

Clinical Differences Among Histological Categories of Sarcoma: Insights from 97,062 Patients


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Population
2.2. Outcomes
2.3. Statistical Analysis
3. Results
3.1. Patient Cohort and Histological Categories
3.2. Clinical Features by Histological Categories
3.3. Survival Outcomes Across Histological Categories
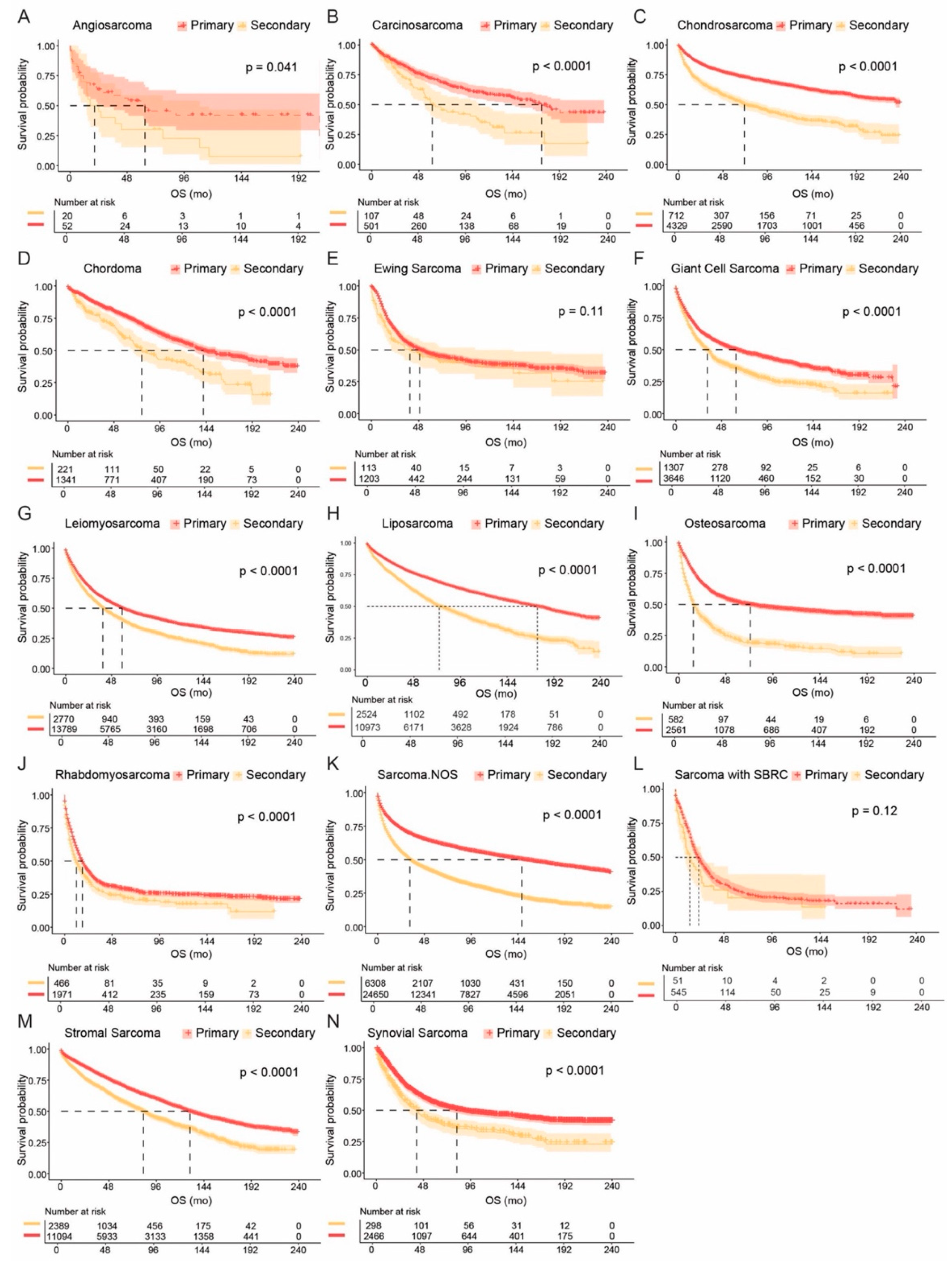
3.4. Comparisons of Primary and Secondary Sarcomas
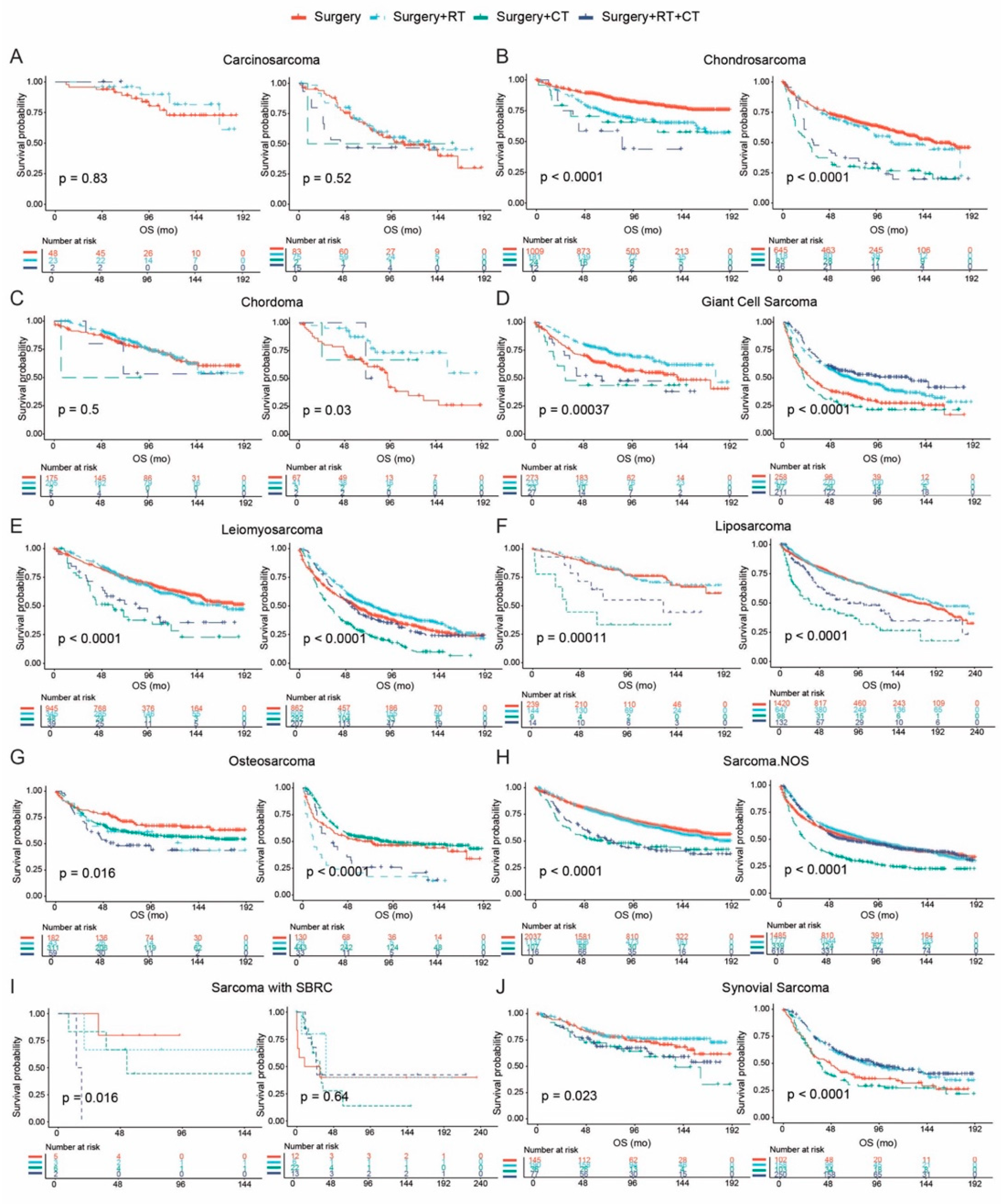
3.5. Treatment Patterns and Survival Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cormier, J.N.; Pollock, R.E. Soft tissue sarcomas. CA Cancer J. Clin. 2004, 54, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Society, A.C. Cancer Facts & Figures 2024; American Cancer Society: Atlanta, GA, USA, 2024. [Google Scholar]
- Katz, D.; Palmerini, E.; Pollack, S.M. More Than 50 Subtypes of Soft Tissue Sarcoma: Paving the Path for Histology-Driven Treatments. In American Society of Clinical Oncology Educational Book; Springer: Berlin/Heidelberg, Germany, 2018; Volume 38, pp. 925–938. [Google Scholar] [CrossRef]
- Eulo, V.; Lesmana, H.; Doyle, L.A.; Nichols, K.E.; Hirbe, A.C. Secondary Sarcomas: Biology, Presentation, and Clinical Care. In American Society of Clinical Oncology Educational Book; Springer: Berlin/Heidelberg, Germany, 2020; Volume 40, pp. 1–12. [Google Scholar]
- Trassard, M.; Le Doussal, V.; Hacène, K.; Terrier, P.; Ranchère, D.; Guillou, L.; Fiche, M.; Collin, F.; Vilain, M.-O.; Bertrand, G.; et al. Prognostic Factors in Localized Primary Synovial Sarcoma: A Multicenter Study of 128 Adult Patients. J. Clin. Oncol. 2001, 19, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Herawi, M.; Epstein, J.I. Specialized Stromal Tumors of the Prostate: A Clinicopathologic Study of 50 Cases. Am. J. Surg. Pathol. 2006, 30, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ortega, D.Y.; Villa-Zepeda, O.; Martinez-Said, H.; Cuellar-Hübbe, M.; Luna-Ortiz, K. Oncology outcomes in Retroperitoneal sarcomas: Prognostic factors in a Retrospective Cohort study. Int. J. Surg. 2016, 32, 45–49. [Google Scholar] [CrossRef]
- Greene, F.L.; Page, D.L.; Fleming, I.D.; Fritz, A.G.; Balch, C.M.; Haller, D.G.; Morrow, M. AJCC Cancer Staging Manual; Springer: New York, NY, USA, 2002. [Google Scholar]
- Sbaraglia, M.; Bellan, E.; Tos, A.P.D. The 2020 WHO Classification of Soft Tissue Tumours: News and perspectives. Pathologica 2021, 113, 70–84. [Google Scholar] [CrossRef]
- Toro, J.R.; Travis, L.B.; Wu, H.J.; Zhu, K.; Fletcher, C.D.; Devesa, S.S. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978–2001: An analysis of 26,758 cases. Int. J. Cancer 2006, 119, 2922–2930. [Google Scholar] [CrossRef]
- Patrichi, A.I.; Gurzu, S. Pathogenetic and molecular classifications of soft tissue and bone tumors: A 2024 update. Pathol.-Res. Pract. 2024, 260, 155406. [Google Scholar] [CrossRef]
- Ferrari, A.; Sultan, I.; Huang, T.T.; Rodriguez-Galindo, C.; Shehadeh, A.; Meazza, C.; Ness, K.K.; Casanova, M.; Spunt, S.L. Soft tissue sarcoma across the age spectrum: A population-based study from the surveillance epidemiology and end results database. Pediatr. Blood Cancer 2011, 57, 943–949. [Google Scholar] [CrossRef]
- Gazendam, A.M.; Popovic, S.; Munir, S.; Parasu, N.; Wilson, D.; Ghert, M. Synovial Sarcoma: A Clinical Review. Curr. Oncol. 2021, 28, 1909–1920. [Google Scholar] [CrossRef]
- Chen, H.-W.; Chen, T.W.-W. Genomic-guided precision therapy for soft tissue sarcoma. ESMO Open 2020, 5, e000626. [Google Scholar] [CrossRef]
- Dufresne, A.; Brahmi, M.; Karanian, M.; Blay, J.-Y. Using biology to guide the treatment of sarcomas and aggressive connective-tissue tumours. Nat. Rev. Clin. Oncol. 2018, 15, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Pestana, R.C.; Groisberg, R.; Roszik, J.; Subbiah, V. Precision Oncology in Sarcomas: Divide and Conquer. JCO Precis. Oncol. 2019, 3, 1–16. [Google Scholar] [CrossRef]
- Vezeridis, M.P.; Moore, R.; Karakousis, C.P. Metastatic Patterns in Soft-Tissue Sarcomas. Arch. Surg. 1983, 118, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Grobmyer, S.R.; Brennan, M.F. Predictive variables detailing the recurrence rate of soft tissue sarcomas. Curr. Opin. Oncol. 2003, 15, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Pisters, P.W.T.; Pollock, R.E.; Lewis, V.O.; Yasko, A.W.; Cormier, J.N.; Respondek, P.M.; Feig, B.W.; Hunt, K.K.; Lin, P.P.; Zagars, G.; et al. Long-term Results of Prospective Trial of Surgery Alone With Selective Use of Radiation for Patients With T1 Extremity and Trunk Soft Tissue Sarcomas. Ann. Surg. 2007, 246, 675–682. [Google Scholar] [CrossRef]
- Gronchi, A.; Miah, A.B.; Dei Tos, A.P.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Blay, J.Y.; Bolle, S.; et al. Soft tissue and visceral sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up(☆). Ann. Oncol. 2021, 32, 1348–1365. [Google Scholar] [CrossRef]
- Judson, I.; Verweij, J.; Gelderblom, H.; Hartmann, J.T.; Schöffski, P.; Blay, J.-Y.; Kerst, J.M.; Sufliarsky, J.; Whelan, J.; Hohenberger, P.; et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: A randomised controlled phase 3 trial. Lancet Oncol. 2014, 15, 415–423. [Google Scholar] [CrossRef]
- Tap, W.D.; Jones, R.L.; Van Tine, B.A.; Chmielowski, B.; Elias, A.D.; Adkins, D.; Agulnik, M.; Cooney, M.M.; Livingston, M.B.; Pennock, G.; et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: An open-label phase 1b and randomised phase 2 trial. Lancet 2016, 388, 488–497. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef]
- Lee, A.T.J.; Jones, R.L.; Huang, P.H. Pazopanib in advanced soft tissue sarcomas. Signal Transduct. Target. Ther. 2019, 4, 16. [Google Scholar] [CrossRef]
- Nascimento, A.G. Dedifferentiated liposarcoma. Semin. Diagn. Pathol. 2001, 18, 263–266. [Google Scholar]
- Thway, K.; Fisher, C. Histopathological Diagnostic Discrepancies in Soft Tissue Tumours Referred to a Specialist Centre. Sarcoma 2009, 2009, 741975. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.Y.; Honoré, C.; Stoeckle, E.; Meeus, P.; Jafari, M.; Gouin, F.; Anract, P.; Ferron, G.; Rochwerger, A.; Ropars, M.; et al. Surgery in reference centers improves survival of sarcoma patients: A nationwide study. Ann. Oncol. 2019, 30, 1143–1153. [Google Scholar] [CrossRef]
- Jain, S.; Kapoor, G. Chemotherapy in Ewing’s sarcoma. Indian J. Orthop. 2010, 44, 369–377. [Google Scholar] [CrossRef]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.-C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol 2015, 33, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Osuna, D.; de Alava, E. Molecular pathology of sarcomas. Rev. Recent Clin. Trials 2009, 4, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Yuen, N.K.; Li, C.-S.; Monjazeb, A.M.; Borys, D.; Bold, R.J.; Canter, R.J. Older Age Modifies Oncologic Outcome Following Radiotherapy in Soft-tissue Sarcoma: A Subtype-specific SEER Analysis. Anticancer Res. 2016, 36, 1745–1750. [Google Scholar]
- Xian, W.; McKeon, F. The challenges and hopes of personalized cancer medicine. Genome Med. 2011, 3, 22. [Google Scholar] [CrossRef]
- Pollack, S.M.; Somaiah, N.; Araujo, D.M.; Druta, M.; Van Tine, B.A.; Burgess, M.A.; Chawla, S.P.; Seetharam, M.; Okuno, S.H.; Bohac, C.; et al. Clinical outcomes of patients with advanced synovial sarcoma or myxoid/round cell liposarcoma treated at major cancer centers in the United States. Cancer Med. 2020, 9, 4593–4602. [Google Scholar] [CrossRef]
- Grünewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med. 2020, 12, e11131. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children’s Oncology Group Study. J. Clin. Oncol. 2017, 35, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Tap, W.D.; Schwartz, G.K.; Carvajal, R.D. Sarcoma Immunotherapy: Past Approaches and Future Directions. Sarcoma 2014, 2014, 391967. [Google Scholar] [CrossRef] [PubMed]
- Bovée, J.V.M.G.; Hogendoorn, P.C.W. Molecular pathology of sarcomas: Concepts and clinical implications. Virchows Arch. 2009, 456, 193–199. [Google Scholar] [CrossRef]
- Dei Tos, A.P. Liposarcoma: New entities and evolving concepts. Ann. Diagn. Pathol. 2000, 4, 252–266. [Google Scholar] [CrossRef]
- Gounder, M.M.; Agaram, N.P.; Trabucco, S.E.; Robinson, V.; Ferraro, R.A.; Millis, S.Z.; Krishnan, A.; Lee, J.; Attia, S.; Abida, W.; et al. Clinical genomic profiling in the management of patients with soft tissue and bone sarcoma. Nat. Commun. 2022, 13, 3406. [Google Scholar] [CrossRef]
- Sayles, L.C.; Breese, M.R.; Koehne, A.L.; Leung, S.G.; Lee, A.G.; Liu, H.-Y.; Spillinger, A.; Shah, A.T.; Tanasa, B.; Straessler, K.; et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov. 2019, 9, 46–63. [Google Scholar] [CrossRef]
- Reimann, E.; Kõks, S.; Ho, X.D.; Maasalu, K.; Märtson, A. Whole exome sequencing of a single osteosarcoma case--integrative analysis with whole transcriptome RNA-seq data. Hum. Genom. 2014, 8, 20. [Google Scholar]
- Ho, X.D.; Nguyen, H.G.; Trinh, L.H.; Reimann, E.; Prans, E.; Kõks, G.; Maasalu, K.; Le, V.Q.; Nguyen, V.H.; Le, N.T.N.; et al. Analysis of the Expression of Repetitive DNA Elements in Osteosarcoma. Front. Genet. 2017, 8, 193. [Google Scholar] [CrossRef]
- Poudel, B.H.; Koks, S. The whole transcriptome analysis using FFPE and fresh tissue samples identifies the molecular fingerprint of osteosarcoma. Exp. Biol. Med. 2024, 249, 10161. [Google Scholar] [CrossRef]
- Denu, R.A.; Dann, A.M.; Keung, E.Z.; Nakazawa, M.S.; Haddad, E.F.N. The Future of Targeted Therapy for Leiomyosarcoma. Cancers 2024, 16, 938. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histology OS (95% CI), Months | Angiosarcoma 46 (23–109) | Carcinosarcoma 144 (114–180) | Chondrosarcoma 234 (200–NA) | Chordoma 133 (122–147) | Ewing Sarcoma 49 (41–58) | Giant Cell Sarcoma 49 (45–55) | Leiomyosarcoma 55 (53–57) | Liposarcoma 148 (142–156) | Osteosarcoma 46 (41–53) | Rhabdomyosarcoma 16 (16–18) | Sarcoma.NOS 105 (100–110) | Sarcoma with SBRC 22 (20–26) | Stromal Sarcoma 122 (117–125) | Synovial Sarcoma 73 (64–86) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Angiosarcoma 46 (23–109) | ||||||||||||||
| Carcinosarcoma 144 (114–180) | <0.0001 | |||||||||||||
| Chondrosarcoma 234 (200–NA) | <0.0001 | 0.039 | ||||||||||||
| Chordoma 133 (122–147) | <0.0001 | 0.520 | 0.008 | |||||||||||
| Ewing sarcoma 49 (41–58) | 0.249 | <0.0001 | <0.0001 | <0.0001 | ||||||||||
| Giant cell sarcoma 49 (45–55) | 0.668 | <0.0001 | <0.0001 | <0.0001 | 0.029 | _ | ||||||||
| Leiomyosarcoma 55 (53–57) | 0.515 | <0.0001 | <0.0001 | <0.0001 | 0.117 | 0.042 | ||||||||
| Liposarcoma 148 (142–156) | <0.0001 | 0.890 | <0.0001 | 0.484 | <0.0001 | <0.0001 | <0.0001 | |||||||
| Osteosarcoma 46 (41–53) | 0.323 | <0.0001 | <0.0001 | <0.0001 | 0.449 | 0.118 | 0.053 | <0.0001 | ||||||
| Rhabdomyosarcoma 16 (16–18) | 0.015 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | |||||
| Sarcoma.NOS 105 (100–110) | 0.022 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | ||||
| Sarcoma with SBRC 22 (20–26) | 0.019 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.129 | <0.0001 | |||
| Stromal sarcoma 122 (117–125) | <0.0001 | 0.199 | <0.0001 | 0.002 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | ||
| Synovial sarcoma 73 (64–86) | 0.023 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.883 | <0.0001 | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Shah, A.; Yao, Y.; Mutter, R.W.; Xu-Welliver, M. Clinical Differences Among Histological Categories of Sarcoma: Insights from 97,062 Patients. Cancers 2025, 17, 1706. https://doi.org/10.3390/cancers17101706
Han Y, Shah A, Yao Y, Mutter RW, Xu-Welliver M. Clinical Differences Among Histological Categories of Sarcoma: Insights from 97,062 Patients. Cancers. 2025; 17(10):1706. https://doi.org/10.3390/cancers17101706
Chicago/Turabian StyleHan, Yiqun, Ahmed Shah, Yuan Yao, Robert W. Mutter, and Meng Xu-Welliver. 2025. "Clinical Differences Among Histological Categories of Sarcoma: Insights from 97,062 Patients" Cancers 17, no. 10: 1706. https://doi.org/10.3390/cancers17101706
APA StyleHan, Y., Shah, A., Yao, Y., Mutter, R. W., & Xu-Welliver, M. (2025). Clinical Differences Among Histological Categories of Sarcoma: Insights from 97,062 Patients. Cancers, 17(10), 1706. https://doi.org/10.3390/cancers17101706