
Unraveling Venetoclax Resistance: Navigating the Future of HMA/Venetoclax-Refractory AML in the Molecular Era



Simple Summary
Abstract
1. Introduction
2. Venetoclax Mechanism of Action
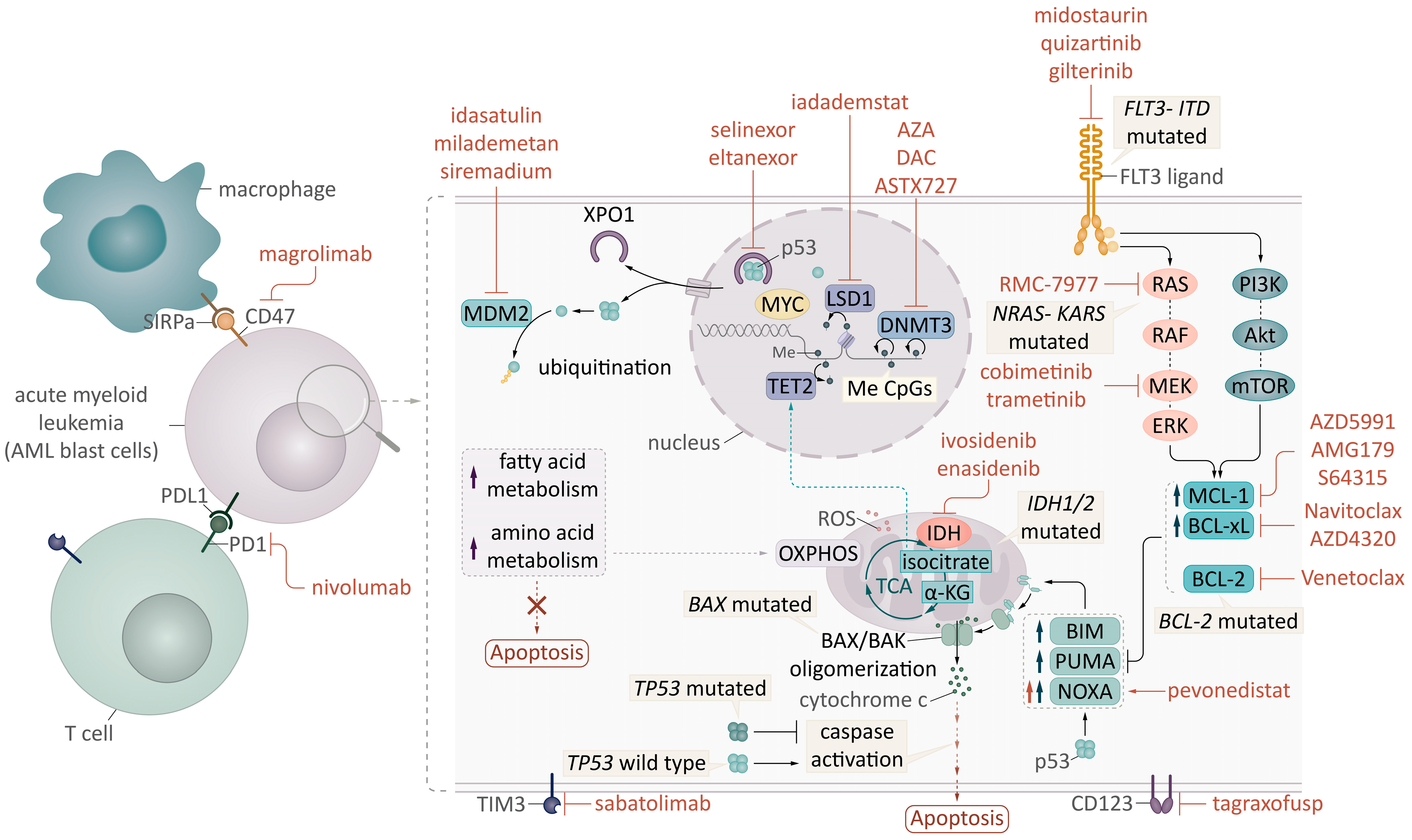
2.1. Intrinsic Apoptosis Mechanism: BCL2 Family Proteins in AML
2.2. Venetoclax-Induced Death in AML
2.3. Combination of Venetoclax with HMAs: The Perfect Match
3. Understanding the Mechanisms of Resistance to Venetoclax
3.1. BLC2 Family Protein Changes: Facilitating Resistance to Apoptosis
3.2. Insights from Clinical Data: Mutation Patterns in HMA/VEN Response and Resistance
3.3. Biological Implications of Mutations Linked to Venetoclax Resistance
3.3.1. TP53 Mutation: The Secrets of the Gatekeeper and Its Partners in Crime
3.3.2. Key Kinases in Venetoclax Resistance: FLT3-ITD and RAS/PTPN11 Mutations
3.4. Impact of AML Differentiation Status on Venetoclax Resistance
3.5. Microenvironment, Metabolism, and Mitochondria in Venetoclax Resistance
4. Strategies to Overcome Resistance with New Targeted Combinations
4.1. Combining BH3 Mimetics: Conquering the Apoptosis Mechanism
4.2. Targeting Epigenetics: Novel Drugs and Approaches
4.3. FLT3 and IDH Inhibitors: Double Attack in the Molecular Era

{kind=link}
| Drug Regimen | Identifier Clinicaltrias.gov | Eligibility Criteria Other than AML | Number of Patients | Response Rates | Study Phase | Reference | |
|---|---|---|---|---|---|---|---|
| Exportin 1 (XPO1) inhibitors | |||||||
| Selinexor + VEN | NCT03955783 | 19 R/R AML | 21% ORR (11% CR, 5% MLFS) | Ib | [108] | ||
| Selinexor + VA (SAV) | NCT05736965 | 20 ndAML | 91.7% ORR (80% CRc) | II | [109] | ||
| HDAC inhibitors | |||||||
| Chidamide + VA (VCA) | NCT05305859 | 53 R/R AML | 81% ORR (CRc 72%) after two cycles | II | [110] | ||
| IDH inhibitors | |||||||
| Ivosidenib + VEN or VA | NCT03471260 | IDH1 mutated myeloid malignancies | 30 AML (8 R/R) | 63% CRc R/R AML, 93% CRc ndAML | Ib/II | [106] | |
| FLT3 inhibitors | |||||||
| Quizartinib + DAC + VEN | NCT03661307 | FLT3mut | 50 (40 R/R) | 69% CR R/R AML | I/II | [104] | |
| Gilteritinib + VEN | NCT03625505 | FLT3mut | 61 R/R AML | 75% mCRc | Ib | [100] | |
| Gilteritinib + VA | NCT04140487 | FLT3mut | 52 (22 R/R) | 96% CRc ndAML, ORR 41% R/R (27% CRc) | I/II | [101] | |
| MDM2 inhibitors | |||||||
| Idasanutlin + VEN | NCT02670044 | 55 R/R or sAML | 26% CRc, 12% MLFS | Ib | [111] | ||
| Menin inhibitors | |||||||
| Revumenib + ASTX727 + VEN | NCT05360160 | KMT2Ar, or NPM1c | 23 R/R AML | 88% ORR (70% CRc) | I/II | [112] | |
| Bleximenib + VA | NCT05453903 | 45 R/R AML | 86% ORR (48% CRc) 82% ORR (36% CRc) with prior VEN exposure | Ib | [113] | ||
| MCL1 inhibitors | |||||||
| AZD5991 + VEN | NCT03218683 | Hematologic malignancies | 33 R/R AML | 0% ORR | [89] | ||
| CDK inhibitors | |||||||
| Alvocidib + VEN | NCT03441555 | 35 R/R AML | ORR 20% (11.4% CRc) | Ib | [114] | ||
| QHRD107 + VA | NCT06532058 | 18 R/R AML | ORR 72.2% (33.3% CRc) | II | [115] | ||
| MEK inhibitors | |||||||
| Trametinib + VA | NCT04487106 | RASmut | 16 R/R AML | 25% ORR (12.5% CRc, 12.5% MLFS) | II | [116] | |
| Cobimetinib + VEN | NCT02670044 | 22 R/R AML | 18% ORR | Ib | [117] | ||
| CD123 targeting molecules | |||||||
| Tagraxofusp + VA | NCT03113643 | AML, MDS, BPDCN | 26 ndAML | 69% ORR (58% CRc) | I | [118] | |
| Enhancing drug activity | |||||||
| ASTX727 + VEN | NCT04746235 | 62 AML (13 R/R) | 64% ORR ndAML, 46% ORR R/R AML | II | [119] | ||
| LDDec + VEN | NCT05184842 | Myeloid neoplasms | 14 AML | 63% ORR (57% CRc) | II | [36] | |
| NEDD8-activating enzyme (NAE) inhibitor | |||||||
| Pevonedistat + VA | NCT03862157 | sAML (MDS/AML) | 32 ndAML | 78% ORR (66% CRc) | I/II | [120] | |
| Pevonedistat + VA | NCT04172844 | 50 R/R AML | 46.7% ORR AML | I | [121] | ||
| Others | |||||||
| Magrolimab + VA | NCT04435691 | 80% ORR ndAML, 34.4% ORR R/R AML (27.5% CRc) 12% ORR R/R AML prior VEN exposure | Ib/II | [122] | |||
| Magrolimab + VA vs. VA | NCT05079230 | ndAML | 39.7% vs. 42.9% CR | III | [123] | ||
4.4. Restoring p53 Activity to Promote Apoptosis
4.5. Alternative Pathways to Trigger Apoptosis
4.6. Other Novel Approaches: The Microenvironment in the Spotlight
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Pratz, K.W.; Jonas, B.A.; Pullarkat, V.A.; Thirman, M.J.; Garcia, J.S.; Fiedler, W.; Yamamoto, K.; Wang, J.; Yoon, S.-S.; Wolach, O.; et al. Long-Term Follow-up of the Phase 3 Viale-a Clinical Trial of Venetoclax Plus Azacitidine for Patients with Untreated Acute Myeloid Leukemia Ineligible for Intensive Chemotherapy. Blood 2022, 140, 529–531. [Google Scholar] [CrossRef]
- Chatzilygeroudi, T.; Darmani, I.; El Gkotmi, N.; Vryttia, P.; Douna, S.; Bouchla, A.; Labropoulou, V.; Kotsopoulou, M.; Symeonidis, A.; Pagoni, M.; et al. Real-Life Multicenter Experience of Venetoclax in Combination with Hypomethylating Agents in Previously Untreated Adult Patients with Acute Myeloid Leukemia in Greece. J. Clin. Med. 2024, 13, 584. [Google Scholar] [CrossRef]
- Todisco, E.; Papayannidis, C.; Fracchiolla, N.; Petracci, E.; Zingaretti, C.; Vetro, C.; Martelli, M.P.; Zappasodi, P.; Di Renzo, N.; Gallo, S.; et al. AVALON: The Italian Cohort Study on Real-life Efficacy of Hypomethylating Agents plus Venetoclax in Newly Diagnosed or Relapsed/Refractory Patients with Acute Myeloid Leukemia. Cancer 2023, 129, 992–1004. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.-P.; Chou, W.-C.; Buckstein, R.; Cermak, J.; et al. Multicenter, Randomized, Open-Label, Phase III Trial of Decitabine versus Patient Choice, with Physician Advice, of Either Supportive Care or Low-Dose Cytarabine for the Treatment of Older Patients with Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International Phase 3 Study of Azacitidine vs Conventional Care Regimens in Older Patients with Newly Diagnosed AML with >30% Blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Maiti, A.; Rausch, C.R.; Cortes, J.E.; Pemmaraju, N.; Daver, N.G.; Ravandi, F.; Garcia-Manero, G.; Borthakur, G.; Naqvi, K.; Ohanian, M.; et al. Outcomes of Relapsed or Refractory Acute Myeloid Leukemia after Frontline Hypomethylating Agent and Venetoclax Regimens. Haematologica 2021, 106, 894–898. [Google Scholar] [CrossRef]
- Wachter, F.; Pikman, Y. Pathophysiology of Acute Myeloid Leukemia. Acta Haematol. 2024, 147, 229–246. [Google Scholar] [CrossRef]
- Ewald, L.; Dittmann, J.; Vogler, M.; Fulda, S. Side-by-Side Comparison of BH3-Mimetics Identifies MCL-1 as a Key Therapeutic Target in AML. Cell Death Dis. 2019, 10, 917. [Google Scholar] [CrossRef]
- Bose, P.; Gandhi, V.; Konopleva, M. Pathways and Mechanisms of Venetoclax Resistance. Leuk. Lymphoma 2017, 58, 2026–2039. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 Family Proteins: Changing Partners in the Dance towards Death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 Inhibition by ABT-199 Causes on-Target Cell Death in Acute Myeloid Leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef]
- Zhu, R.; Li, L.; Nguyen, B.; Seo, J.; Wu, M.; Seale, T.; Levis, M.; Duffield, A.; Hu, Y.; Small, D. FLT3 Tyrosine Kinase Inhibitors Synergize with BCL-2 Inhibition to Eliminate FLT3/ITD Acute Leukemia Cells through BIM Activation. Sig. Transduct. Target Ther. 2021, 6, 186. [Google Scholar] [CrossRef]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High Expression of Bcl-2 Protein in Acute Myeloid Leukemia Cells Is Associated with Poor Response to Chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar] [CrossRef]
- Delia, D.; Aiello, A.; Soligo, D.; Fontanella, E.; Melani, C.; Pezzella, F.; Pierotti, M.A.; Della Porta, G. Bcl-2 Proto-Oncogene Expression in Normal and Neoplastic Human Myeloid Cells. Blood 1992, 79, 1291–1298. [Google Scholar] [CrossRef]
- Lauria, F.; Raspadori, D.; Rondelli, D.; Ventura, M.A.; Fiacchini, M.; Visani, G.; Forconi, F.; Tura, S. High Bcl-2 Expression in Acute Myeloid Leukemia Cells Correlates with CD34 Positivity and Complete Remission Rate. Leukemia 1997, 11, 2075–2078. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell. 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An Inhibitor of Bcl-2 Family Proteins Induces Regression of Solid Tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Park, C.-M.; Bruncko, M.; Adickes, J.; Bauch, J.; Ding, H.; Kunzer, A.; Marsh, K.C.; Nimmer, P.; Shoemaker, A.R.; Song, X.; et al. Discovery of an Orally Bioavailable Small Molecule Inhibitor of Prosurvival B-Cell Lymphoma 2 Proteins. J. Med. Chem. 2008, 51, 6902–6915. [Google Scholar] [CrossRef]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.-L.; et al. Navitoclax, a Targeted High-Affinity Inhibitor of BCL-2, in Lymphoid Malignancies: A Phase 1 Dose-Escalation Study of Safety, Pharmacokinetics, Pharmacodynamics, and Antitumour Activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity While Sparing Platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Waclawiczek, A.; Leppä, A.-M.; Renders, S.; Stumpf, K.; Reyneri, C.; Betz, B.; Janssen, M.; Shahswar, R.; Donato, E.; Karpova, D.; et al. Combinatorial BCL2 Family Expression in Acute Myeloid Leukemia Stem Cells Predicts Clinical Response to Azacitidine/Venetoclax. Cancer Discov. 2023, 13, 1408–1427. [Google Scholar] [CrossRef]
- Lee, J.B.; Khan, D.H.; Hurren, R.; Xu, M.; Na, Y.; Kang, H.; Mirali, S.; Wang, X.; Gronda, M.; Jitkova, Y.; et al. Venetoclax Enhances T Cell-Mediated Antileukemic Activity by Increasing ROS Production. Blood 2021, 138, 234–245. [Google Scholar] [CrossRef]
- Corradi, G.; Forte, D.; Cristiano, G.; Polimeno, A.; Ciciarello, M.; Salvestrini, V.; Bandini, L.; Robustelli, V.; Ottaviani, E.; Cavo, M.; et al. Ex Vivo Characterization of Acute Myeloid Leukemia Patients Undergoing Hypomethylating Agents and Venetoclax Regimen Reveals a Venetoclax-Specific Effect on Non-Suppressive Regulatory T Cells and Bona Fide PD-1+TIM3+ Exhausted CD8+ T Cells. Front. Immunol. 2024, 15, 1386517. [Google Scholar] [CrossRef]
- Bogenberger, J.M.; Kornblau, S.M.; Pierceall, W.E.; Lena, R.; Chow, D.; Shi, C.-X.; Mantei, J.; Ahmann, G.; Gonzales, I.M.; Choudhary, A.; et al. BCL-2 Family Proteins as 5-Azacytidine-Sensitizing Targets and Determinants of Response in Myeloid Malignancies. Leukemia 2014, 28, 1657–1665. [Google Scholar] [CrossRef]
- Bogenberger, J.M.; Delman, D.; Hansen, N.; Valdez, R.; Fauble, V.; Mesa, R.A.; Tibes, R. Ex Vivo Activity of BCL-2 Family Inhibitors ABT-199 and ABT-737 Combined with 5-Azacytidine in Myeloid Malignancies. Leuk. Lymphoma 2015, 56, 226–229. [Google Scholar] [CrossRef]
- Tsao, T.; Shi, Y.; Kornblau, S.; Lu, H.; Konoplev, S.; Antony, A.; Ruvolo, V.; Qiu, Y.H.; Zhang, N.; Coombes, K.R.; et al. Concomitant Inhibition of DNA Methyltransferase and BCL-2 Protein Function Synergistically Induce Mitochondrial Apoptosis in Acute Myelogenous Leukemia Cells. Ann. Hematol. 2012, 91, 1861–1870. [Google Scholar] [CrossRef]
- Sharon, D.; Cathelin, S.; Mirali, S.; Di Trani, J.M.; Yanofsky, D.J.; Keon, K.A.; Rubinstein, J.L.; Schimmer, A.D.; Ketela, T.; Chan, S.M. Inhibition of Mitochondrial Translation Overcomes Venetoclax Resistance in AML through Activation of the Integrated Stress Response. Sci. Transl. Med. 2019, 11, eaax2863. [Google Scholar] [CrossRef]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with Azacitidine Disrupts Energy Metabolism and Targets Leukemia Stem Cells in Patients with Acute Myeloid Leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Hu, X.; Li, L.; Nkwocha, J.; Sharma, K.; Zhou, L.; Grant, S. Synergistic Interactions between the Hypomethylating Agent Thio-Deoxycytidine and Venetoclax in Myelodysplastic Syndrome Cells. Hematol. Rep. 2023, 15, 91–100. [Google Scholar] [CrossRef]
- Nguyen, L.X.T.; Troadec, E.; Kalvala, A.; Kumar, B.; Hoang, D.H.; Viola, D.; Zhang, B.; Nguyen, D.Q.; Aldoss, I.; Ghoda, L.; et al. The Bcl-2 Inhibitor Venetoclax Inhibits Nrf2 Antioxidant Pathway Activation Induced by Hypomethylating Agents in AML. J. Cell. Physiol. 2019, 234, 14040–14049. [Google Scholar] [CrossRef]
- Kamachi, K.; Ureshino, H.; Watanabe, T.; Yoshida-Sakai, N.; Fukuda-Kurahashi, Y.; Kawasoe, K.; Hoshiko, T.; Yamamoto, Y.; Kurahashi, Y.; Kimura, S. Combination of a New Oral Demethylating Agent, OR2100, and Venetoclax for Treatment of Acute Myeloid Leukemia. Cancer Res. Commun. 2023, 3, 297–308. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax Combined with Decitabine or Azacitidine in Treatment-Naive, Elderly Patients with Acute Myeloid Leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Levitz, D.; Saunthararajah, Y.; Fedorov, K.; Shapiro, L.C.; Mantzaris, I.; Shastri, A.; Kornblum, N.; Sica, R.A.; Shah, N.; Konopleva, M.; et al. A Metabolically Optimized, Noncytotoxic Low-Dose Weekly Decitabine/Venetoclax in MDS and AML. Clin. Cancer Res. 2023, 29, 2774–2780. [Google Scholar] [CrossRef]
- Lin, K.H.; Winter, P.S.; Xie, A.; Roth, C.; Martz, C.A.; Stein, E.M.; Anderson, G.R.; Tingley, J.P.; Wood, K.C. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci. Rep. 2016, 6, 27696. [Google Scholar] [CrossRef]
- Zhang, H.; Nakauchi, Y.; Köhnke, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majeti, R.; Tyner, J.W. Integrated Analysis of Patient Samples Identifies Biomarkers for Venetoclax Efficacy and Combination Strategies in Acute Myeloid Leukemia. Nat. Cancer 2020, 1, 826–839. [Google Scholar] [CrossRef]
- Bisaillon, R.; Moison, C.; Thiollier, C.; Krosl, J.; Bordeleau, M.-E.; Lehnertz, B.; Lavallée, V.-P.; MacRae, T.; Mayotte, N.; Labelle, C.; et al. Genetic Characterization of ABT-199 Sensitivity in Human AML. Leukemia 2020, 34, 63–74. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, J.; Ma, J.; Xie, C.; Edwards, H.; Wang, G.; Caldwell, J.T.; Xiang, S.; Zhang, X.; Chu, R.; et al. Binding of Released Bim to Mcl-1 Is a Mechanism of Intrinsic Resistance to ABT-199 Which Can Be Overcome by Combination with Daunorubicin or Cytarabine in AML Cells. Clin. Cancer Res. 2016, 22, 4440–4451. [Google Scholar] [CrossRef]
- Zhang, Q.; Riley-Gillis, B.; Han, L.; Jia, Y.; Lodi, A.; Zhang, H.; Ganesan, S.; Pan, R.; Konoplev, S.N.; Sweeney, S.R.; et al. Activation of RAS/MAPK Pathway Confers MCL-1 Mediated Acquired Resistance to BCL-2 Inhibitor Venetoclax in Acute Myeloid Leukemia. Signal Transduct. Target Ther. 2022, 7, 51. [Google Scholar] [CrossRef]
- Bhatt, S.; Pioso, M.S.; Olesinski, E.A.; Yilma, B.; Ryan, J.A.; Mashaka, T.; Leutz, B.; Adamia, S.; Zhu, H.; Kuang, Y.; et al. Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 872–890.e6. [Google Scholar] [CrossRef]
- Anderson, M.A.; Deng, J.; Seymour, J.F.; Tam, C.; Kim, S.Y.; Fein, J.; Yu, L.; Brown, J.R.; Westerman, D.; Si, E.G.; et al. The BCL2 Selective Inhibitor Venetoclax Induces Rapid Onset Apoptosis of CLL Cells in Patients via a TP53-Independent Mechanism. Blood 2016, 127, 3215–3224. [Google Scholar] [CrossRef]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Döhner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax Resistance and Acquired BCL2 Mutations in Chronic Lymphocytic Leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef]
- Birkinshaw, R.W.; Gong, J.-N.; Luo, C.S.; Lio, D.; White, C.A.; Anderson, M.A.; Blombery, P.; Lessene, G.; Majewski, I.J.; Thijssen, R.; et al. Structures of BCL-2 in Complex with Venetoclax Reveal the Molecular Basis of Resistance Mutations. Nat. Commun. 2019, 10, 2385. [Google Scholar] [CrossRef]
- Blombery, P.; Anderson, M.A.; Gong, J.-N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef]
- Weiss, J.; Peifer, M.; Herling, C.D.; Frenzel, L.P.; Hallek, M. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia (Comment to Tausch et al.). Haematologica 2019, 104, e540. [Google Scholar] [CrossRef]
- Nachmias, B.; Aumann, S.; Haran, A.; Schimmer, A.D. Venetoclax Resistance in Acute Myeloid Leukaemia-Clinical and Biological Insights. Br. J. Haematol. 2024, 204, 1146–1158. [Google Scholar] [CrossRef]
- Brown, F.C.; Wang, X.; Birkinshaw, R.; Chua, C.C.; Morley, T.; Kasapgil, S.; Pomilio, G.; Blombery, P.; Huang, D.C.S.; Czabotar, P.; et al. Acquired BCL2 Variants Associated with Venetoclax Resistance in Acute Myeloid Leukemia. Blood Adv. 2025, 9, 127–131. [Google Scholar] [CrossRef]
- Blombery, P.; Lew, T.E.; Dengler, M.A.; Thompson, E.R.; Lin, V.S.; Chen, X.; Nguyen, T.; Panigrahi, A.; Handunnetti, S.M.; Carney, D.A.; et al. Clonal Hematopoiesis, Myeloid Disorders and BAX-Mutated Myelopoiesis in Patients Receiving Venetoclax for CLL. Blood 2022, 139, 1198–1207. [Google Scholar] [CrossRef]
- Moujalled, D.M.; Brown, F.C.; Chua, C.C.; Dengler, M.A.; Pomilio, G.; Anstee, N.S.; Litalien, V.; Thompson, E.; Morley, T.; MacRaild, S.; et al. Acquired Mutations in BAX Confer Resistance to BH3-Mimetic Therapy in Acute Myeloid Leukemia. Blood 2023, 141, 634–644. [Google Scholar] [CrossRef]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Döhner, H.; Pratz, K.W.; DiNardo, C.D.; Wei, A.H.; Jonas, B.A.; Pullarkat, V.A.; Thirman, M.J.; Récher, C.; Schuh, A.C.; Babu, S.; et al. Genetic Risk Stratification and Outcomes among Treatment-Naive Patients with AML Treated with Venetoclax and Azacitidine. Blood 2024, 144, 2211–2222. [Google Scholar] [CrossRef]
- Bataller, A.; Bazinet, A.; DiNardo, C.D.; Maiti, A.; Borthakur, G.; Daver, N.G.; Short, N.J.; Jabbour, E.J.; Issa, G.C.; Pemmaraju, N.; et al. Prognostic Risk Signature in Patients with Acute Myeloid Leukemia Treated with Hypomethylating Agents and Venetoclax. Blood Adv. 2024, 8, 927–935. [Google Scholar] [CrossRef]
- Sango, J.; Carcamo, S.; Sirenko, M.; Maiti, A.; Mansour, H.; Ulukaya, G.; Tomalin, L.E.; Cruz-Rodriguez, N.; Wang, T.; Olszewska, M.; et al. RAS-Mutant Leukaemia Stem Cells Drive Clinical Resistance to Venetoclax. Nature 2024, 636, 241–250. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular Patterns of Response and Treatment Failure after Frontline Venetoclax Combinations in Older Patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- Venugopal, S.; Shoukier, M.; Konopleva, M.; Dinardo, C.D.; Ravandi, F.; Short, N.J.; Andreeff, M.; Borthakur, G.; Daver, N.; Pemmaraju, N.; et al. Outcomes in Patients with Newly Diagnosed TP53-Mutated Acute Myeloid Leukemia with or without Venetoclax-Based Therapy. Cancer 2021, 127, 3541–3551. [Google Scholar] [CrossRef]
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch, C.R.; Furudate, K.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; et al. Outcomes of TP53-Mutant Acute Myeloid Leukemia with Decitabine and Venetoclax. Cancer 2021, 127, 3772–3781. [Google Scholar] [CrossRef]
- Konopleva, M.; Thirman, M.J.; Pratz, K.W.; Garcia, J.S.; Recher, C.; Pullarkat, V.; Kantarjian, H.M.; DiNardo, C.D.; Dail, M.; Duan, Y.; et al. Impact of FLT3 Mutation on Outcomes after Venetoclax and Azacitidine for Patients with Treatment-Naïve Acute Myeloid Leukemia. Clin. Cancer Res. 2022, 28, 2744–2752. [Google Scholar] [CrossRef]
- Chyla, B.J.; Harb, J.; Mantis, C.; Riehm, J.J.; Ross, J.A.; Sun, Y.; Huang, X.; Jiang, Q.; Dail, M.; Peale, F.V.; et al. Response to Venetoclax in Combination with Low Intensity Therapy (LDAC or HMA) in Untreated Patients with Acute Myeloid Leukemia Patients with IDH, FLT3 and Other Mutations and Correlations with BCL2 Family Expression. Blood 2019, 134, 546. [Google Scholar] [CrossRef]
- Daver, N.G.; Iqbal, S.; Huang, J.; Renard, C.; Lin, J.; Pan, Y.; Williamson, M.; Ramsingh, G. Clinical Characteristics and Overall Survival among Acute Myeloid Leukemia Patients with TP53 Gene Mutation or Chromosome 17p Deletion. Am. J. Hematol. 2023, 98, 1176–1184. [Google Scholar] [CrossRef]
- Daver, N.G.; Iqbal, S.; Renard, C.; Chan, R.J.; Hasegawa, K.; Hu, H.; Tse, P.; Yan, J.; Zoratti, M.J.; Xie, F.; et al. Treatment Outcomes for Newly Diagnosed, Treatment-Naïve TP53-Mutated Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. J. Hematol. Oncol. 2023, 16, 19. [Google Scholar] [CrossRef]
- Chua, C.C.; Reynolds, J.; Salmon, J.M.; Fong, C.; Ting, S.B.; Tiong, I.S.; Fleming, S.; MacRaild, S.; Moujalled, D.M.; Pomilio, G.; et al. Anti-Leukemic Activity of Single Agent Venetoclax in Newly Diagnosed Acute Myeloid Leukemia: A Sub-Set Analysis of the Caveat Study. Blood 2019, 134, 462. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Thijssen, R.; Diepstraten, S.T.; Moujalled, D.; Chew, E.; Flensburg, C.; Shi, M.X.; Dengler, M.A.; Litalien, V.; MacRaild, S.; Chen, M.; et al. Intact TP-53 Function Is Essential for Sustaining Durable Responses to BH3-Mimetic Drugs in Leukemias. Blood 2021, 137, 2721–2735. [Google Scholar] [CrossRef]
- Findley, H.W.; Gu, L.; Yeager, A.M.; Zhou, M. Expression and Regulation of Bcl-2, Bcl-Xl, and Bax Correlate with P53 Status and Sensitivity to Apoptosis in Childhood Acute Lymphoblastic Leukemia. Blood 1997, 89, 2986–2993. [Google Scholar] [CrossRef]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional Genomic Landscape of Acute Myeloid Leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Chyla, B.; Daver, N.; Doyle, K.; McKeegan, E.; Huang, X.; Ruvolo, V.; Wang, Z.; Chen, K.; Souers, A.; Leverson, J.; et al. Genetic Biomarkers of Sensitivity and Resistance to Venetoclax Monotherapy in Patients with Relapsed Acute Myeloid Leukemia. Am. J. Hematol. 2018, 93, E202–E205. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, S.; Qiao, X.; Knight, T.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Wang, G.; et al. Inhibition of Bcl-2 Synergistically Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6815–6826. [Google Scholar] [CrossRef]
- Young, A.I.; Timpson, P.; Gallego-Ortega, D.; Ormandy, C.J.; Oakes, S.R. Myeloid Cell Leukemia 1 (MCL-1), an Unexpected Modulator of Protein Kinase Signaling during Invasion. Cell Adhes. Migr. 2018, 12, 513–523. [Google Scholar] [CrossRef]
- Dumon, S.; Santos, S.C.R.; Debierre-Grockiego, F.; Gouilleux-Gruart, V.; Cocault, L.; Boucheron, C.; Mollat, P.; Gisselbrecht, S.; Gouilleux, F. IL-3 Dependent Regulation of Bcl-xL Gene Expression by STAT5 in a Bone Marrow Derived Cell Line. Oncogene 1999, 18, 4191–4199. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-Regulates MCL-1 to Promote Survival of Stem Cells in Acute Myeloid Leukemia via FLT3-ITD–Specific STAT5 Activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef]
- Janssen, M.; Schmidt, C.; Bruch, P.-M.; Blank, M.F.; Rohde, C.; Waclawiczek, A.; Heid, D.; Renders, S.; Göllner, S.; Vierbaum, L.; et al. Venetoclax Synergizes with Gilteritinib in FLT3 Wild-Type High-Risk Acute Myeloid Leukemia by Suppressing MCL-1. Blood 2022, 140, 2594–2610. [Google Scholar] [CrossRef]
- Kuusanmäki, H.; Leppä, A.-M.; Pölönen, P.; Kontro, M.; Dufva, O.; Deb, D.; Yadav, B.; Brück, O.; Kumar, A.; Everaus, H.; et al. Phenotype-Based Drug Screening Reveals Association between Venetoclax Response and Differentiation Stage in Acute Myeloid Leukemia. Haematologica 2020, 105, 708–720. [Google Scholar] [CrossRef]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef]
- Kuusanmäki, H.; Dufva, O.; Vähä-Koskela, M.; Leppä, A.-M.; Huuhtanen, J.; Vänttinen, I.; Nygren, P.; Klievink, J.; Bouhlal, J.; Pölönen, P.; et al. Erythroid/Megakaryocytic Differentiation Confers BCL-XL Dependency and Venetoclax Resistance in Acute Myeloid Leukemia. Blood 2023, 141, 1610–1625. [Google Scholar] [CrossRef] [PubMed]
- Garciaz, S.; Guirguis, A.A.; Müller, S.; Brown, F.C.; Chan, Y.-C.; Motazedian, A.; Rowe, C.L.; Kuzich, J.A.; Chan, K.L.; Tran, K.; et al. Pharmacologic Reduction of Mitochondrial Iron Triggers a Noncanonical BAX/BAK-Dependent Cell Death. Cancer Discov. 2022, 12, 774–791. [Google Scholar] [CrossRef]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef]
- Hege Hurrish, K.; Qiao, X.; Li, X.; Su, Y.; Carter, J.; Ma, J.; Kalpage, H.A.; Hüttemann, M.; Edwards, H.; Wang, G.; et al. Co-Targeting of HDAC, PI3K, and Bcl-2 Results in Metabolic and Transcriptional Reprogramming and Decreased Mitochondrial Function in Acute Myeloid Leukemia. Biochem. Pharmacol. 2022, 205, 115283. [Google Scholar] [CrossRef]
- Roca-Portoles, A.; Rodriguez-Blanco, G.; Sumpton, D.; Cloix, C.; Mullin, M.; Mackay, G.M.; O’Neill, K.; Lemgruber, L.; Luo, X.; Tait, S.W.G. Venetoclax Causes Metabolic Reprogramming Independent of BCL-2 Inhibition. Cell Death Dis. 2020, 11, 616. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty Acid Metabolism Underlies Venetoclax Resistance in Acute Myeloid Leukemia Stem Cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Naji, N.S.; Sathish, M.; Karantanos, T. Inflammation and Related Signaling Pathways in Acute Myeloid Leukemia. Cancers 2024, 16, 3974. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Reville, P.K.; Yassouf, M.Y.; Jelloul, F.Z.; Ly, C.; Desai, P.N.; Wang, Z.; Borges, P.; Veletic, I.; Dasdemir, E.; et al. Comprehensive Characterization of IFNγ Signaling in Acute Myeloid Leukemia Reveals Prognostic and Therapeutic Strategies. Nat. Commun. 2024, 15, 1821. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Zhang, Q.; Ruvolo, V.; Kuruvilla, V.M.; Wang, X.; Mak, D.H.; Battula, V.L.; Konopleva, M.; et al. Maximal Activation of Apoptosis Signaling by Cotargeting Antiapoptotic Proteins in BH3 Mimetic-Resistant AML and AML Stem Cells. Mol. Cancer Ther. 2022, 21, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Teh, T.-C.; Nguyen, N.-Y.; Moujalled, D.M.; Segal, D.; Pomilio, G.; Rijal, S.; Jabbour, A.; Cummins, K.; Lackovic, K.; Blombery, P.; et al. Enhancing Venetoclax Activity in Acute Myeloid Leukemia by Co-Targeting MCL1. Leukemia 2018, 32, 303–312. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Ayoub, E.; Ostermann, L.B.; Huang, X.; Loghavi, S.; Boettcher, S.; Nishida, Y.; Ruvolo, V.; et al. Combined Inhibition of BCL-2 and MCL-1 Overcomes BAX Deficiency-Mediated Resistance of TP53-Mutant Acute Myeloid Leukemia to Individual BH3 Mimetics. Blood Cancer J. 2023, 13, 57. [Google Scholar] [CrossRef]
- Desai, P.; Lonial, S.; Cashen, A.; Kamdar, M.; Flinn, I.; O’Brien, S.; Garcia, J.S.; Korde, N.; Moslehi, J.; Wey, M.; et al. A Phase 1 First-in-Human Study of the MCL-1 Inhibitor AZD5991 in Patients with Relapsed/Refractory Hematologic Malignancies. Clin. Cancer Res. 2024, 30, 4844–4855. [Google Scholar] [CrossRef]
- Balachander, S.B.; Criscione, S.W.; Byth, K.F.; Cidado, J.; Adam, A.; Lewis, P.; Macintyre, T.; Wen, S.; Lawson, D.; Burke, K.; et al. AZD4320, A Dual Inhibitor of Bcl-2 and Bcl-xL, Induces Tumor Regression in Hematologic Cancer Models without Dose-Limiting Thrombocytopenia. Clin. Cancer Res. 2020, 26, 6535–6549. [Google Scholar] [CrossRef]
- Lv, D.; Pal, P.; Liu, X.; Jia, Y.; Thummuri, D.; Zhang, P.; Hu, W.; Pei, J.; Zhang, Q.; Zhou, S.; et al. Development of a BCL-xL and BCL-2 Dual Degrader with Improved Anti-Leukemic Activity. Nat. Commun. 2021, 12, 6896. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; McCloskey, J.; Griffiths, E.A.; Yee, K.W.L.; Zeidan, A.M.; Al-Kali, A.; Deeg, H.J.; Patel, P.A.; Sabloff, M.; Keating, M.-M.; et al. Oral Decitabine-Cedazuridine versus Intravenous Decitabine for Myelodysplastic Syndromes and Chronic Myelomonocytic Leukaemia (ASCERTAIN): A Registrational, Randomised, Crossover, Pharmacokinetics, Phase 3 Study. Lancet Haematol. 2024, 11, e15–e26. [Google Scholar] [CrossRef]
- Wei, A.H.; Döhner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.-H.; Porkka, K.; et al. Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N. Engl. J. Med. 2020, 383, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-R.; Wan, C.-L.; Liu, S.-B.; Qiu, Q.-C.; Wu, T.-M.; Wang, J.; Li, Y.-Y.; Ge, S.-S.; Qiu, Y.; Shen, X.-D.; et al. A Combined Histone Deacetylases Targeting Strategy to Overcome Venetoclax Plus Azacitidine Regimen Resistance in Acute Myeloid Leukaemia: Three Case Reports. Front. Oncol. 2021, 11, 797941. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, J.; Zhou, M.; Li, J.-L.; Qiu, Q.-C.; Fu, J.-H.; Xue, S.-L.; Qiu, H.-Y. Epigenetic Therapy with Chidamide Alone or Combined with 5-azacitidine Exerts Antitumour Effects on Acute Myeloid Leukaemia Cells in Vitro. Oncol. Rep. 2022, 47, 66. [Google Scholar] [CrossRef]
- Maes, T.; Mascaró, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdones, A.; Lufino, M.M.P.; Somervaille, T.C.P.; et al. ORY-1001, a Potent and Selective Covalent KDM1A Inhibitor, for the Treatment of Acute Leukemia. Cancer Cell 2018, 33, 495–511.e12. [Google Scholar] [CrossRef] [PubMed]
- Salamero, O.; Montesinos, P.; Willekens, C.; Pérez-Simón, J.A.; Pigneux, A.; Récher, C.; Popat, R.; Carpio, C.; Molinero, C.; Mascaró, C.; et al. First-in-Human Phase I Study of Iadademstat (ORY-1001): A First-in-Class Lysine-Specific Histone Demethylase 1A Inhibitor, in Relapsed or Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2020, 38, 4260–4273. [Google Scholar] [CrossRef]
- Jalte, M.; Abbassi, M.; El Mouhi, H.; Daha Belghiti, H.; Ahakoud, M.; Bekkari, H. FLT3 Mutations in Acute Myeloid Leukemia: Unraveling the Molecular Mechanisms and Implications for Targeted Therapies. Cureus 2023, 15, e45765. [Google Scholar] [CrossRef]
- Seipel, K.; Graber, C.; Flückiger, L.; Bacher, U.; Pabst, T. Rationale for a Combination Therapy with the STAT5 Inhibitor AC-4-130 and the MCL1 Inhibitor S63845 in the Treatment of FLT3-Mutated or TET2-Mutated Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 8092. [Google Scholar] [CrossRef]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.; McCloskey, J.; Smith, C.C.; Schiller, G.; Bradley, T.; et al. Venetoclax Plus Gilteritinib for FLT3-Mutated Relapsed/Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2022, 40, 4048–4059. [Google Scholar] [CrossRef]
- Short, N.J.; Daver, N.; Dinardo, C.D.; Kadia, T.; Nasr, L.F.; Macaron, W.; Yilmaz, M.; Borthakur, G.; Montalban-Bravo, G.; Garcia-Manero, G.; et al. Azacitidine, Venetoclax, and Gilteritinib in Newly Diagnosed and Relapsed or Refractory FLT3-Mutated AML. J. Clin. Oncol. 2024, 42, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Kantarjian, H.; Short, N.J.; Reville, P.; Konopleva, M.; Kadia, T.; DiNardo, C.; Borthakur, G.; Pemmaraju, N.; Maiti, A.; et al. Hypomethylating Agent and Venetoclax with FLT3 Inhibitor “Triplet” Therapy in Older/Unfit Patients with FLT3 Mutated AML. Blood Cancer J. 2022, 12, 77. [Google Scholar] [CrossRef]
- Maiti, A.; DiNardo, C.D.; Daver, N.G.; Rausch, C.R.; Ravandi, F.; Kadia, T.M.; Pemmaraju, N.; Borthakur, G.; Bose, P.; Issa, G.C.; et al. Triplet Therapy with Venetoclax, FLT3 Inhibitor and Decitabine for FLT3-Mutated Acute Myeloid Leukemia. Blood Cancer J. 2021, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Muftuoglu, M.; DiNardo, C.D.; Kadia, T.M.; Konopleva, M.Y.; Borthakur, G.; Pemmaraju, N.; Short, N.J.; Alvarado Valero, Y.; Maiti, A.; et al. Phase I/II Study of Quizartinib, Venetoclax, and Decitabine Triple Combination in FLT3-ITD Mutated AML. Blood 2023, 142, 158. [Google Scholar] [CrossRef]
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycka, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.-P.; et al. Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N. Engl. J. Med. 2022, 386, 1519–1531. [Google Scholar] [CrossRef]
- Lachowiez, C.A.; Loghavi, S.; Zeng, Z.; Tanaka, T.; Kim, Y.J.; Uryu, H.; Turkalj, S.; Jakobsen, N.A.; Luskin, M.R.; Duose, D.Y.; et al. A Phase Ib/II Study of Ivosidenib with Venetoclax ± Azacitidine in IDH1-Mutated Myeloid Malignancies. Blood Cancer Discov. 2023, 4, 276–293. [Google Scholar] [CrossRef]
- Venugopal, S.; Takahashi, K.; Daver, N.; Maiti, A.; Borthakur, G.; Loghavi, S.; Short, N.J.; Ohanian, M.; Masarova, L.; Issa, G.; et al. Efficacy and Safety of Enasidenib and Azacitidine Combination in Patients with IDH2 Mutated Acute Myeloid Leukemia and Not Eligible for Intensive Chemotherapy. Blood Cancer J. 2022, 12, 10. [Google Scholar] [CrossRef]
- Ball, S.; Byrne, M.T.; Fischer, M.A.; Awan, F.T.; Tomlinson, B.K.; Stopczynski, T.; Patil, L.; Gu, C.J.; Leonardi, C.; Walsh, K.J.; et al. Application of a Phenotype-Based Predictive Analytic in a Phase Ib Study of Selinexor and Venetoclax in Patients with Relapsed or Refractory Acute Myeloid Leukemia. Blood 2024, 144, 6012. [Google Scholar] [CrossRef]
- Yang, L.; Chen, F.; Liang, H.; Bai, Y.; Wu, W.; Yan, X.; Ren, J.; Li, S.; Yu, Y.; Tong, L.; et al. Selinexor in Combination with Venetoclax and Azacitidine for Newly Diagnosed (ND) Unfit Acute Myeloid Leukemia (AML): A Multicenter, Open-Label Prospective Study. Blood 2023, 142, 55. [Google Scholar] [CrossRef]
- Zha, J.; Wang, Y.; Li, Z.; Lin, Z.; Cai, Y.; Gong, T.; Yang, W.; Xie, S.; Huang, Z.; Shi, P.; et al. Update Results of a Phase II Trial of Venetoclax in Combination with Azacitidine and Chidamide in Relapsed/Refractory Acute Myeloid Leukemia. Blood 2023, 142, 4274. [Google Scholar] [CrossRef]
- Daver, N.G.; Dail, M.; Garcia, J.S.; Jonas, B.A.; Yee, K.W.L.; Kelly, K.R.; Vey, N.; Assouline, S.; Roboz, G.J.; Paolini, S.; et al. Venetoclax and Idasanutlin in Relapsed/Refractory AML: A Nonrandomized, Open-Label Phase 1b Trial. Blood 2023, 141, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Cuglievan, B.; Daver, N.; DiNardo, C.D.; Farhat, A.; Short, N.J.; McCall, D.; Pike, A.; Tan, S.; Kammerer, B.; et al. Phase I/II Study of the All-Oral Combination of Revumenib (SNDX-5613) with Decitabine/Cedazuridine (ASTX727) and Venetoclax (SAVE) in R/R AML. Blood 2024, 144, 216. [Google Scholar] [CrossRef]
- Wei, A.H.; Searle, E.; Aldoss, I.; Alfonso-Piérola, A.; Alonso-Dominguez, J.M.; Curtis, M.; Daskalakis, N.; Della Porta, M.G.; Döhner, H.; D’Souza, A.; et al. A Phase 1b Study of the Menin-Kmt2a Inhibitor jnj-75276617 in Combination with Venetoclax and Azacitidine in Relapsed/Refractory Acute Myeloid Leukemia with Alterations in kmt2a or npm1; EHA Abstr. Book: Hague, The Netherlands, 2024. [Google Scholar]
- Jonas, B.A.; Hou, J.-Z.; Roboz, G.J.; Alvares, C.L.; Jeyakumar, D.; Edwards, J.R.; Erba, H.P.; Kelly, R.J.; Röllig, C.; Fiedler, W.; et al. A Phase 1b Study of Venetoclax and Alvocidib in Patients with Relapsed/Refractory Acute Myeloid Leukemia. Hematol. Oncol. 2023, 41, 743–752. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, Y.; Wu, W.; Ge, Z.; Liu, L.; Ji, C.; Gu, W.; Wang, C.; Zhang, Y.; Yang, W.; et al. High Remission Rates in Relapsed/Refractory Acute Myeloid Leukemia with QHRD107 (CDK9 Inhibitor), Venetoclax, and Azacitidine Combination Therapy (107VA Regimen): Preliminary Results of a Phase 2a Study. Blood 2024, 144, 2882. [Google Scholar] [CrossRef]
- Desikan, S.P.; Ravandi, F.; Pemmaraju, N.; Konopleva, M.; Loghavi, S.; Jabbour, E.J.; Daver, N.; Jain, N.; Chien, K.S.; Maiti, A.; et al. A Phase II Study of Azacitidine, Venetoclax, and Trametinib in Relapsed or Refractory Acute Myeloid Leukemia Harboring RAS Pathway-Activating Mutations. Acta Haematol. 2022, 145, 529–536. [Google Scholar] [CrossRef]
- Daver, N.; Pollyea, D.A.; Yee, K.W.L.; Fenaux, P.; Brandwein, J.M.; Vey, N.; Martinelli, G.; Kelly, K.R.; Roboz, G.J.; Garcia, J.S.; et al. Preliminary Results from a Phase Ib Study Evaluating BCL-2 Inhibitor Venetoclax in Combination with MEK Inhibitor Cobimetinib or MDM2 Inhibitor Idasanutlin in Patients with Relapsed or Refractory (R/R) AML. Blood 2017, 130, 813. [Google Scholar] [CrossRef]
- Lane, A.A.; Garcia, J.S.; Raulston, E.G.; Garzon, J.L.; Galinsky, I.; Baxter, E.W.; Leonard, R.; DeAngelo, D.J.; Luskin, M.R.; Reilly, C.R.; et al. Phase 1b Trial of Tagraxofusp in Combination with Azacitidine with or without Venetoclax in Acute Myeloid Leukemia. Blood Adv. 2024, 8, 591–602. [Google Scholar] [CrossRef]
- Bazinet, A.; Garcia-Manero, G.; Short, N.; Alvarado, Y.; Bataller, A.; Abuasab, T.; Islam, R.; Montalbano, K.; Issa, G.; Maiti, A.; et al. Oral Decitabine and Cedazuridine plus Venetoclax for Older or Unfit Patients with Acute Myeloid Leukaemia: A Phase 2 Study. Lancet Haematol. 2024, 11, e276–e286. [Google Scholar] [CrossRef]
- Short, N.J.; Muftuoglu, M.; Ong, F.; Nasr, L.; Macaron, W.; Montalban-Bravo, G.; Alvarado, Y.; Basyal, M.; Daver, N.; Dinardo, C.D.; et al. A Phase 1/2 Study of Azacitidine, Venetoclax and Pevonedistat in Newly Diagnosed Secondary AML and in MDS or CMML after Failure of Hypomethylating Agents. J. Hematol. Oncol. 2023, 16, 73. [Google Scholar] [CrossRef]
- Murthy, G.S.G.; Saliba, A.N.; Szabo, A.; Harrington, A.; Abedin, S.; Carlson, K.; Michaelis, L.; Runaas, L.; Baim, A.; Hinman, A.; et al. A Phase I Study of Pevonedistat, Azacitidine, and Venetoclax in Patients with Relapsed/Refractory Acute Myeloid Leukemia. Haematologica 2024, 109, 2864–2872. [Google Scholar] [CrossRef]
- Daver, N.; Konopleva, M.; Maiti, A.; Kadia, T.M.; DiNardo, C.D.; Loghavi, S.; Pemmaraju, N.; Jabbour, E.J.; Montalban-Bravo, G.; Tang, G.; et al. Phase I/II Study of Azacitidine (AZA) with Venetoclax (VEN) and Magrolimab (Magro) in Patients (Pts) with Newly Diagnosed Older/Unfit or High-Risk Acute Myeloid Leukemia (AML) and Relapsed/Refractory (R/R) AML. Blood 2021, 138, 371. [Google Scholar] [CrossRef]
- Daver, N.; Vyas, P.; Huls, G.; Döhner, H.; Maury, S.; Novák, J.; Papayannidis, C.; Chamorro, C.M.; Montesinos, P.; Niroula, R.; et al. Magrolimab vs Placebo in Combination with Venetoclax and Azacitidine in Previously Untreated Patients with Acute Myeloid Leukemia Who Are Ineligible for Intensive Chemotherapy: The Enhance-3 Study; EHA Abstr. Book: Hague, The Netherlands, 2024. [Google Scholar]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 Inhibition: An Important Step Forward in Cancer Therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and P53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748–760.e6. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Wei, A.H.; Stein, E.; DeAngelo, D.J.; Pathak, D.; Xu, Y.; Grzesiak, S.; Venditti, A. Siremadlin in Combination with Venetoclax (VEN) Plus Azacitidine (AZA) in Adult Patients with Acute Myeloid Leukemia (AML) Who Are Ineligible for Intensive Chemotherapy: A Phase Ib/II Trial. Blood 2022, 140, 11625–11627. [Google Scholar] [CrossRef]
- Fischer, M.A.; Friedlander, S.Y.; Arrate, M.P.; Chang, H.; Gorska, A.E.; Fuller, L.D.; Ramsey, H.E.; Kashyap, T.; Argueta, C.; Debler, S.; et al. Venetoclax Response Is Enhanced by Selective Inhibitor of Nuclear Export Compounds in Hematologic Malignancies. Blood Adv. 2020, 4, 586–598. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, Y.; Liu, D.; Wang, X.; Zhu, Y.; Tong, J.; Chen, E.; Xue, L.; Zhao, N.; Liang, T.; et al. Selinexor Synergistically Promotes the Antileukemia Activity of Venetoclax in Acute Myeloid Leukemia by Inhibiting Glycolytic Function and Downregulating the Expression of DNA Replication Genes. Immunotargets Ther. 2023, 12, 135–147. [Google Scholar] [CrossRef]
- Nishida, Y.; Ishizawa, J.; Ayoub, E.; Montoya, R.H.; Ostermann, L.B.; Muftuoglu, M.; Ruvolo, V.R.; Patsilevas, T.; Scruggs, D.A.; Khazaei, S.; et al. Enhanced TP53 Reactivation Disrupts MYC Transcriptional Program and Overcomes Venetoclax Resistance in Acute Myeloid Leukemias. Sci. Adv. 2023, 9, eadh1436. [Google Scholar] [CrossRef]
- Bogenberger, J.; Whatcott, C.; Hansen, N.; Delman, D.; Shi, C.-X.; Kim, W.; Haws, H.; Soh, K.; Lee, Y.S.; Peterson, P.; et al. Combined Venetoclax and Alvocidib in Acute Myeloid Leukemia. Oncotarget 2017, 8, 107206–107222. [Google Scholar] [CrossRef]
- Knorr, K.L.B.; Schneider, P.A.; Meng, X.W.; Dai, H.; Smith, B.D.; Hess, A.D.; Karp, J.E.; Kaufmann, S.H. MLN4924 Induces Noxa Upregulation in Acute Myelogenous Leukemia and Synergizes with Bcl-2 Inhibitors. Cell Death Differ. 2015, 22, 2133–2142. [Google Scholar] [CrossRef]
- Han, L.; Zhang, Q.; Dail, M.; Shi, C.; Cavazos, A.; Ruvolo, V.R.; Zhao, Y.; Kim, E.; Rahmani, M.; Mak, D.H.; et al. Concomitant Targeting of BCL2 with Venetoclax and MAPK Signaling with Cobimetinib in Acute Myeloid Leukemia Models. Haematologica 2020, 105, 697–707. [Google Scholar] [CrossRef]
- Popescu, B.; Stieglitz, E.; Smith, C.C. RAS MULTI(ON) Inhibitor RMC-7977 Targets Oncogenic RAS Mutations and Overcomes RAS/MAPK-Mediated Resistance to FLT3 Inhibitors in AML Models. Blood 2023, 142, 2793. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi, A.T.; Odenike, O.; Roboz, G.J.; Sweet, K.; Miller, C.; Wennborg, A.; et al. Eprenetapopt Combined with Venetoclax and Azacitidine in TP53-Mutated Acute Myeloid Leukaemia: A Phase 1, Dose-Finding and Expansion Study. Lancet Haematol. 2023, 10, e272–e283. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Munoz-Sagredo, L.; Streule, K.; Muschong, P.; Bayer, E.; Walter, R.J.; Gutjahr, J.C.; Greil, R.; Concha, M.L.; Müller-Tidow, C.; et al. CD44 Loss of Function Sensitizes AML Cells to the BCL-2 Inhibitor Venetoclax by Decreasing CXCL12-Driven Survival Cues. Blood 2021, 138, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
| Drug Regimen | Identifier Clinicaltrias.gov | Eligibility Criteria Other than AML | Treatment Category | Study Phase | Year of Initiation |
|---|---|---|---|---|---|
| Exportin 1 (XPO1) inhibitors | |||||
| Selinexor + VEN | NCT03955783 | AML or DLBCL | Salvage | Ib | 2019 |
| Selinexor + VA (SAV) | NCT05736965 | Frontline | II | 2023 | |
| SAV | NCT06449482 | Frontline | I/II | 2023 | |
| Eltanexor + VEN | NCT06399640 | AML or MDS | Salvage | I | 2024 |
| HDAC inhibitors | |||||
| Chidamide + VA (VCA) | NCT05305859 | Salvage | II | 2022 | |
| VCA | NCT05566054 | Frontline | II | 2022 | |
| VCA | NCT06220162 | Failed response to VA | Salvage | II | 2024 |
| VCA | NCT06386302 | Frontline | II | 2024 | |
| IDH inhibitors | |||||
| Ivosidenib + VEN or VA | NCT03471260 | IDH1 mutated myeloid malignancies | Salvage | Ib/II | 2018 |
| ASTX727 + VEN + IDH inhibitor | NCT04774393 | Salvage | Ib/II | 2021 | |
| Ivosidenib + VEN | NCT06611839 | IDH1 mutation | Both | I/II | 2024 |
| FLT3 inhibitors | |||||
| Quizartinib + DAC + VEN | NCT03661307 | Both | I/II | 2018 | |
| Gilteritinib + VA | NCT04140487 | FLT3mut CMML, MDS/MPN, AML | Salvage | I/II | 2019 |
| Quizartinib +VA or (LDAC + VEN) (VEN-A-QUI trial) | NCT04687761 | Frontline | I/II | 2020 | |
| ASTX727 + gilteritinib + VEN | NCT05010122 | FLT3mut | Both | I/II | 2021 |
| Gilteritinib + VA | NCT05520567 | FLT3mut | Frontline | I/II | 2023 |
| Gilteritinib + VA vs. VA | NCT06317649 | FLT3mut | Frontline | II | 2024 |
| Gilteritinib + VA | NCT06696183 | FLT3mut | Frontline | II | 2025 |
| MDM2 inhibitors | |||||
| KRT-232 + DAC + VEN | NCT03041688 | Both | Ib | 2018 | |
| Siremadlin + VA | NCT05155709 | Both | Ib/II | 2022 | |
| Menin inhibitors | |||||
| Revumenib + ASTX727 + VEN | NCT05360160 | KMT2Ar, NUP98r, or NPM1c | Both | I/II | 2022 |
| Revumenib + VA | NCT06652438 | KMT2Ar, or NPM1c | Frontline | III | 2024 |
| Bleximenib + VA | NCT05453903 | Frontline | Ib | 2022 | |
| MCL1 inhibitors | |||||
| AMG176 +/− VEN | NCT02675452 | AML or MM | Salvage | I | 2016 |
| S64315 + VEN | NCT03672695 | Salvage | I | 2018 | |
| CDK inhibitors | |||||
| Alvocidib + VEN | NCT03441555 | Salvage | Ib | 2018 | |
| CYC065 + VEN | NCT04017546 | Salvage | I | 2019 | |
| QHRD107 + VA | NCT06532058 | Salvage | II | 2023 | |
| RVU120 + VEN (RIVER-81 trial) | NCT06191263 | Salvage | II | 2024 | |
| PD1 inhibitors | |||||
| Pembrolizumab + DAC +/− VEN | NCT03969446 | AML or MDS | Both | Ib | 2020 |
| AK117 + VA | NCT06387420 | Both | I | 2024 | |
| Tislelizumab + VA | NCT06536959 | AML or MDS | Salvage | II | 2024 |
| CD123 targeting molecules | |||||
| IMGN632 + VA | NCT04086264 | Salvage | Ib/II | 2019 | |
| Tagraxofusp + VA | NCT06456463 | CD123+ | Frontline | II | 2022 |
| Tagraxofusp + VA | NCT05442216 | sAML post HMA | Salvage | II | 2024 |
| APVO436 + VEN (RAINIER trial) | NCT06634394 | CD123+ AML | Frontline | Ib/II | 2024 |
| Tagraxofusp + VA | NCT06456463 | CD123+ AML | Frontline | II | 2025 |
| Enhancing drug activity | |||||
| ASTX727 + VEN | NCT04746235 | Both | II | 2021 | |
| ASTX727 + VEN | NCT04657081 | Frontline | I/II | 2021 | |
| Cobicistat + VA | NCT06014489 | Frontline | II | 2024 | |
| NEDD8-activating enzyme (NAE) inhibitor | |||||
| Pevonedistat + VA vs. VA | NCT04266795 | Frontline | II | 2020 | |
| Others | |||||
| Lintuzumab + VEN | NCT03867682 | Salvage | I/II | 2020 | |
| Cusatuzumab + VA | NCT04150887 | Frontline | Ib | 2019 | |
| Cusatuzumab + VA | NCT06384261 | Frontline | II | 2024 | |
| evorpacept + VA (ASPEN-05) | NCT04755244 | Both | Ι/ΙΙ | 2021 | |
| Navitoclax + VA | NCT05222984 | VEN treated AML | Salvage | I | 2022 |
| Iadademstat + VA | NCT06357182 | Frontline | Ib | 2024 | |
| Iadademstat + VA | NCT06514261 | Frontline | I | 2025 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatzilygeroudi, T.; Karantanos, T.; Pappa, V. Unraveling Venetoclax Resistance: Navigating the Future of HMA/Venetoclax-Refractory AML in the Molecular Era. Cancers 2025, 17, 1586. https://doi.org/10.3390/cancers17091586
Chatzilygeroudi T, Karantanos T, Pappa V. Unraveling Venetoclax Resistance: Navigating the Future of HMA/Venetoclax-Refractory AML in the Molecular Era. Cancers. 2025; 17(9):1586. https://doi.org/10.3390/cancers17091586
Chicago/Turabian StyleChatzilygeroudi, Theodora, Theodoros Karantanos, and Vasiliki Pappa. 2025. "Unraveling Venetoclax Resistance: Navigating the Future of HMA/Venetoclax-Refractory AML in the Molecular Era" Cancers 17, no. 9: 1586. https://doi.org/10.3390/cancers17091586
APA StyleChatzilygeroudi, T., Karantanos, T., & Pappa, V. (2025). Unraveling Venetoclax Resistance: Navigating the Future of HMA/Venetoclax-Refractory AML in the Molecular Era. Cancers, 17(9), 1586. https://doi.org/10.3390/cancers17091586