Modeling the Transitional Phase of Epithelial Cells Reveals Prognostic and Therapeutic Targets in Pancreatic Ductal Adenocarcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Single Cell Analysis
2.2. Differentiation Analysis
2.3. Differential Expression Analysis
2.4. Continuous Expressed Gene Pattern Analysis Across PDAC Stages
2.5. Functional Enrichment Analysis
2.6. Lasso Cox Regression Model Construction and Validation
2.7. Evaluation of Association Between Risk Score and Clinical Outcomes
2.8. Cell–Cell Communication Analysis
2.9. Immune Cell Infiltration Analysis
2.10. Quantitative Polymerase Chain Reaction (Qpcr)
3. Results
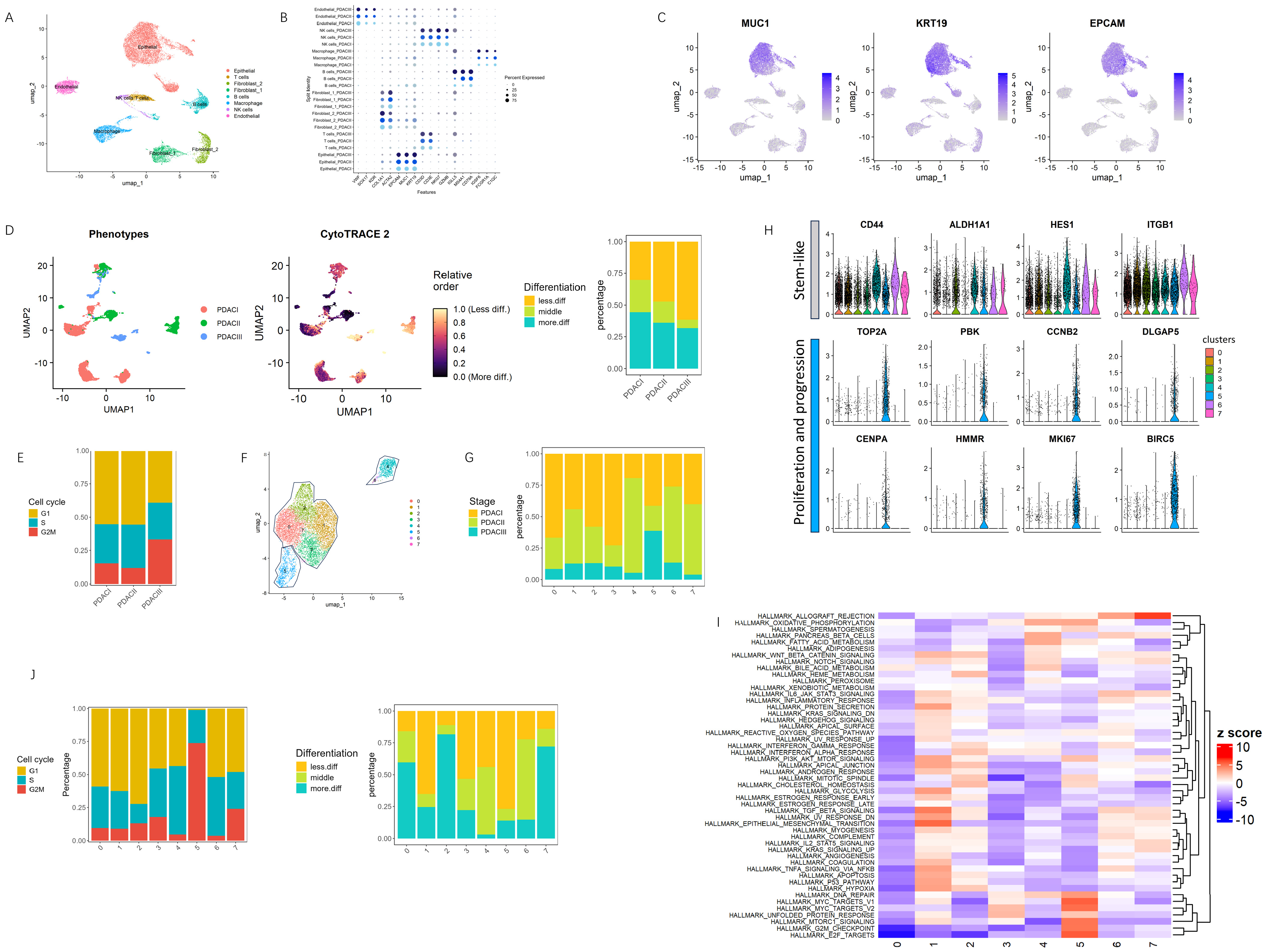
3.1. Identification of a Highly Proliferative Epithelial Subpopulation Driving Early Tumorigenesis in PDAC
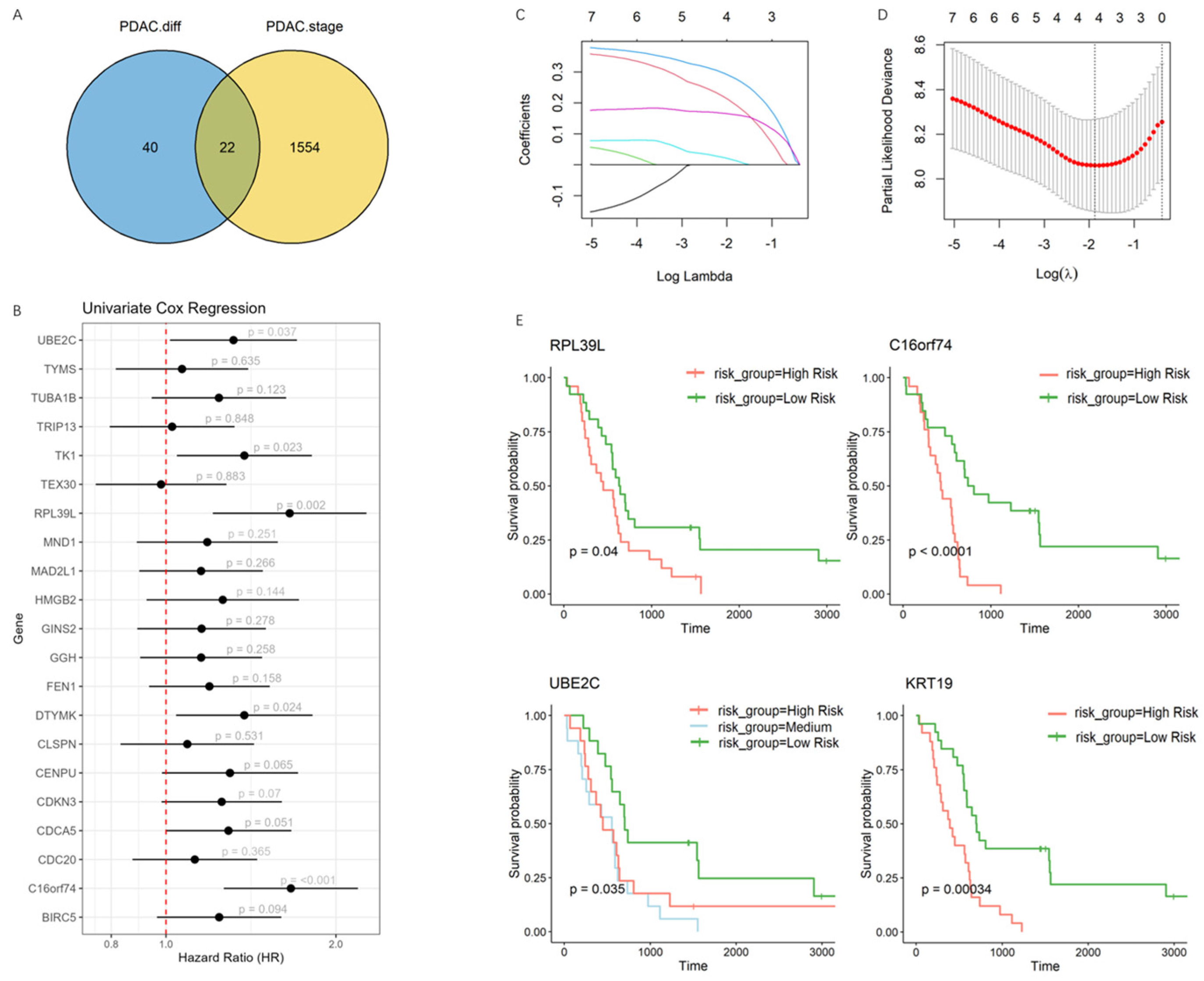
3.2. Development of a Prognostic Model Capturing the Transitional Phase of PDAC Progression
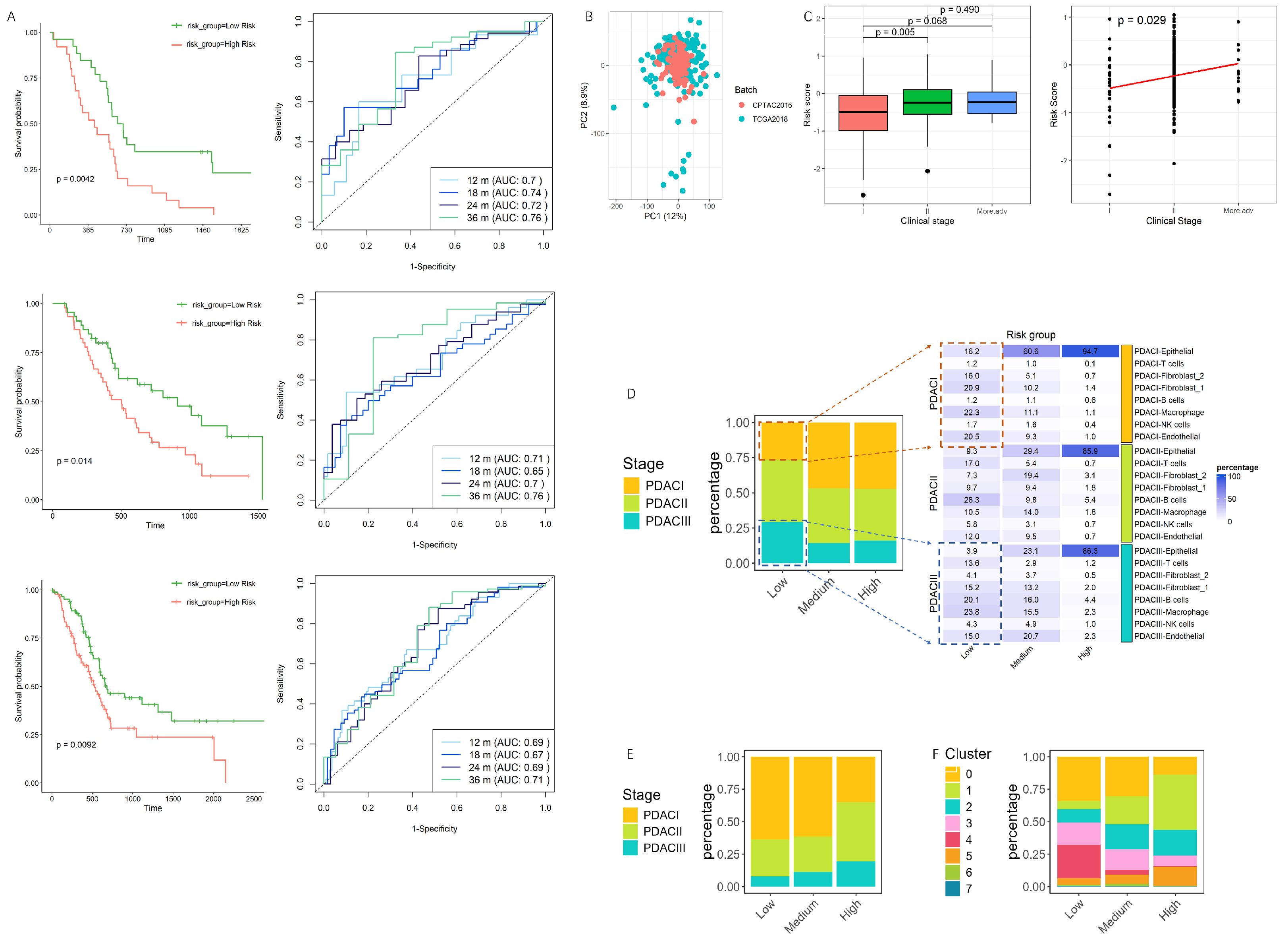
3.3. Validation and Performance Evaluation of the Prognostic Model in PDAC Progression and Single-Cell Risk Stratification
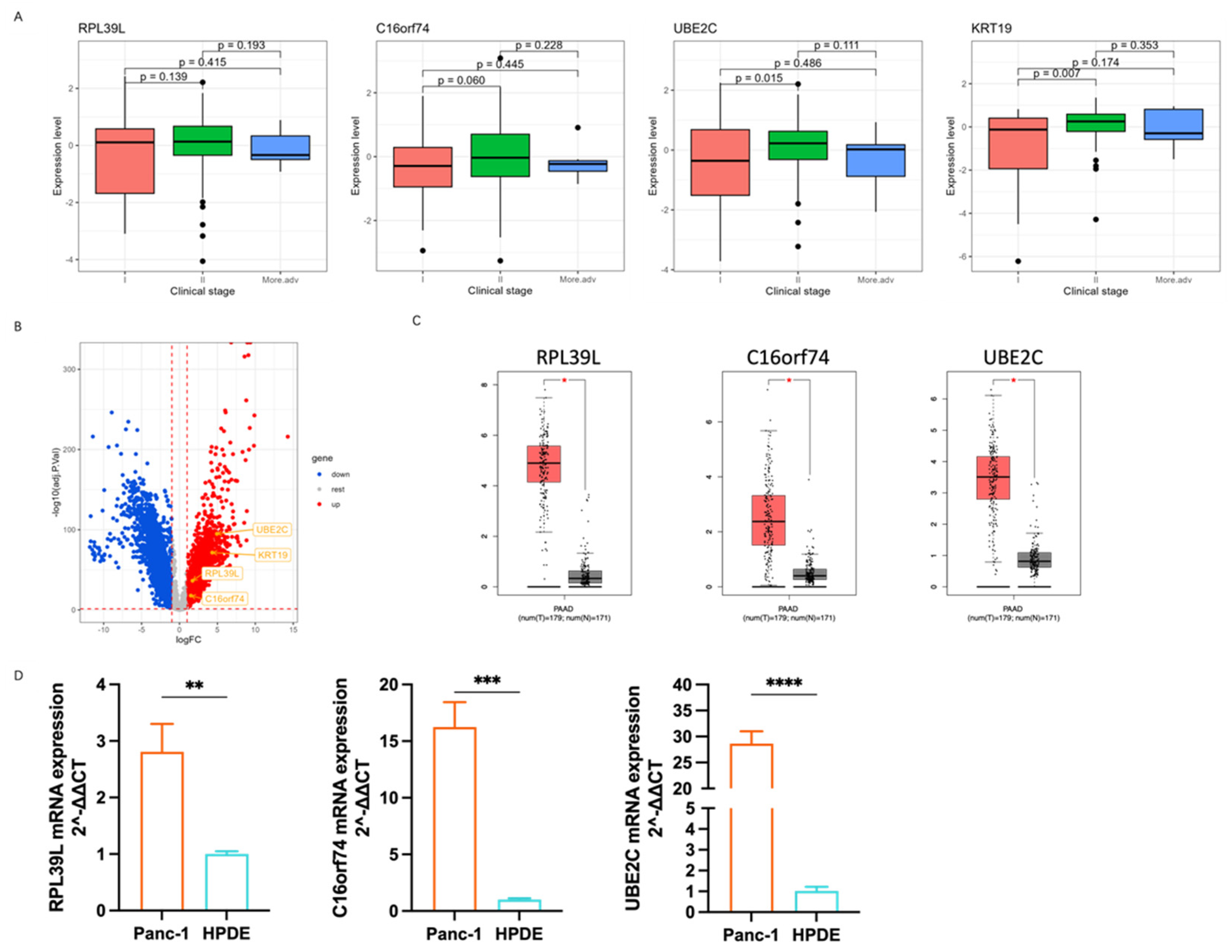
3.4. The Correlation of Marker Genes Expression with PDAC Progress
3.5. Cell–Cell Communication Network Analysis Highlights Key Signaling Interactions and Centrality in Collagen Pathways
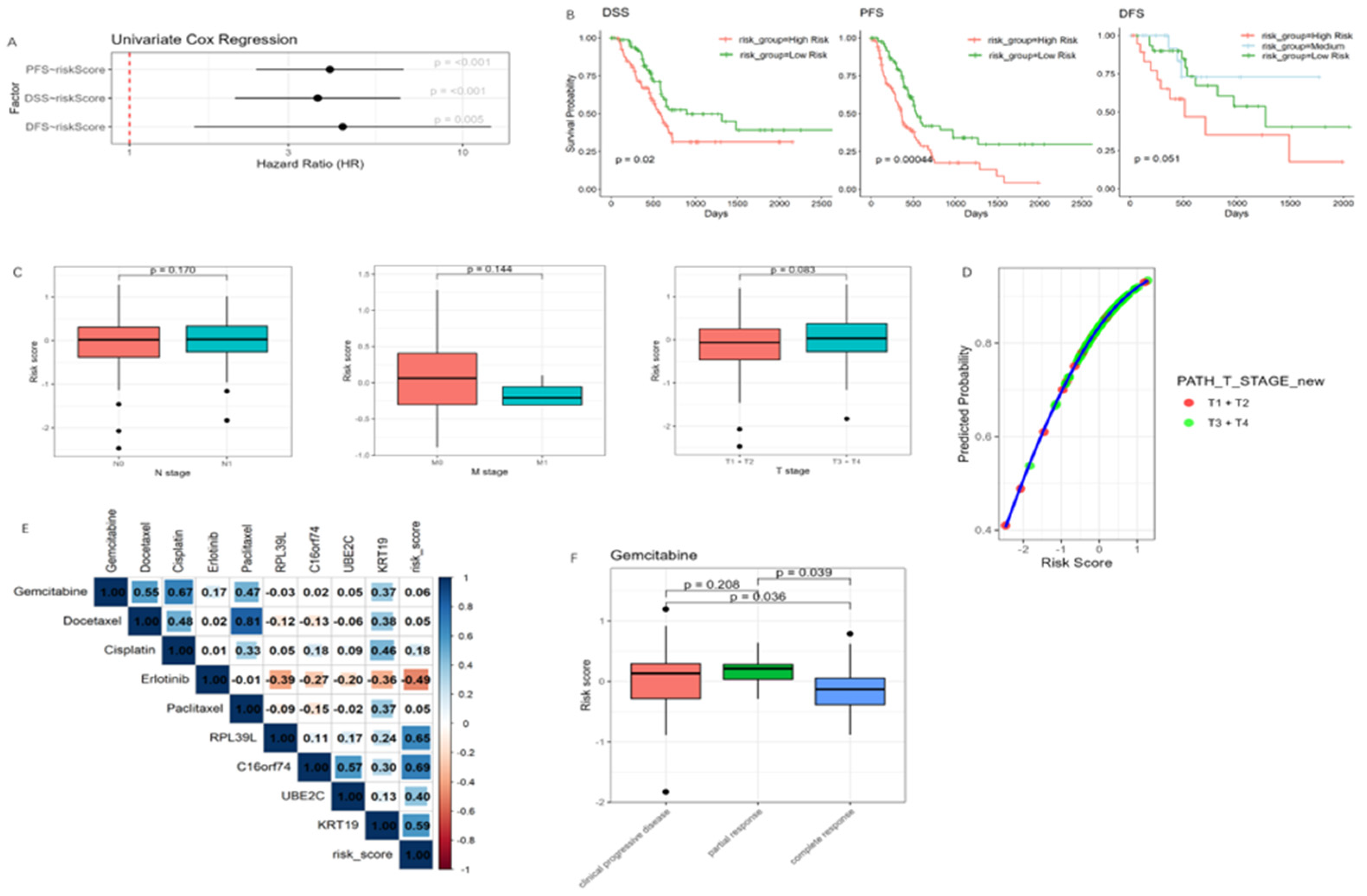
3.6. Association of the Risk Model with Clinical Outcomes, Tumor Invasiveness, and Drug Response
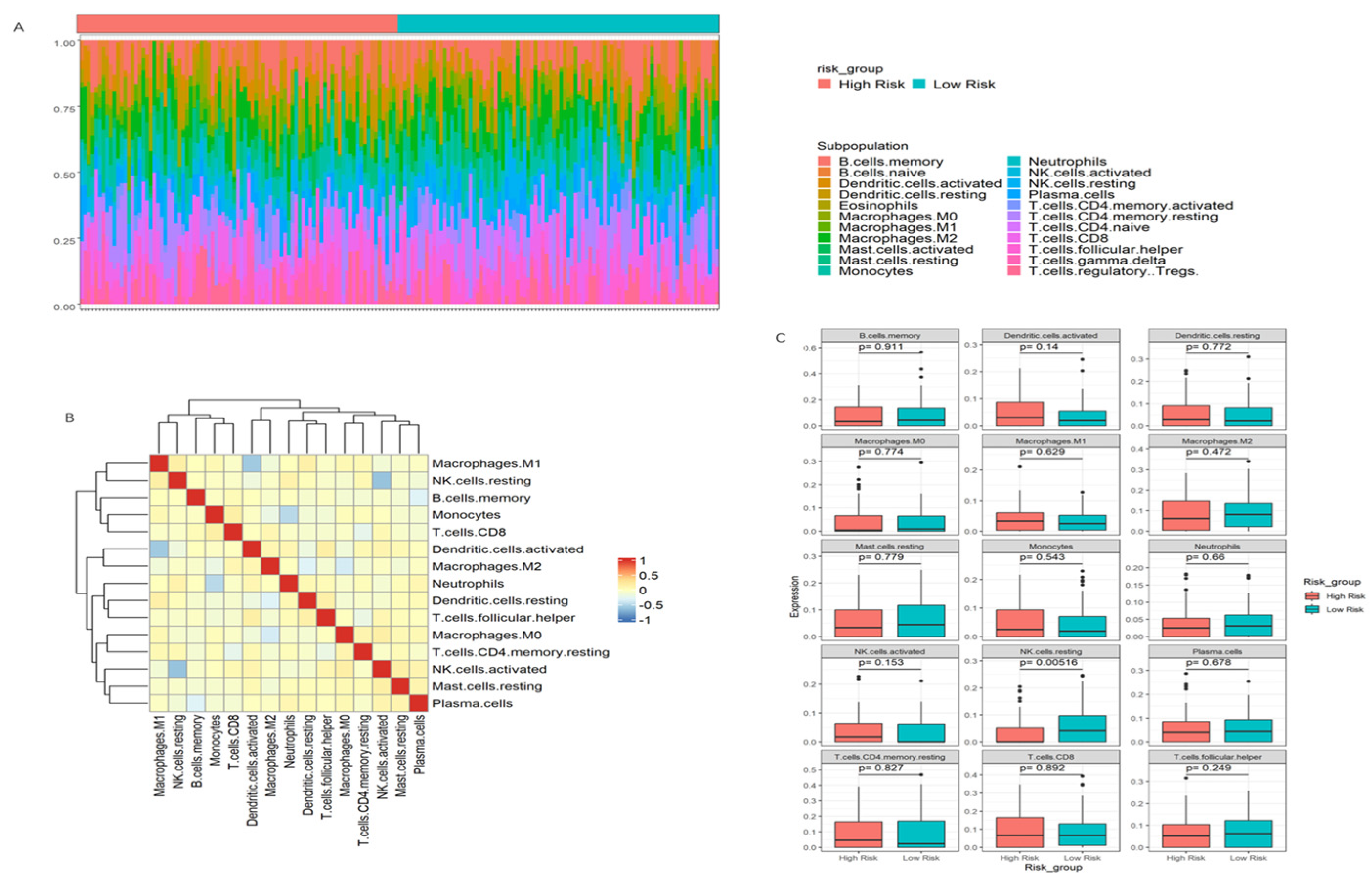
3.7. Immune Infiltration Patterns and Their Association with Risk Scores in PDAC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PDAC | Pancreatic ductal adenocarcinoma |
| PAAD | Pancreatic adenocarcinoma |
| PCa | Pancreatic cancer |
| ECM | Extracellular matrix |
| TME | Tumor microenvironment |
| HPDE | Human pancreatic duct epithelial |
| qPCR | quantitative polymerase chain reaction |
| NK cells | Natural killer cells |
| Adjusted p-value | p.adj |
References
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer Statistics, 2025. CA Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef] [PubMed]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, A.G.; Karamouzis, M.V. Pancreatic Ductal Adenocarcinoma: Treatment Hurdles, Tumor Microenvironment and Immunotherapy. World J. Gastrointest. Oncol. 2020, 12, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.I.; O’Reilly, E.M. Therapeutic Developments in Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 7–24. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Yamada, R.; Tsuboi, J.; Murashima, Y.; Tanaka, T.; Nose, K.; Nakagawa, H. Advances in the Early Diagnosis of Pancreatic Ductal Adenocarcinoma and Premalignant Pancreatic Lesions. Biomedicines 2023, 11, 1687. [Google Scholar] [CrossRef]
- Yang, J.; Xu, R.; Wang, C.; Qiu, J.; Ren, B.; You, L. Early Screening and Diagnosis Strategies of Pancreatic Cancer: A Comprehensive Review. Cancer Commun. 2021, 41, 1257–1274. [Google Scholar] [CrossRef]
- Ren, S.; Qian, L.-C.; Cao, Y.-Y.; Daniels, M.J.; Song, L.-N.; Tian, Y.; Wang, Z.-Q. Computed Tomography-Based Radiomics Diagnostic Approach for Differential Diagnosis between Early- and Late-Stage Pancreatic Ductal Adenocarcinoma. World J. Gastrointest. Oncol. 2024, 16, 1256–1267. [Google Scholar] [CrossRef]
- Schmidtlein, P.M.; Volz, C.; Braun, R.; Thürling, I.; Lapshyna, O.; Wellner, U.F.; Konukiewitz, B.; Lehnert, H.; Marquardt, J.-U.; Ungefroren, H. A Comparative Endocrine Trans-Differentiation Approach to Pancreatic Ductal Adenocarcinoma Cells with Different EMT Phenotypes Identifies Quasi-Mesenchymal Tumor Cells as Those with Highest Plasticity. Cancers 2021, 13, 4663. [Google Scholar] [CrossRef]
- Nacusi, L.P.; Sheaff, R.J. Deregulation of Cell Cycle Machinery in Pancreatic Cancer. Pancreatology 2007, 7, 373–377. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef]
- Glaviano, A.; Singh, S.K.; Lee, E.H.C.; Okina, E.; Lam, H.Y.; Carbone, D.; Reddy, E.P.; O’Connor, M.J.; Koff, A.; Singh, G.; et al. Cell Cycle Dysregulation in Cancer. Pharmacol. Rev. 2025, 77, 100030. [Google Scholar] [CrossRef] [PubMed]
- Oshi, M.; Newman, S.; Tokumaru, Y.; Yan, L.; Matsuyama, R.; Endo, I.; Katz, M.H.G.; Takabe, K. High G2M Pathway Score Pancreatic Cancer Is Associated with Worse Survival, Particularly after Margin-Positive (R1 or R2) Resection. Cancers 2020, 12, 2871. [Google Scholar] [CrossRef] [PubMed]
- Karamitopoulou, E. Tumour Microenvironment of Pancreatic Cancer: Immune Landscape Is Dictated by Molecular and Histopathological Features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef]
- Hingorani, S.R. Epithelial and Stromal Co-Evolution and Complicity in Pancreatic Cancer. Nat. Rev. Cancer 2023, 23, 57–77. [Google Scholar] [CrossRef]
- Maneshi, P.; Mason, J.; Dongre, M.; Öhlund, D. Targeting Tumor-Stromal Interactions in Pancreatic Cancer: Impact of Collagens and Mechanical Traits. Front. Cell Dev. Biol. 2021, 9, 787485. [Google Scholar] [CrossRef]
- Zeltz, C.; Primac, I.; Erusappan, P.; Alam, J.; Noel, A.; Gullberg, D. Cancer-Associated Fibroblasts in Desmoplastic Tumors: Emerging Role of Integrins. Semin. Cancer Biol. 2020, 62, 166–181. [Google Scholar] [CrossRef]
- Prakash, J.; Shaked, Y. The Interplay between Extracellular Matrix Remodeling and Cancer Therapeutics. Cancer Discov. 2024, 14, 1375–1388. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Q.; Wang, J.; Lou, Y.; Hong, Z.; Wei, S.; Sun, K.; Wang, J.; Chen, Y.; Sheng, J.; et al. Dynamic Profiling of Immune Microenvironment during Pancreatic Cancer Development Suggests Early Intervention and Combination Strategy of Immunotherapy. eBioMedicine 2022, 78, 103958. [Google Scholar] [CrossRef]
- Muller, M.; Haghnejad, V.; Schaefer, M.; Gauchotte, G.; Caron, B.; Peyrin-Biroulet, L.; Bronowicki, J.-P.; Neuzillet, C.; Lopez, A. The Immune Landscape of Human Pancreatic Ductal Carcinoma: Key Players, Clinical Implications, and Challenges. Cancers 2022, 14, 995. [Google Scholar] [CrossRef]
- Fincham, R.E.A.; Delvecchio, F.R.; Goulart, M.R.; Yeong, J.P.S.; Kocher, H.M. Natural Killer Cells in Pancreatic Cancer Stroma. World J. Gastroenterol. 2021, 27, 3483–3501. [Google Scholar] [CrossRef] [PubMed]
- Marcon, F.; Zuo, J.; Pearce, H.; Nicol, S.; Margielewska-Davies, S.; Farhat, M.; Mahon, B.; Middleton, G.; Brown, R.; Roberts, K.J.; et al. NK Cells in Pancreatic Cancer Demonstrate Impaired Cytotoxicity and a Regulatory IL-10 Phenotype. Oncoimmunology 2020, 9, 1845424. [Google Scholar] [CrossRef]
- Hoshikawa, M.; Aoki, T.; Matsushita, H.; Karasaki, T.; Hosoi, A.; Odaira, K.; Fujieda, N.; Kobayashi, Y.; Kambara, K.; Ohara, O.; et al. NK Cell and IFN Signatures Are Positive Prognostic Biomarkers for Resectable Pancreatic Cancer. Biochem. Biophys. Res. Commun. 2018, 495, 2058–2065. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.-G.; Long, E.O. Activation, Co–Activation, and Co–Stimulation of Resting Human NK Cells. Immunol. Rev. 2006, 214, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Malchiodi, Z.X.; Suter, R.K.; Deshpande, A.; Peran, I.; Harris, B.T.; Duttargi, A.; Chien, M.-J.; Hariharan, S.; Wetherill, L.; Jablonski, S.A.; et al. Natural Killer Cells Associate with Epithelial Cells in the Pancreatic Ductal Adenocarcinoma Tumor Microenvironment. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kang, M.; Armenteros, J.J.A.; Gulati, G.S.; Gleyzer, R.; Avagyan, S.; Brown, E.L.; Zhang, W.; Usmani, A.; Earland, N.; Wu, Z.; et al. Mapping Single-Cell Developmental Potential in Health and Disease with Interpretable Deep Learning. bioRxiv 2024. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision Weights Unlock Linear Model Analysis Tools for RNA-Seq Read Counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Fan, Y.; Li, L.; Sun, S. Powerful and Accurate Detection of Temporal Gene Expression Patterns from Multi-Sample Multi-Stage Single-Cell Transcriptomics Data with TDEseq. Genome Biol. 2024, 25, 96. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for Systematic Analysis of Cell-Cell Communication from Single-Cell and Spatially Resolved Transcriptomics. Nat. Protoc. 2025, 20, 180–219. [Google Scholar] [CrossRef]
- Pei, Y.F.; Yin, X.M.; Liu, X.Q. TOP2A Induces Malignant Character of Pancreatic Cancer through Activating β-Catenin Signaling Pathway. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2018, 1864, 197–207. [Google Scholar] [CrossRef]
- Koshino, A.; Nagano, A.; Ota, A.; Hyodo, T.; Ueki, A.; Komura, M.; Sugimura-Nagata, A.; Ebi, M.; Ogasawara, N.; Kasai, K.; et al. PBK Enhances Cellular Proliferation with Histone H3 Phosphorylation and Suppresses Migration and Invasion with CDH1 Stabilization in Colorectal Cancer. Front. Pharmacol. 2022, 12, 772926. [Google Scholar] [CrossRef] [PubMed]
- Aljohani, A.I.; Toss, M.S.; El-Sharawy, K.A.; Mirza, S.; Ball, G.R.; Green, A.R.; Rakha, E.A. Upregulation of Cyclin B2 (CCNB2) in Breast Cancer Contributes to the Development of Lymphovascular Invasion. Am. J. Cancer Res. 2022, 12, 469–489. [Google Scholar]
- Janostiak, R.; Mačinga, P.; Csergeová, L.; Fabián, O.; Sticová, E.; Zahradník, J.; Květoň, M.; Hucl, T. Abstract B037: Disks Large-Associated Protein 5 (DLGAP5) Is a Putative Prognostic Biomarker for Intraductal Papillary Mucinous Neoplasm (IPMN) of Pancreas. Clin. Cancer Res. 2024, 30, B037. [Google Scholar] [CrossRef]
- Zheng, C.; Zhang, T.; Li, D.; Huang, C.; Tang, H.; Ni, X.-F.; Chen, B. Upregulation of CENPM Facilitates Tumor Metastasis via the mTOR/p70S6K Signaling Pathway in Pancreatic Cancer. Oncol. Rep. 2020, 44, 1003–1012. [Google Scholar] [CrossRef]
- Lin, A.; Feng, J.; Chen, X.; Wang, D.; Wong, M.; Zhang, G.; Na, J.; Zhang, T.; Chen, Z.; Chen, Y.-T.; et al. High Levels of Truncated RHAMM Cooperate with Dysfunctional P53 to Accelerate the Progression of Pancreatic Cancer. Cancer Lett. 2021, 514, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Liang, X.; Wang, H.; Guo, M.; Shen, H.; Shi, Y.; Liu, Q.; Sun, Y.; Yang, L.; Zhan, M. The RNA Methyltransferase NSUN6 Suppresses Pancreatic Cancer Development by Regulating Cell Proliferation. eBioMedicine 2021, 63, 103195. [Google Scholar] [CrossRef]
- Boidot, R.; Vegran, F.; Jacob, D.; Chevrier, S.; Gangneux, N.; Taboureau, J.; Oudin, C.; Rainville, V.; Mercier, L.; Lizard-Nacol, S. The Expression of BIRC5 Is Correlated with Loss of Specific Chromosomal Regions in Breast Carcinomas. Genes. Chromosomes Cancer 2008, 47, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Ujiki, M.B.; Ding, X.-Z.; Salabat, M.R.; Bentrem, D.J.; Golkar, L.; Milam, B.; Talamonti, M.S.; Bell, R.H.; Iwamura, T.; Adrian, T.E. Apigenin Inhibits Pancreatic Cancer Cell Proliferation through G2/M Cell Cycle Arrest. Mol. Cancer 2006, 5, 76. [Google Scholar] [CrossRef]
- Gires, O.; Pan, M.; Schinke, H.; Canis, M.; Baeuerle, P.A. Expression and Function of Epithelial Cell Adhesion Molecule EpCAM: Where Are We after 40 Years? Cancer Metastasis Rev. 2020, 39, 969–987. [Google Scholar] [CrossRef]
- Barriere, G.; Fici, P.; Gallerani, G.; Fabbri, F.; Zoli, W.; Rigaud, M. Circulating Tumor Cells and Epithelial, Mesenchymal and Stemness Markers: Characterization of Cell Subpopulations. Ann. Transl. Med. 2014, 2, 109. [Google Scholar] [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3–CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [PubMed]
- Mezzapelle, R.; Leo, M.; Caprioglio, F.; Colley, L.S.; Lamarca, A.; Sabatino, L.; Colantuoni, V.; Crippa, M.P.; Bianchi, M.E. CXCR4/CXCL12 Activities in the Tumor Microenvironment and Implications for Tumor Immunotherapy. Cancers 2022, 14, 2314. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Ménard, C.; Martin, F.; Zitvogel, L. The Role of Regulatory T Cells in the Control of Natural Killer Cells: Relevance during Tumor Progression. Immunol. Rev. 2006, 214, 229–238. [Google Scholar] [CrossRef]
- Wang, X.; Xue, X.; Pang, M.; Yu, L.; Qian, J.; Li, X.; Tian, M.; Lyu, A.; Lu, C.; Liu, Y. Epithelial–Mesenchymal Plasticity in Cancer: Signaling Pathways and Therapeutic Targets. MedComm 2024, 5, e659. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The Epigenetics of Epithelial-Mesenchymal Plasticity in Cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Ye, L.; Sanders, A.J.; Lane, J.; Jiang, W.G. Cancer Invasion and Metastasis: Molecular and Cellular Perspective. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial–Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Li, Y.; Liu, F.; Cai, Q.; Deng, L.; Ouyang, Q.; Zhang, X.H.-F.; Zheng, J. Invasion and Metastasis in Cancer: Molecular Insights and Therapeutic Targets. Signal Transduct. Target. Ther. 2025, 10, 57. [Google Scholar] [CrossRef]
- Nussinov, R.; Yavuz, B.R.; Jang, H. Molecular Principles Underlying Aggressive Cancers. Signal Transduct. Target. Ther. 2025, 10, 42. [Google Scholar] [CrossRef]
- Yao, W.; Rose, J.L.; Wang, W.; Seth, S.; Jiang, H.; Taguchi, A.; Liu, J.; Yan, L.; Kapoor, A.; Hou, P.; et al. Syndecan1 Is a Critical Mediator of Macropinocytosis in Pancreatic Cancer. Nature 2019, 568, 410–414. [Google Scholar] [CrossRef]
- Zhu, Y.; Zheng, D.; Lei, L.; Cai, K.; Xie, H.; Zheng, J.; Yu, C. High Expression of Syndecan-4 Is Related to Clinicopathological Features and Poor Prognosis of Pancreatic Adenocarcinoma. BMC Cancer 2022, 22, 1042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shen, Q.; Liu, F.; Yang, F.; Gao, M.; Jiang, X.; Li, Y.; Zhang, X.; En, G.; Pan, X.; et al. SDC1 and ITGA2 as Novel Prognostic Biomarkers for PDAC Related to IPMN. Sci. Rep. 2023, 13, 18727. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.; McMillan, R.H.; Chang, Y.-T.; Penchev, V.R.; Rajeshkumar, N.V.; Maitra, A.; Goggins, M.G.; Eshelman, J.R.; Wolfgang, C.L.; Rasheed, Z.A.; et al. Direct Interactions with Cancer-Associated Fibroblasts Lead to Enhanced Pancreatic Cancer Stem Cell Function. Pancreas 2019, 48, 329–334. [Google Scholar] [CrossRef]
- Zhang, T.; Ren, Y.; Yang, P.; Wang, J.; Zhou, H. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma. Cell Death Dis. 2022, 13, 897. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.G.; Carpenter, E.S.; Kemp, S.B.; Sirihorachai, V.; The, S.; Delrosario, L.; Lazarus, J.; Amir, E.D.; Gunchick, V.; Espinoza, C.; et al. Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat. Cancer 2020, 1, 1097–1112. [Google Scholar] [CrossRef]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor Immunoevasion by the Conversion of Effector NK Cells into Type 1 Innate Lymphoid Cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef]
- Cózar, B.; Greppi, M.; Carpentier, S.; Narni-Mancinelli, E.; Chiossone, L.; Vivier, E. Tumor-Infiltrating Natural Killer Cells. Cancer Discov. 2021, 11, 34–44. [Google Scholar] [CrossRef]
- Witalisz-Siepracka, A.; Klein, K.; Zdársky, B.; Stoiber, D. The Multifaceted Role of STAT3 in NK-Cell Tumor Surveillance. Front. Immunol. 2022, 13, 947568. [Google Scholar] [CrossRef]
- Mao, C.; Chen, Y.; Xing, D.; Zhang, T.; Lin, Y.; Long, C.; Yu, J.; Luo, Y.; Ming, T.; Xie, W.; et al. Resting Natural Killer Cells Promote the Progress of Colon Cancer Liver Metastasis by Elevating Tumor-Derived sSCF. eLife 2024, 13, RP97201. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, L.; Chen, Z.; Zhang, J.; Li, Q. Modeling the Transitional Phase of Epithelial Cells Reveals Prognostic and Therapeutic Targets in Pancreatic Ductal Adenocarcinoma. Cancers 2025, 17, 1813. https://doi.org/10.3390/cancers17111813
Ye L, Chen Z, Zhang J, Li Q. Modeling the Transitional Phase of Epithelial Cells Reveals Prognostic and Therapeutic Targets in Pancreatic Ductal Adenocarcinoma. Cancers. 2025; 17(11):1813. https://doi.org/10.3390/cancers17111813
Chicago/Turabian StyleYe, Linhan, Zongyao Chen, Jingcheng Zhang, and Qiaolin Li. 2025. "Modeling the Transitional Phase of Epithelial Cells Reveals Prognostic and Therapeutic Targets in Pancreatic Ductal Adenocarcinoma" Cancers 17, no. 11: 1813. https://doi.org/10.3390/cancers17111813
APA StyleYe, L., Chen, Z., Zhang, J., & Li, Q. (2025). Modeling the Transitional Phase of Epithelial Cells Reveals Prognostic and Therapeutic Targets in Pancreatic Ductal Adenocarcinoma. Cancers, 17(11), 1813. https://doi.org/10.3390/cancers17111813