variants
2.5. Association Analysis Method
2.6. Splice Analysis Method
2.7. Statistical Analysis
3. Results
3.1. Distribution of Variants After Classification
3.2. Association Analysis
3.3. Splice Analysis
4. Discussion
Strength and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Information About the Danish Breast Cancer Screening Programme. 2023. Available online: https://www.sst.dk/-/media/Udgivelser/2023/Kraeft/Screening/UK-Pjece-brystkraeft.ashx (accessed on 17 December 2024).
- Gottschau, M.; Mellemkjaer, L.; Hannibal, C.G.; Kjaer, S.K. Ovarian and tubal cancer in Denmark: An update on incidence and survival. Acta Obs. Gynecol. Scand. 2016, 95, 1181–1189. [Google Scholar] [CrossRef]
- Rosendahl, M.; Jensen, A.N.; Hansen, B.L.; Sengeløv, L. Ovariecancer. 2020. Available online: https://www.sundhed.dk/sundhedsfaglig/laegehaandbogen/gynaekologi/tilstande-og-sygdomme/svulster-og-dysplasi/ovariecancer/ (accessed on 28 May 2024).
- Family History of Breast Cancer and Inherited Genes. 2023. Available online: https://www.cancerresearchuk.org/about-cancer/breast-cancer/risks-causes/family-history-and-inherited-genes (accessed on 17 December 2024).
- DBCG. Kliniske Retningslinjer: Arvelig Mammacancer—Henvisningskriterier til Genetisk Rådgivning, Indikation for Tilbud om Surveillance og Profylaktisk Kirurgi (Version 2). 2023. Available online: https://www.dmcg.dk/siteassets/kliniske-retningslinjer---skabeloner-og-vejledninger/kliniske-retningslinjer-opdelt-pa-dmcg/dbcg/dbcg_arvelig-cancer-mamma_v.2.0_admgodk_02012023.pdf (accessed on 24 November 2024).
- Dilmac, S.; Ozpolat, B. Mechanisms of PARP-Inhibitor-Resistance in BRCA-Mutated Breast Cancer and New Therapeutic Approaches. Cancers 2023, 15, 3642. [Google Scholar] [CrossRef]
- Morganti, S.; Marra, A.; De Angelis, C.; Toss, A.; Licata, L.; Giugliano, F.; Taurelli Salimbeni, B.; Berton Giachetti, P.P.M.; Esposito, A.; Giordano, A.; et al. PARP Inhibitors for Breast Cancer Treatment: A Review. JAMA Oncol. 2024, 10, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, A.M.; Cruger, D.G.; Thomassen, M.; Kruse, T.A. Evaluation of two different models to predict BRCA1 and BRCA2 mutations in a cohort of Danish hereditary breast and/or ovarian cancer families. Clin. Genet. 2006, 69, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Thomassen, M.; Hansen, T.V.; Borg, A.; Lianee, H.T.; Wikman, F.; Pedersen, I.S.; Bisgaard, M.L.; Nielsen, F.C.; Kruse, T.A.; Gerdes, A.M. BRCA1 and BRCA2 mutations in Danish families with hereditary breast and/or ovarian cancer. Acta Oncol. 2008, 47, 772–777. [Google Scholar] [CrossRef]
- Hoffman-Andrews, L. The known unknown: The challenges of genetic variants of uncertain significance in clinical practice. J. Law Biosci. 2017, 4, 648–657. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hedge, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- DBCG. Retningslinjer 2010: 19 Genetisk Udredning, Rådgivning og Molekylærgenetisk Analyse. 2006. Available online: https://dbcg.dk/PDF%20Filer/Retningslinier%202010%20Kap%2019%20010410.pdf (accessed on 9 September 2024).
- DBCG. Retningslinjer 2014: 19 Arvelig Cancer Mammae—Ovarii. 2014. Available online: https://www.dbcg.dk/PDF%20Filer/Retningslinier_Kap19_Genetisk_udredning_030214.pdf (accessed on 9 September 2024).
- DBCG. Retningslinjer 2016: 19 Arvelig Cancer Mammae—Ovarii. 2016. Available online: https://dbcg.dk/PDF%20Filer/Kapitel_19_HBOC_23.09.2016.pdf (accessed on 9 September 2024).
- DBCG. Kliniske Retningslinjer: Arvelig Mammacancer—Henvisningskriterier til Genetisk Rådgivning, Indikation for Tilbud om Surveillance og Profylaktisk Kirurgi Arvelig Mammacancer (Version 1.0). 2020. Available online: https://www.dmcg.dk/siteassets/kliniske-retningslinjer---skabeloner-og-vejledninger/kliniske-retningslinjer-opdelt-pa-dmcg/dbcg/dbcg_arvelig-cancer-mamma_admgodk161220.docx.pdf (accessed on 9 September 2024).
- Møller, N.B.; Boonen, D.S.; Feldner, E.S.; Hao, Q.; Larsen, M.; Lænkholm, A.V.; Borg, Å.; Kvist, A.; Törngren, T.; Jensen, U.B.; et al. Validation of the BOADICEA model for predicting the likelihood of carrying pathogenic variants in eight breast and ovarian cancer susceptibility genes. Sci. Rep. 2023, 13, 8536. [Google Scholar] [CrossRef]
- Öfverholm, A.; Törngren, T.; Rosén, A.; Arver, B.; Einbeigi, Z.; Haraldsson, K.; Ståhlbom, A.K.; Kuchinskava, E.; Lindblom, A.; Melin, B.; et al. Extended genetic analysis and tumor characteristics in over 4600 women with suspected hereditary breast and ovarian cancer. BMC Cancer 2023, 23, 738. [Google Scholar] [CrossRef]
- ClinGen. ClinGen Hereditary Breast, Ovarian and Pancreatic Cancer Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for ATM Version 1.3.0. 2024. Available online: https://cspec.genome.network/cspec/ui/svi/doc/GN020 (accessed on 28 May 2024).
- ClinGen. ClinGen ENIGMA BRCA1 and BRCA2 Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for BRCA1 Version 1.2.0. 2023. Available online: https://cspec.genome.network/cspec/ui/svi/doc/GN092 (accessed on 28 May 2024).
- ClinGen. ClinGen ENIGMA BRCA1 and BRCA2 Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for BRCA2 Version 1.2.0. 2023. Available online: https://cspec.genome.network/cspec/ui/svi/doc/GN097 (accessed on 28 May 2024).
- ClinGen. ClinGen CDH1 Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 3.1. 2022. Available online: https://cspec.genome.network/cspec/ui/svi/doc/GN007 (accessed on 28 May 2024).
- ClinGen. ClinGen Hereditary Breast, Ovarian and Pancreatic Cancer Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for PALB2 Version 1.1.0. 2023. Available online: https://cspec.genome.network/cspec/ui/svi/doc/GN077 (accessed on 28 May 2024).
- ClinGen. ClinGen PTEN Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for PTEN Version 3.1.0. 2024. Available online: https://cspec.genome.network/cspec/ui/svi/doc/GN003 (accessed on 28 May 2024).
- ClinGen. ClinGen TP53 Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines for TP53 Version 1.4.0. 2023. Available online: https://cspec.genome.network/cspec/ui/svi/doc/GN009 (accessed on 28 May 2024).
- ClinGen. ClinGen Sequence Variant Interpretation Recommendation for PM2—Version 1.0. 2020. Available online: https://clinicalgenome.org/site/assets/files/5182/pm2_-_svi_recommendation_-_approved_sept2020.pdf (accessed on 28 May 2024).
- Reuter, K.; Biehl, A.; Koch, L.; Helms, V. PreTIS: A Tool to Predict Non-canonical 5′ UTR Translational Initiation Sites in Human and Mouse. PLoS Comput. Biol. 2016, 12, e1005170. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Walker, L.C.; Hoya, M.; Wiggins, G.A.R.; Lindy, A.; Vincent, L.M.; Parsons, M.T.; Canson, D.M.; Bis-Brewer, D.; Cass, A.; Tchourbanov, A.; et al. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am. J. Hum. Genet. 2023, 110, 1046–1067. [Google Scholar] [CrossRef] [PubMed]
- Davy, G.; Rousselin, A.; Goardon, N.; Castéra, L.; Harter, V.; Legros, A.; Muller, E.; Fouillet, R.; Brault, B.; Smirnova, A.S.; et al. Detecting splicing patterns in genes involved in hereditary breast and ovarian cancer. Eur. J. Hum. Genet. 2017, 25, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, I.S.; Schmidt, A.Y.; Bertelsen, B.; Ernst, A.; Andersen, C.L.T.; Kruse, T.; Rossing, M.; Thomassen, M. A Danish national effort of BRCA1/2 variant classification. Acta Oncol. 2018, 57, 159–162. [Google Scholar] [CrossRef]
- Sanoguera-Miralles, L.; Valenzuela-Palomo, A.; Bueno-Martínez, E.; Esteban-Sánchez, A.; Lorca, V.; Llinares-Burguet, I.; García-Álvarez, A.; Pérez-Segura, P.; Infante, M.; Easton, D.F.; et al. Systematic Minigene-Based Splicing Analysis and Tentative Clinical Classification of 52 CHEK2 Splice-Site Variants. Clin. Chem. 2024, 70, 319–338. [Google Scholar] [CrossRef]
- Dabney, J.; Meyer, M. Length and GC-biases during sequencing library amplification: A comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques 2012, 52, 87–94. [Google Scholar] [CrossRef]
- Bueno-Martínez, E.; Sanoguera-Miralles, L.; Valenzuela-Palomo, A.; Esteban-Sánchez, A.; Lorca, V.; Llinares-Burguet, I.; Allen, J.; García-Álvarez, A.; Pérez-Segura, P.; Durán, M.; et al. Minigene-based splicing analysis and ACMG/AMP-based tentative classification of 56 ATM variants. J. Pathol. 2022, 258, 83–101. [Google Scholar] [CrossRef]
- ClinGen. Variant: NM_000051.4(ATM):c.1066-6T>G. 2022. Available online: https://erepo.clinicalgenome.org/evrepo/ui/interpretation/bccd9f16-4086-4c14-8d78-04c52ad58630 (accessed on 5 December 2024).
- Schröder, S.; Wieland, B.; Ohlenbusch, A.; Yigit, G.; Altmüller, J.; Boltshauser, E.; Dörk, T.; Brockmann, K. Evidence of pathogenicity for the leaky splice variant c.1066-6T>G in ATM. Am. J. Med. Genet. A 2020, 182, 2971–2975. [Google Scholar] [CrossRef]
- Dörk, T.; Bendix, R.; Bremer, M.; Rades, D.; Klöpper, K.; Nicke, M.; Skawran, B.; Hector, A.; Yamini, P.; Steinmann, D.; et al. Spectrum of ATM gene mutations in a hospital-based series of unselected breast cancer patients. Cancer Res. 2001, 61, 7608–7615. [Google Scholar]
- Broeks, A.; Urbanus, J.H.; Floore, A.N.; Dahler, E.C.; Klijn, J.G.; Rutgers, E.J.; Devilee, P.; Russell, N.S.; Leeuwen, F.E.; Veer, L.J. ATM-heterozygous germline mutations contribute to breast cancer-susceptibility. Am. J. Hum. Genet. 2000, 66, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Fiévet, A.; Bellanger, D.; Rieunier, G.; Dubois d’Enghien, C.; Sophie, J.; Calvas, P.; Carriere, J.P.; Anheim, M.; Castrioto, A.; Flabeau, O.; et al. Functional classification of ATM variants in ataxia-telangiectasia patients. Hum. Mutat. 2019, 40, 1713–1730. [Google Scholar] [CrossRef]
- Austen, B.; Barone, G.; Reiman, A.; Byrd, P.J.; Baker, C.; Starczynski, J.; Nobbs, M.C.; Murphy, R.P.; Enright, H.; Chaila, E.; et al. Pathogenic ATM mutations occur rarely in a subset of multiple myeloma patients. Br. J. Haematol. 2008, 142, 925–933. [Google Scholar] [CrossRef]
- Bernstein, J.L.; Teraoka, S.; Southey, M.C.; Jenkins, M.A.; Andrulis, I.L.; Knight, J.A.; John, E.M.; Lapinski, R.; Wolitzer, A.L.; Whittemore, A.S.; et al. Population-based estimates of breast cancer risks associated with ATM gene variants c.7271T>G and c.1066-6T>G (IVS10-6T>G) from the Breast Cancer Family Registry. Hum. Mutat. 2006, 27, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Mao, C.; Li, S.M.; Liu, Q.; Lin, L.; Chen, Q. Lack of association between ATM C.1066-6T > G mutation and breast cancer risk: A meta-analysis of 8831 cases and 4957 controls. Breast Cancer Res. Treat. 2011, 125, 473–477. [Google Scholar] [CrossRef]
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Hauke, J.; Horvath, J.; Groß, E.; Gehrig, A.; Honisch, E.; Hackmann, K.; Schmidt, G.; Arnold, N.; Faust, U.; Sutter, C.; et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018, 7, 1349–1358. [Google Scholar] [CrossRef]
- Cheng, H.H.; Shevach, J.W.; Castro, E.; Couch, F.J.; Domchek, S.M.; Eeles, R.A.; Giri, V.N.; Hall, M.J.; King, M.C.; Lin, D.W.; et al. BRCA1, BRCA2, and Associated Cancer Risks and Management for Male Patients: A Review. JAMA Oncol. 2024, 10, 1272–1281. [Google Scholar] [CrossRef]
- Harrison, S.M.; Biesecker, L.G.; Rehm, H.L. Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr. Protoc. Hum. Genet. 2019, 103, e93. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Harrison, S.M. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet. Med. 2018, 20, 1687–1688. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number (%) | |
|---|---|
| Sex, female | 5467 (92.3%) |
| Cancer a | 4598 (77.6%) |
| Breast cancer b | 3706 (62.6%) |
| Bilateral breast cancer | 564 (9.5%) |
| Ovarian cancer | 890 (15.0%) |
| Prostate cancer | 106 (1.8%) |
| Pancreatic cancer | 34 (0.6%) |
| Average age breast cancer patients c | 51.4 years |
| Average age ovarian cancer patients d | 61.8 years |
| ER, positive e | 2588 (69.9%) |
| ER, negative e | 782 (21.1%) |
| ER, unspecified e | 336 (9.1%) |
| HER2, positive e | 517 (14.0%) |
| HER2, negative e | 2595 (70.0%) |
| HER2, unspecified e | 594 (16.0%) |
| Total Number of | VUS Number | LP/P Number |
|---|---|---|
| Identified variants | 1892 | 658 |
| Carriers | 1606 | 630 |
| Distinct variants | 645 | 241 |
| Variant types | VUS Count of total variants (count of distinct variants) | LP/P Count of total variants (count of distinct variants) |
| Loss of function | 3 (3) | 546 (187) |
| In-frame deletion/insertion | 212 (17) | 2 (2) |
| Missense | 773 (383) | 83 (33) |
| Silent | 36 (19) | 2 (2) |
| Splice region | 108 (58) | 7 (6) |
| Intronic | 695 (126) | |
| 5-UTR | 56 (35) | |
| Copy number | 9 (4) | 18 (11 distinct) |
| Gene | VUS Count of total variants (count of distinct variants, percentages compared to the total population) | LP/P Count of total variants (count of distinct variants, percentages compared to the total population) |
| ATM | 365 (197, 6.16%) | 51 (27, 0.86%) |
| BARD1 | 522 (53, 8.81%) | 5 (4, 0.08%) |
| BRCA1 | 96 (38, 1.62%) | 210 (66, 3.55%) |
| BRCA2 | 48 (32, 0.81%) | 171 (68, 2.89%) |
| BRIP1 | 217 (60, 3.66%) | 25 (6, 0.42%) |
| CDH1 | 60 (40, 1.01%) | 2 (2, 0.03%) |
| CHEK2 | 236 (56, 3.98%) | 126 (10, 2.13%) |
| CHEK2 c.1100delC | 99 (1.67%) | |
| PALB2 | 34 (24, 0.57%) | 31 (20, 0.52%) |
| PTEN | 27 (8, 0.46%) | 5 (5, 0.08%) |
| RAD51C | 45 (23, 0.76%) | 6 (3, 0.10%) |
| RAD51D | 64 (26, 1.08%) | 7 (3, 0.12%) |
| STK11 | 152 (34, 2.57%) | |
| TP53 | 26 (15, 0.44%) | 19 (16, 0.32%) |
| Number of Samples and Frequencies | Association Study: gnomAD 2.1 Swedish vs. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study Population | Genome Aggregation Database (gnomAD) Groups | BC | OC | |||||||
| Gene | Sequence Ontology | HGVSc. | BC (n = 3706) | OC (n = 890) | gnomAD2.1 (Sweden) (n = 13,067) | gnomAD2.1 (Sweden) Frequency | gnomAD2.1 (NFE) Frequency (n = 56,885) | gnomAD4.0 (NFE) Frequency (n = 590,031) | OR (95%CI); FDR | OR (95% CI); FDR |
| ATM | Missense | NM_000051.4:c.3519G>C | 2 | 3 | 0 | 0 | 0 | 3.39 × 10−6 | ∞(0.66–∞); 0.36 | ∞(6.08–∞); 0.041 |
| ATM | Synonymous | NM_000051.4:c.7521C>T | 1 | 4 | 0 | 0 | 8.79 × 10−5 | 0.00015 | ∞(0.09–∞); 0.36 | ∞(9.71–∞); 0.0039 |
| ATM | Missense | NM_000051.4:c.8428A>C | 7 | 0 | 0 | 0 | 0.00012 | 7.80 × 10−5 | ∞(5.09–∞); 0.0041 | NA |
| BARD1 | Splice_region | NM_000465.4:c.1569-13C>G | 5 | 2 | 0 | 0 | 8.79 × 10−5 | 2.034 × 10−5 | ∞(3.23–∞); 0.028 | ∞(2.76–∞); 0.21 |
| BARD1 | Missense | NM_000465.4:c.1915T>C | 6 | 0 | 0 | 0 | 8.79 × 10−5 | 0.00011 | ∞(4.16–∞); 0.0079 | NA |
| CHEK2 | Missense | NM_007194.4:c.433C>T | 6 | 1 | 0 | 0 | 0.00011 | 0.00011 | ∞(4.16–∞); 0.0079 | ∞(0.38–∞); 0.48 |
| CHEK2 | Missense | NM_007194.4:c.715G>A | 6 | 2 | 0 | 0 | 0.00011 | 0.00023 | ∞(4.16–∞); 0.0079 | ∞(2.76–∞); 0.21 |
| CHEK2 | Missense | NM_007194.4:c.1183G>C | 8 | 2 | 3 | 0.00023 | 8.79 × 10−5 | 5.59 × 10−5 | 9.42(2.26–55.12); 0.028 | 9.8(0.82–85.67); 0.48 |
| CHEK2 | Missense | NM_007194.4:c.1427C>T | 22 | 2 | 14 | 0.0011 | 0.0010 | 0.00098 | 5.57(2.72–11.78); 0.00024 | 2.1(0.23–9.16); 1 |
| BRCA1 | Splice_region | NM_007294.4:c.4096+3A>G | 8 | 5 | 0 | 0 | 0 | 5.084 × 10−6 | ∞(6.03–∞); 0.0013 | ∞(13.50–∞); 0.00005 |
| BRIP1 | In-frame_ deletion | NM_032043.3:c.1687_1689delGAT | 9 | 2 | 2 | 0.00015 | 3.52 × 10−5 | 0 | 15.9(3.29–151.18); 0.0052 | 14.7(1.06–202.8); 0.48 |
| Variant | Prior Classification | Criteria Used for Prior Classification | Criteria After RNA Sequencing | Samples gnomAD 4.0 (NFE) | Samples gnomAD 2.1 (NFE) | Samples gnomAD 2.1 (Swedish) | ClinVar | SpliceAI Delta Acceptor Loss (Position) | SpliceAI Delta Acceptor Gain (Position) | SpliceAI Delta Donor Loss (Position) | SpliceAI Delta Donor Gain (Position) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ATM c.1066-6T>G | C1 | BS1BP2_strong | PVS1 (RNA) | 3044 | 253 | 43 | C1: 12 C2: 11 C3: 7 | 0.62 (6) | 0.00 (1) | 0.00 (5) | 0.00 (−1) |
| ATM c.3078-10T>G | C3 | PM2_supporting PP3 | 0 | 0 | 0 | C3: 2 | 0.70 (10) | 0.00 (1) | 0.00 (43) | 0.00 (−1) | |
| ATM c.6007-1581A>G | C2 | BS1 | BP7_ strong | 235 | 0 | 0 | NA | 0.00 (284) | 0.23 (−105) | 0.05 (143) | 0.64 (0) |
| ATM c.3994-2A>G | C4 | PVS1PM2_supporting | 6 | 4 | 4 | C4: 6 C5: 3 | 0.99 (2) | 0.42 (22) | 0.00 (20) | 0.00 (−40) | |
| BRIP1 c.2493-10T>A | C3 | PM2_supportingPP3 | 1 | 1 | 1 | C3: 2 C2: 2 | 0.00 (−2) | 0.30 (−10) | 0.00 (−11) | 0.00 (−1) | |
| CHEK2 c.1461+5G>T | C3 | PM2_supportingPP3 | 0 | 0 | 0 | C4: 1 C3: 4 | 0.00 (33) | 0.00 (−21) | 0.90 (5) | 0.00 (50) | |
| RAD51C c.705+3152C>T | C3 | PM2_supportingPP3 | 2 | 0 | 0 | NA | 0.00 (127) | 0.46 (8) | 0.00 (−464) | 0.34 (127) | |
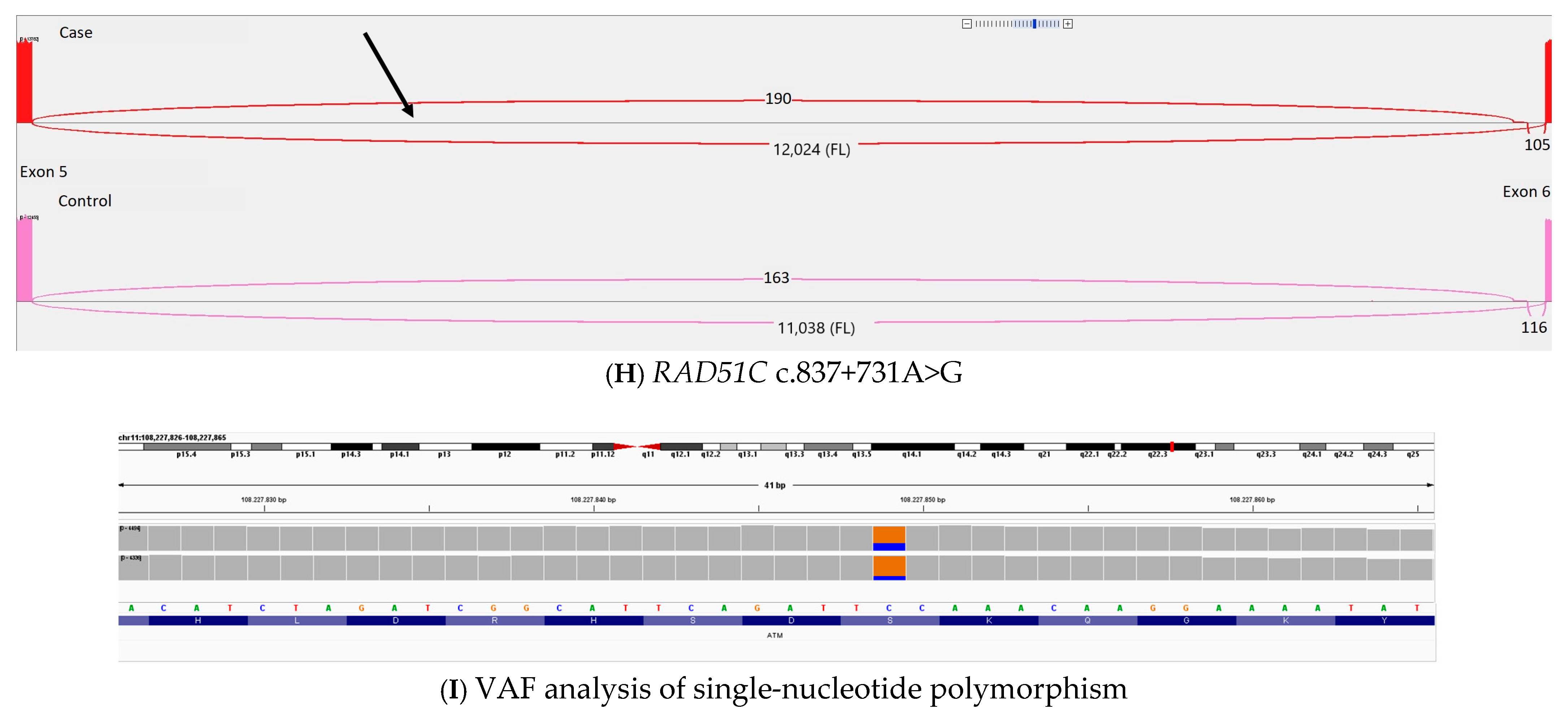
| RAD51C c.837+731A>G | C3 | PM2_supporting PP3 | BP7_ strong | 0 | 0 | 0 | NA | 0.00 (58) | 0.23 (−125) | 0.06 (−69) | 0.66 (−5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munch, A.K.; Feldner, E.S.; Bækgaard, C.H.; Larsen, M.B.; Slemming-Adamsen, N.; Boonen, D.S.; Møller, N.B.; Pedersen, I.S.; Hansen, T.V.O.; Terkelsen, T.; et al. Classification of Gene Variants in a Danish Population with Suspected Predisposition to Hereditary Breast and/or Ovarian Cancer. Cancers 2025, 17, 1819. https://doi.org/10.3390/cancers17111819
Munch AK, Feldner ES, Bækgaard CH, Larsen MB, Slemming-Adamsen N, Boonen DS, Møller NB, Pedersen IS, Hansen TVO, Terkelsen T, et al. Classification of Gene Variants in a Danish Population with Suspected Predisposition to Hereditary Breast and/or Ovarian Cancer. Cancers. 2025; 17(11):1819. https://doi.org/10.3390/cancers17111819
Chicago/Turabian StyleMunch, Anne K., Elisabeth S. Feldner, Caroline H. Bækgaard, Mie B. Larsen, Naja Slemming-Adamsen, Desirée S. Boonen, Nanna B. Møller, Inge S. Pedersen, Thomas V. O. Hansen, Thorkild Terkelsen, and et al. 2025. "Classification of Gene Variants in a Danish Population with Suspected Predisposition to Hereditary Breast and/or Ovarian Cancer" Cancers 17, no. 11: 1819. https://doi.org/10.3390/cancers17111819
APA StyleMunch, A. K., Feldner, E. S., Bækgaard, C. H., Larsen, M. B., Slemming-Adamsen, N., Boonen, D. S., Møller, N. B., Pedersen, I. S., Hansen, T. V. O., Terkelsen, T., Burton, M., Hao, Q., Boonen, S. E., & Thomassen, M. (2025). Classification of Gene Variants in a Danish Population with Suspected Predisposition to Hereditary Breast and/or Ovarian Cancer. Cancers, 17(11), 1819. https://doi.org/10.3390/cancers17111819