
Innate Immunity and Platelets: Unveiling Their Role in Chronic Pancreatitis and Pancreatic Cancer
 ,
,  ,
,
Simple Summary
Abstract
1. Introduction
2. Innate Immune Cells in Chronic Pancreatitis and PDAC
2.1. Natural Killer (NK) Cells
2.1.1. Key Physiological Functions
2.1.2. Pathophysiology in PDAC
2.1.3. Clinical Implications in PDAC
2.1.4. Therapeutic Strategies
2.2. Macrophages
2.2.1. Key Physiological Functions
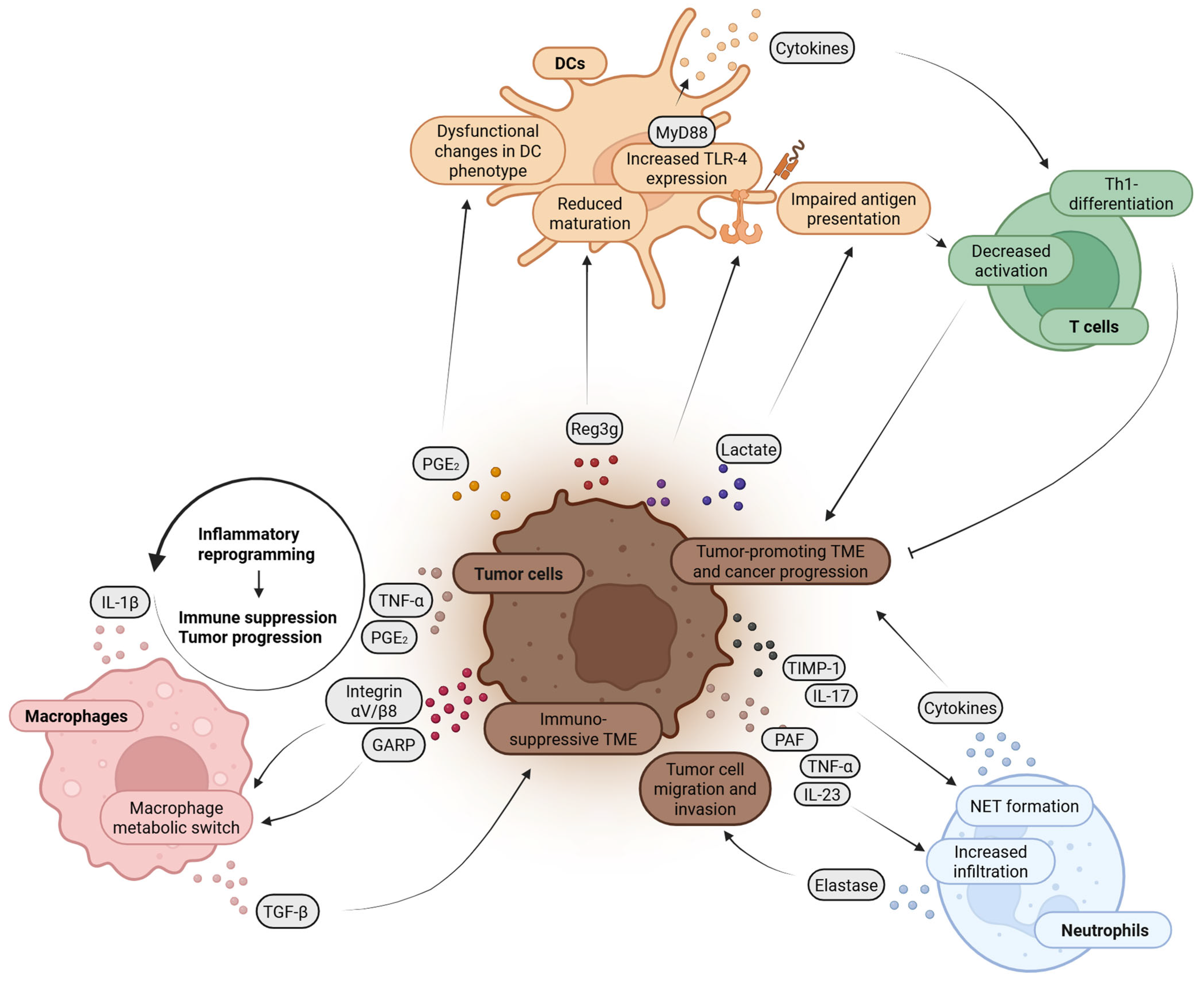
2.2.2. Pathophysiology in Chronic Pancreatitis or PDAC
2.2.3. Clinical Implications and Therapeutic Strategies
2.3. Dendritic Cells
2.3.1. Key Physiological Functions
2.3.2. Pathophysiology in PDAC
2.3.3. Clinical Implications in PDAC
2.3.4. Therapeutic Strategies
2.4. Mast Cells
2.4.1. Key Physiological Functions
2.4.2. Pathophysiology and Clinical Implications in PDAC
2.4.3. Therapeutic Strategies
2.5. Neutrophils
2.5.1. Key Physiological Functions
2.5.2. Pathophysiology and Clinical Implications in PDAC
2.6. Myeloid-Derived Suppressor Cells (MDSCs)
2.6.1. Key Physiological Functions
2.6.2. Pathophysiology and Clinical Implications in PDAC
2.6.3. Therapeutic Strategies
3. Platelets as Regulators of Tumor Growth and Immunity
3.1. Key Physiological Functions
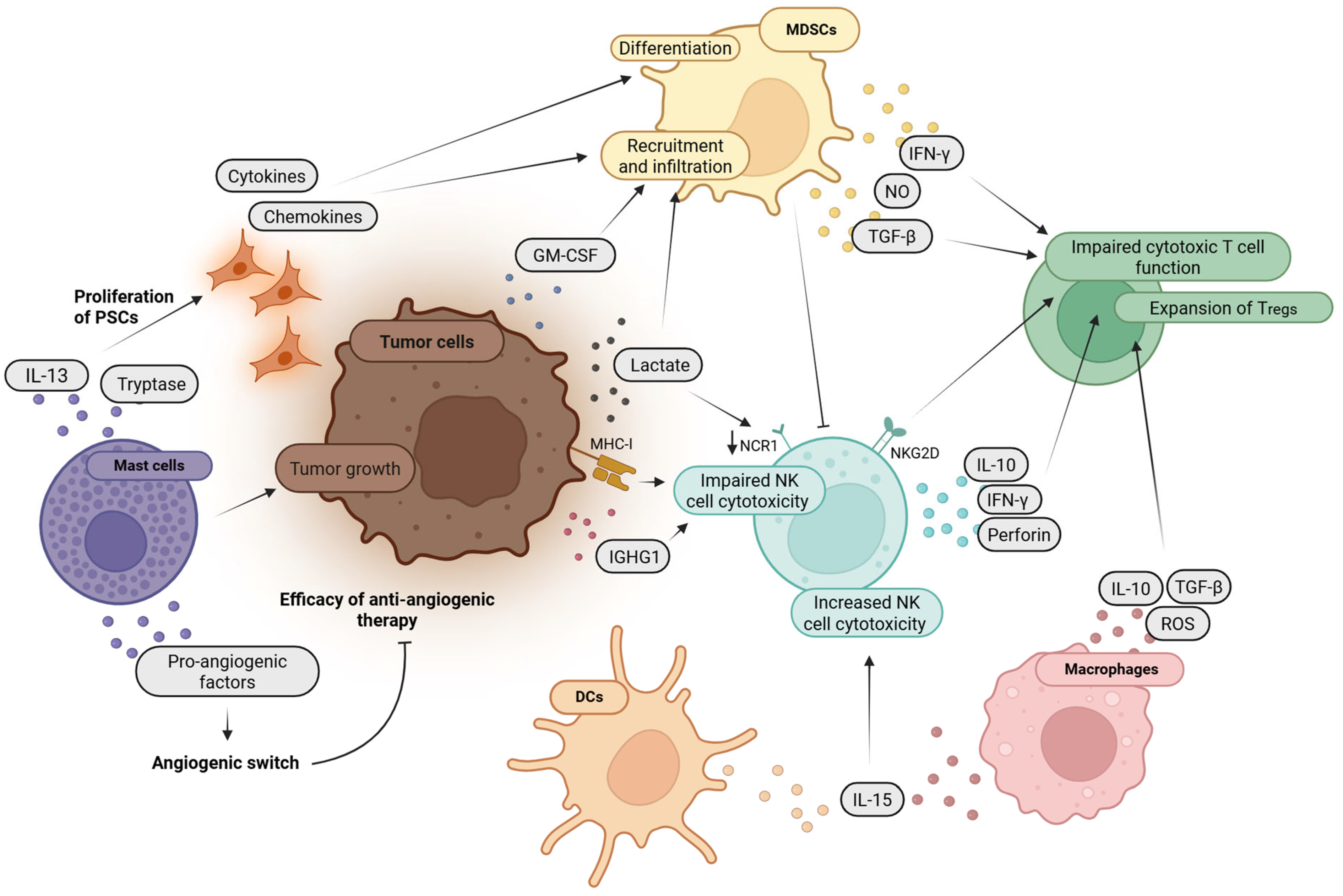
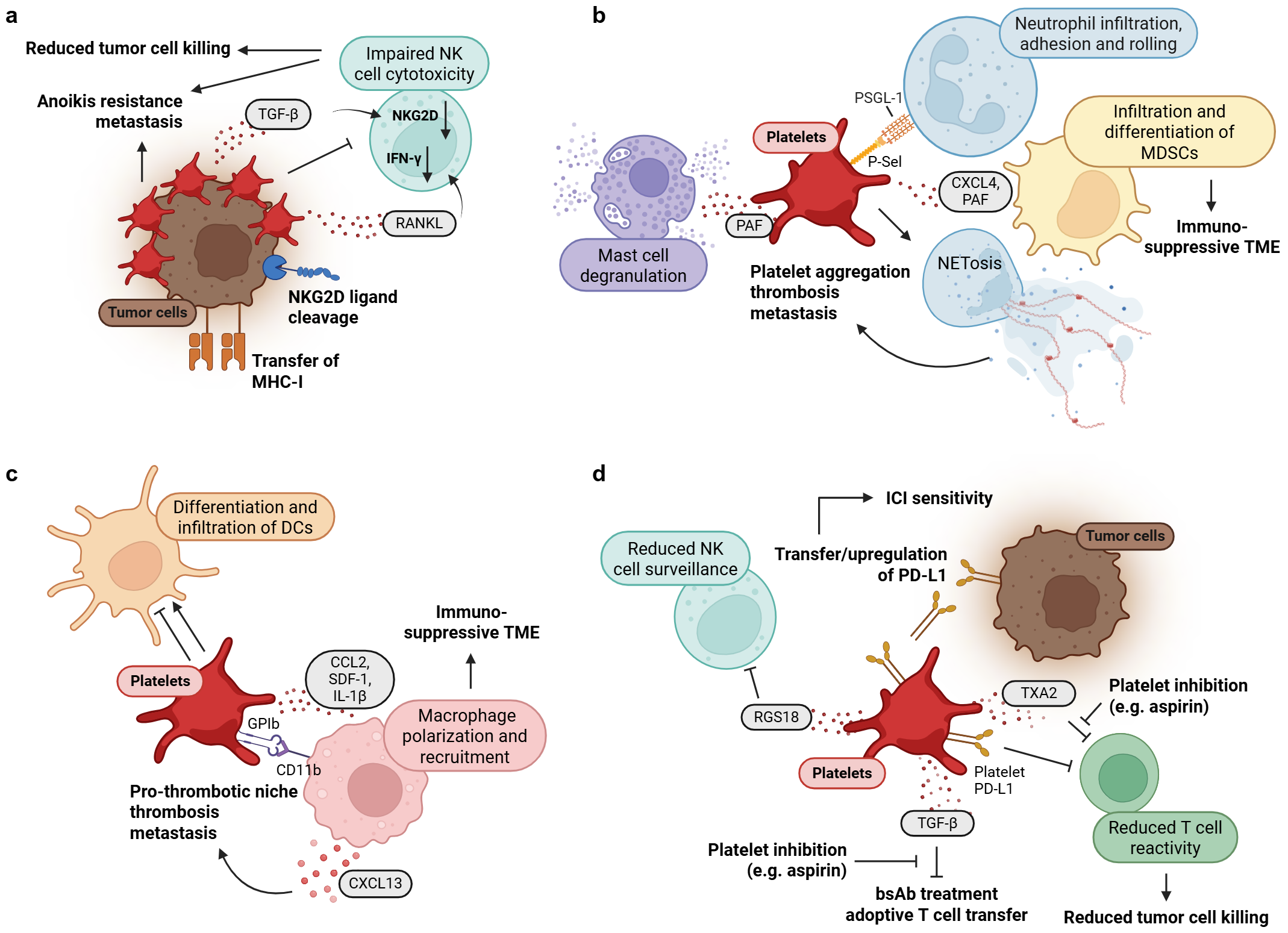
3.2. Impact of Platelets on Immune Cells of the Innate Immune System
3.3. Role of Platelets in Immune Evasion
3.4. Pathophysiology and Clinical Implications in PDAC
3.5. Therapeutic Strategies
4. Future Directions and Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Xiao, A.Y.; Tan, M.L.Y.; Wu, L.M.; Asrani, V.M.; Windsor, J.A.; Yadav, D.; Petrov, M.S. Global incidence and mortality of pancreatic diseases: A systematic review, meta-analysis, and meta-regression of population-based cohort studies. Lancet Gastroenterol. Hepatol. 2016, 1, 45–55. [Google Scholar] [CrossRef]
- Malka, D.; Hammel, P.; Maire, F.; Rufat, P.; Madeira, I.; Pessione, F.; Lévy, P.; Ruszniewski, P. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut 2002, 51, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Chen, Y.; Tan, C.; Ke, N.; Du, B.; Liu, X. Risk of pancreatic cancer in patients undergoing surgery for chronic pancreatitis. BMC Surg. 2019, 19, 83. [Google Scholar] [CrossRef] [PubMed]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andrén-Sandberg, A.; Domellöf, L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Coté, G.A.; Yadav, D.; Slivka, A.; Hawes, R.H.; Anderson, M.A.; Burton, F.R.; Brand, R.E.; Banks, P.A.; Lewis, M.D.; Disario, J.A.; et al. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin. Gastroenterol. Hepatol. 2011, 9, 266–273.e27. [Google Scholar] [CrossRef]
- Lin, Y.; Tamakoshi, A.; Hayakawa, T.; Ogawa, M.; Ohno, Y. Associations of alcohol drinking and nutrient intake with chronic pancreatitis: Findings from a case-control study in Japan. Am. J. Gastroenterol. 2001, 96, 2622–2627. [Google Scholar] [CrossRef]
- Rosendahl, J.; Bödeker, H.; Mössner, J.; Teich, N. Hereditary chronic pancreatitis. Orphanet J. Rare Dis. 2007, 2, 1. [Google Scholar] [CrossRef]
- Klöppel, G. Chronic pancreatitis, pseudotumors and other tumor-like lesions. Mod. Pathol. 2007, 20 (Suppl. 1), S113–S131. [Google Scholar] [CrossRef]
- Shrikhande, S.V.; Martignoni, M.E.; Shrikhande, M.; Kappeler, A.; Ramesh, H.; Zimmermann, A.; Büchler, M.W.; Friess, H. Comparison of histological features and inflammatory cell reaction in alcoholic, idiopathic and tropical chronic pancreatitis. Br. J. Surg. 2003, 90, 1565–1572. [Google Scholar] [CrossRef]
- Gandhi, S.; de la Fuente, J.; Murad, M.H.; Majumder, S. Chronic Pancreatitis Is a Risk Factor for Pancreatic Cancer, and Incidence Increases With Duration of Disease: A Systematic Review and Meta-analysis. Clin. Transl. Gastroenterol. 2022, 13, e00463. [Google Scholar] [CrossRef]
- Le Cosquer, G.; Maulat, C.; Bournet, B.; Cordelier, P.; Buscail, E.; Buscail, L. Pancreatic Cancer in Chronic Pancreatitis: Pathogenesis and Diagnostic Approach. Cancers 2023, 15, 761. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Zhao, Y.; Cao, Y.; Li, Y.; Yang, M.; Tian, Y.; Dai, J.; Song, L.; Ren, S.; Wang, Z. Exploring the key genetic association between chronic pancreatitis and pancreatic ductal adenocarcinoma through integrated bioinformatics. Front. Genet. 2023, 14, 1115660. [Google Scholar] [CrossRef]
- Li, H.; Hao, C.; Yang, Q.; Gao, W.; Ma, B.; Xue, D. Identification of hub genes in chronic pancreatitis and analysis of association with pancreatic cancer via bioinformatic analysis. Gen. Physiol. Biophys. 2022, 41, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Wang, S.; Chao, X.; Jiang, X.; Wang, T.; Rodriguez, Y.; Yang, L.; Pacher, P.; Ni, H.-M.; Ding, W.-X. Loss of acinar cell VMP1 triggers spontaneous pancreatitis in mice. Autophagy 2022, 18, 1572–1582. [Google Scholar] [CrossRef]
- Morris, J.P.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Raphael, B.J. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Shi, C.; Hong, S.-M.; Lim, P.; Kamiyama, H.; Khan, M.; Anders, R.A.; Goggins, M.; Hruban, R.H.; Eshleman, J.R. KRAS2 mutations in human pancreatic acinar-ductal metaplastic lesions are limited to those with PanIN: Implications for the human pancreatic cancer cell of origin. Mol. Cancer Res. 2009, 7, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.-M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733.e9. [Google Scholar] [CrossRef] [PubMed]
- Braxton, A.M.; Kiemen, A.L.; Grahn, M.P.; Forjaz, A.; Parksong, J.; Mahesh Babu, J.; Lai, J.; Zheng, L.; Niknafs, N.; Jiang, L.; et al. 3D genomic mapping reveals multifocality of human pancreatic precancers. Nature 2024, 629, 679–687. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef] [PubMed]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, W.; Collins, M.A.; Bednar, F.; Rakshit, S.; Zetter, B.R.; Stanger, B.Z.; Chung, I.; Rhim, A.D.; Di Magliano, M.P. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013, 73, 6359–6374. [Google Scholar] [CrossRef]
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef]
- Lin, Y.; Pu, S.; Wang, J.; Wan, Y.; Wu, Z.; Guo, Y.; Feng, W.; Ying, Y.; Ma, S.; Meng, X.J.; et al. Pancreatic STAT5 activation promotes KrasG12D-induced and inflammation-induced acinar-to-ductal metaplasia and pancreatic cancer. Gut 2024, 73, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Ceron-Hernandez, J.; Martinez-Navajas, G.; Sanchez-Manas, J.M.; Molina, M.P.; Xie, J.; Aznar-Peralta, I.; Garcia-Diaz, A.; Perales, S.; Torres, C.; Serrano, M.J.; et al. Oncogenic KRASG12D Transfer from Platelet-like Particles Enhances Proliferation and Survival in Non-Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2025, 26, 3264. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liao, C.; Tan, J. KRAS-mutant colorectal cancer cell lines cause a prothrombotic state through the upregulation of thrombin: Experimental study. Ann. Med. Surg. 2024, 86, 850–855. [Google Scholar] [CrossRef]
- Strasenburg, W.; Jóźwicki, J.; Durślewicz, J.; Kuffel, B.; Kulczyk, M.P.; Kowalewski, A.; Grzanka, D.; Drewa, T.; Adamowicz, J. Tumor Cell-Induced Platelet Aggregation as an Emerging Therapeutic Target for Cancer Therapy. Front. Oncol. 2022, 12, 909767. [Google Scholar] [CrossRef]
- Inman, K.S.; Francis, A.A.; Murray, N.R. Complex role for the immune system in initiation and progression of pancreatic cancer. World J. Gastroenterol. 2014, 20, 11160–11181. [Google Scholar] [CrossRef]
- Yousuf, S.; Qiu, M.; von Voith Voithenberg, L.; Hulkkonen, J.; Macinkovic, I.; Schulz, A.R.; Hartmann, D.; Mueller, F.; Mijatovic, M.; Ibberson, D.; et al. Spatially Resolved Multi-Omics Single-Cell Analyses Inform Mechanisms of Immune Dysfunction in Pancreatic Cancer. Gastroenterology 2023, 165, 891–908.e14. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Q.; Wang, J.; Lou, Y.; Hong, Z.; Wei, S.; Sun, K.; Wang, J.; Chen, Y.; Sheng, J.; et al. Dynamic profiling of immune microenvironment during pancreatic cancer development suggests early intervention and combination strategy of immunotherapy. EBioMedicine 2022, 78, 103958. [Google Scholar] [CrossRef]
- Zwart, E.S.; van Ee, T.; Affandi, A.J.; Boyd, L.N.C.; Rodriguez, E.; den Haan, J.M.M.; Farina, A.; van Grieken, N.C.T.; Meijer, L.L.; van Kooyk, Y.; et al. Spatial immune composition of tumor microenvironment in patients with pancreatic cancer. Cancer Immunol. Immunother. 2023, 72, 4385–4397. [Google Scholar] [CrossRef]
- Wang, J.; He, P.; Gaida, M.; Yang, S.; Schetter, A.J.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.; Lee, D.; et al. Inducible nitric oxide synthase enhances disease aggressiveness in pancreatic cancer. Oncotarget 2016, 7, 52993–53004. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef]
- Xiang, H.; Yang, R.; Tu, J.; Xi, Y.; Yang, S.; Lv, L.; Zhai, X.; Zhu, Y.; Dong, D.; Tao, X. Metabolic reprogramming of immune cells in pancreatic cancer progression. Biomed. Pharmacother. 2023, 157, 113992. [Google Scholar] [CrossRef] [PubMed]
- Schmied, L.; Höglund, P.; Meinke, S. Platelet-Mediated Protection of Cancer Cells From Immune Surveillance—Possible Implications for Cancer Immunotherapy. Front. Immunol. 2021, 12, 640578. [Google Scholar] [CrossRef] [PubMed]
- Tuerhong, N.; Yang, Y.; Wang, C.; Huang, P.; Li, Q. Interactions between platelets and the cancer immune microenvironment. Crit. Rev. Oncol. Hematol. 2024, 199, 104380. [Google Scholar] [CrossRef]
- Ward, M.P.; Kane, L.E.; Norris, L.A.; Mohamed, B.M.; Kelly, T.; Bates, M.; Clarke, A.; Brady, N.; Martin, C.M.; Brooks, R.D.; et al. Platelets, immune cells and the coagulation cascade; friend or foe of the circulating tumour cell? Mol. Cancer 2021, 20, 59. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef]
- Guillerey, C. NK Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1273, 69–90. [Google Scholar] [CrossRef]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef]
- Reschner, A.; Hubert, P.; Delvenne, P.; Boniver, J.; Jacobs, N. Innate lymphocyte and dendritic cell cross-talk: A key factor in the regulation of the immune response. Clin. Exp. Immunol. 2008, 152, 219–226. [Google Scholar] [CrossRef]
- Koehl, U.; Toubert, A.; Pittari, G. Editorial: Tailoring NK Cell Receptor-Ligand Interactions: An Art in Evolution. Front. Immunol. 2018, 9, 351. [Google Scholar] [CrossRef]
- Kärre, K. NK cells, MHC class I molecules and the missing self. Scand. J. Immunol. 2002, 55, 221–228. [Google Scholar] [CrossRef]
- Ljunggren, H.G.; Kärre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Parham, P. MHC class I molecules and KIRs in human history, health and survival. Nat. Rev. Immunol. 2005, 5, 201–214. [Google Scholar] [CrossRef]
- Kumar, V.; McNerney, M.E. A new self: MHC-class-I-independent natural-killer-cell self-tolerance. Nat. Rev. Immunol. 2005, 5, 363–374. [Google Scholar] [CrossRef] [PubMed]
- López-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef]
- Ferretti, E.; Carlomagno, S.; Pesce, S.; Muccio, L.; Obino, V.; Greppi, M.; Solari, A.; Setti, C.; Marcenaro, E.; Della Chiesa, M.; et al. Role of the Main Non HLA-Specific Activating NK Receptors in Pancreatic, Colorectal and Gastric Tumors Surveillance. Cancers 2020, 12, 3705. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-P.; Zhu, Y.; Zhang, J.-J.; Xu, Z.-K.; Qian, Z.-Y.; Dai, C.-C.; Jiang, K.-R.; Wu, J.-L.; Gao, W.-T.; Li, Q.; et al. Comprehensive analysis of the percentage of surface receptors and cytotoxic granules positive natural killer cells in patients with pancreatic cancer, gastric cancer, and colorectal cancer. J. Transl. Med. 2013, 11, 262. [Google Scholar] [CrossRef]
- Kaur, K.; Chang, H.-H.; Topchyan, P.; Cook, J.M.; Barkhordarian, A.; Eibl, G.; Jewett, A. Deficiencies in Natural Killer Cell Numbers, Expansion, and Function at the Pre-Neoplastic Stage of Pancreatic Cancer by KRAS Mutation in the Pancreas of Obese Mice. Front. Immunol. 2018, 9, 1229. [Google Scholar] [CrossRef]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef]
- Lim, S.A.; Kim, J.; Jeon, S.; Shin, M.H.; Kwon, J.; Kim, T.-J.; Im, K.; Han, Y.; Kwon, W.; Kim, S.-W.; et al. Defective Localization With Impaired Tumor Cytotoxicity Contributes to the Immune Escape of NK Cells in Pancreatic Cancer Patients. Front. Immunol. 2019, 10, 496. [Google Scholar] [CrossRef]
- Bähr, I.; Spielmann, J.; Quandt, D.; Kielstein, H. Obesity-Associated Alterations of Natural Killer Cells and Immunosurveillance of Cancer. Front. Immunol. 2020, 11, 245. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Fu, T.; Jiang, Y.-Z.; Shao, Z.-M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Investig. 2019, 129, 3499–3510. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Villagrana, R.D.; Albores-García, D.; Cervantes-Villagrana, A.R.; García-Acevez, S.J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct. Target. Ther. 2020, 5, 99. [Google Scholar] [CrossRef]
- Marcon, F.; Zuo, J.; Pearce, H.; Nicol, S.; Margielewska-Davies, S.; Farhat, M.; Mahon, B.; Middleton, G.; Brown, R.; Roberts, K.J.; et al. NK cells in pancreatic cancer demonstrate impaired cytotoxicity and a regulatory IL-10 phenotype. Oncoimmunology 2020, 9, 1845424. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ni, R.; Chen, J.; Liu, Z.; Xiao, M.; Jiang, F.; Lu, C. The presence of IGHG1 in human pancreatic carcinomas is associated with immune evasion mechanisms. Pancreas 2011, 40, 753–761. [Google Scholar] [CrossRef]
- He, C.; Wang, D.; Shukla, S.K.; Hu, T.; Thakur, R.; Fu, X.; King, R.J.; Kollala, S.S.; Attri, K.S.; Murthy, D.; et al. Vitamin B6 Competition in the Tumor Microenvironment Hampers Antitumor Functions of NK Cells. Cancer Discov. 2024, 14, 176–193. [Google Scholar] [CrossRef]
- Janakiram, N.B.; Mohammed, A.; Bryant, T.; Ritchie, R.; Stratton, N.; Jackson, L.; Lightfoot, S.; Benbrook, D.M.; Asch, A.S.; Lang, M.L.; et al. Loss of natural killer T cells promotes pancreatic cancer in LSL-KrasG12D/+ mice. Immunology 2017, 152, 36–51. [Google Scholar] [CrossRef]
- Kobayashi, H.; Dubois, S.; Sato, N.; Sabzevari, H.; Sakai, Y.; Waldmann, T.A.; Tagaya, Y. Role of trans-cellular IL-15 presentation in the activation of NK cell-mediated killing, which leads to enhanced tumor immunosurveillance. Blood 2005, 105, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.-G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed]
- Placke, T.; Örgel, M.; Schaller, M.; Jung, G.; Rammensee, H.-G.; Kopp, H.-G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef]
- Hoshikawa, M.; Aoki, T.; Matsushita, H.; Karasaki, T.; Hosoi, A.; Odaira, K.; Fujieda, N.; Kobayashi, Y.; Kambara, K.; Ohara, O.; et al. NK cell and IFN signatures are positive prognostic biomarkers for resectable pancreatic cancer. Biochem. Biophys. Res. Commun. 2018, 495, 2058–2065. [Google Scholar] [CrossRef]
- van Audenaerde, J.R.M.; Roeyen, G.; Darcy, P.K.; Kershaw, M.H.; Peeters, M.; Smits, E.L.J. Natural killer cells and their therapeutic role in pancreatic cancer: A systematic review. Pharmacol. Ther. 2018, 189, 31–44. [Google Scholar] [CrossRef]
- Brooks, J.; Fleischmann-Mundt, B.; Woller, N.; Niemann, J.; Ribback, S.; Peters, K.; Demir, I.E.; Armbrecht, N.; Ceyhan, G.O.; Manns, M.P.; et al. Perioperative, Spatiotemporally Coordinated Activation of T and NK Cells Prevents Recurrence of Pancreatic Cancer. Cancer Res. 2018, 78, 475–488. [Google Scholar] [CrossRef]
- Koh, C.Y.; Blazar, B.R.; George, T.; Welniak, L.A.; Capitini, C.M.; Raziuddin, A.; Murphy, W.J.; Bennett, M. Augmentation of antitumor effects by NK cell inhibitory receptor blockade in vitro and in vivo. Blood 2001, 97, 3132–3137. [Google Scholar] [CrossRef] [PubMed]
- McKenna, D.H.; Sumstad, D.; Bostrom, N.; Kadidlo, D.M.; Fautsch, S.; McNearney, S.; Dewaard, R.; McGlave, P.B.; Weisdorf, D.J.; Wagner, J.E.; et al. Good manufacturing practices production of natural killer cells for immunotherapy: A six-year single-institution experience. Transfusion 2007, 47, 520–528. [Google Scholar] [CrossRef]
- van den Eynde, A.; Gehrcken, L.; Verhezen, T.; Lau, H.W.; Hermans, C.; Lambrechts, H.; Flieswasser, T.; Quatannens, D.; Roex, G.; Zwaenepoel, K.; et al. IL-15-secreting CAR natural killer cells directed toward the pan-cancer target CD70 eliminate both cancer cells and cancer-associated fibroblasts. J. Hematol. Oncol. 2024, 17, 8. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. TAMing pancreatic cancer: Combat with a double edged sword. Mol. Cancer 2019, 18, 48. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the Immune Reaction to Pancreatic Cancer from Inception to Invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Väyrynen, S.A.; Zhang, J.; Yuan, C.; Väyrynen, J.P.; Dias Costa, A.; Williams, H.; Morales-Oyarvide, V.; Lau, M.C.; Rubinson, D.A.; Dunne, R.F.; et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1069–1081. [Google Scholar] [CrossRef]
- Baer, J.M.; Zuo, C.; Kang, L.-I.; de la Lastra, A.A.; Borcherding, N.C.; Knolhoff, B.L.; Bogner, S.J.; Zhu, Y.; Yang, L.; Laurent, J.; et al. Fibrosis induced by resident macrophages has divergent roles in pancreas inflammatory injury and PDAC. Nat. Immunol. 2023, 24, 1443–1457. [Google Scholar] [CrossRef]
- Caronni, N.; La Terza, F.; Vittoria, F.M.; Barbiera, G.; Mezzanzanica, L.; Cuzzola, V.; Barresi, S.; Pellegatta, M.; Canevazzi, P.; Dunsmore, G.; et al. IL-1β+ macrophages fuel pathogenic inflammation in pancreatic cancer. Nature 2023, 623, 415–422. [Google Scholar] [CrossRef]
- Liu, M.; Ren, Y.; Zhou, Z.; Yang, J.; Shi, X.; Cai, Y.; Arreola, A.X.; Luo, W.; Fung, K.-M.; Xu, C.; et al. The crosstalk between macrophages and cancer cells potentiates pancreatic cancer cachexia. Cancer Cell 2024, 42, 885–903.e4. [Google Scholar] [CrossRef]
- Crezee, T.; Rabold, K.; de Jong, L.; Jaeger, M.; Netea-Maier, R.T. Metabolic programming of tumor associated macrophages in the context of cancer treatment. Ann. Transl. Med. 2020, 8, 1028. [Google Scholar] [CrossRef]
- Hörhold, F.; Eisel, D.; Oswald, M.; Kolte, A.; Röll, D.; Osen, W.; Eichmüller, S.B.; König, R. Reprogramming of macrophages employing gene regulatory and metabolic network models. PLoS Comput. Biol. 2020, 16, e1007657. [Google Scholar] [CrossRef]
- Tan, Z.; Xie, N.; Cui, H.; Moellering, D.R.; Abraham, E.; Thannickal, V.J.; Liu, G. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. J. Immunol. 2015, 194, 6082–6089. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, X.; Fujiwara, K.; Jurcak, N.; Muth, S.; Zhou, J.; Xiao, Q.; Li, A.; Che, X.; Li, Z.; et al. Pancreatic cancer cells render tumor-associated macrophages metabolically reprogrammed by a GARP and DNA methylation-mediated mechanism. Signal Transduct. Target. Ther. 2021, 6, 366. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.-J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: An open-label, multicentre, phase 1b study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R. Autoimmunity: From Bench to Bedside; Editorial Universidad del Rosario: Bogotá, Colombia, 2013; ISBN 978-958-738-366-9. [Google Scholar]
- Jagannathan-Bogdan, M.; Zon, L.I. Hematopoiesis. Development 2013, 140, 2463–2467. [Google Scholar] [CrossRef]
- Shortman, K.; Liu, Y. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef]
- Collin, M.; Milne, P. Langerhans cell origin and regulation. Curr. Opin. Hematol. 2016, 23, 28–35. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Arlt, E.; Kindermann, A.; Fritsche, A.-K.; Navarrete Santos, A.; Kielstein, H.; Bazwinsky-Wutschke, I. A Flow Cytometry-Based Examination of the Mouse White Blood Cell Differential in the Context of Age and Sex. Cells 2024, 13, 1583. [Google Scholar] [CrossRef]
- Balan, S.; Saxena, M.; Bhardwaj, N. Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol. 2019, 348, 1–68. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Du, J.; Cao, H.; Liu, X.; Xu, Q.; Xiang, M. Reg3g Promotes Pancreatic Carcinogenesis in a Murine Model of Chronic Pancreatitis. Dig. Dis. Sci. 2015, 60, 3656–3668. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, Z.; Cheng, Q.; Wang, H.; Cao, H.; Xu, Q.; Tuo, Y.; Jiang, L.; Zou, Y.; Ren, H.; et al. Acceleration of pancreatic tumorigenesis under immunosuppressive microenvironment induced by Reg3g overexpression. Cell Death Dis. 2017, 8, e3033. [Google Scholar] [CrossRef]
- Ochi, A.; Nguyen, A.H.; Bedrosian, A.S.; Mushlin, H.M.; Zarbakhsh, S.; Barilla, R.; Zambirinis, C.P.; Fallon, N.C.; Rehman, A.; Pylayeva-Gupta, Y.; et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J. Exp. Med. 2012, 209, 1671–1687. [Google Scholar] [CrossRef]
- Kim, H.-J.; Kim, H.; Lee, J.-H.; Hwangbo, C. Toll-like receptor 4 (TLR4): New insight immune and aging. Immun. Ageing 2023, 20, 67. [Google Scholar] [CrossRef]
- Lin, J.H.; Huffman, A.P.; Wattenberg, M.M.; Walter, D.M.; Carpenter, E.L.; Feldser, D.M.; Beatty, G.L.; Furth, E.E.; Vonderheide, R.H. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J. Exp. Med. 2020, 217, e20190673. [Google Scholar] [CrossRef]
- Falcomatà, C.; Bärthel, S.; Schneider, G.; Rad, R.; Schmidt-Supprian, M.; Saur, D. Context-Specific Determinants of the Immunosuppressive Tumor Microenvironment in Pancreatic Cancer. Cancer Discov. 2023, 13, 278–297. [Google Scholar] [CrossRef]
- Justus, C.R.; Dong, L.; Yang, L.V. Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front. Physiol. 2013, 4, 354. [Google Scholar] [CrossRef]
- Zhang, B.; Ohuchida, K.; Tsutsumi, C.; Shimada, Y.; Mochida, Y.; Oyama, K.; Iwamoto, C.; Sheng, N.; Fei, S.; Shindo, K.; et al. Dynamic glycolytic reprogramming effects on dendritic cells in pancreatic ductal adenocarcinoma. J. Exp. Clin. Cancer Res. 2024, 43, 271. [Google Scholar] [CrossRef]
- Jang, J.-E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef]
- Kenkel, J.A.; Tseng, W.W.; Davidson, M.G.; Tolentino, L.L.; Choi, O.; Bhattacharya, N.; Seeley, E.S.; Winer, D.A.; Reticker-Flynn, N.E.; Engleman, E.G. An Immunosuppressive Dendritic Cell Subset Accumulates at Secondary Sites and Promotes Metastasis in Pancreatic Cancer. Cancer Res. 2017, 77, 4158–4170. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.; Fogar, P.; Falconi, M.; Fadi, E.; Sperti, C.; Frasson, C.; Greco, E.; Tamburrino, D.; Teolato, S.; Moz, S.; et al. Pancreatic tumors and immature immunosuppressive myeloid cells in blood and spleen: Role of inhibitory co-stimulatory molecules PDL1 and CTLA4. An in vivo and in vitro study. PLoS ONE 2013, 8, e54824. [Google Scholar] [CrossRef] [PubMed]
- Tjomsland, V.; Niklasson, L.; Sandström, P.; Borch, K.; Druid, H.; Bratthäll, C.; Messmer, D.; Larsson, M.; Spångeus, A. The desmoplastic stroma plays an essential role in the accumulation and modulation of infiltrated immune cells in pancreatic adenocarcinoma. Clin. Dev. Immunol. 2011, 2011, 212810. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhou, B.P.; Tao, M.; Liu, J.; Li, W. The Role of Stromal Components in Pancreatic Cancer Progression. Anti-Cancer Agents Med. Chem. 2016, 16, 1117–1124. [Google Scholar] [CrossRef]
- Mahadevan, K.K.; Dyevoich, A.M.; Chen, Y.; Li, B.; Sugimoto, H.; Sockwell, A.M.; McAndrews, K.M.; Sthanam, L.K.; Wang, H.; Shalapour, S.; et al. Type I conventional dendritic cells facilitate immunotherapy in pancreatic cancer. Science 2024, 384, eadh4567. [Google Scholar] [CrossRef]
- Hu, Z.I.; O’Reilly, E.M. Therapeutic developments in pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 7–24. [Google Scholar] [CrossRef]
- Cappello, P.; Tomaino, B.; Chiarle, R.; Ceruti, P.; Novarino, A.; Castagnoli, C.; Migliorini, P.; Perconti, G.; Giallongo, A.; Milella, M.; et al. An integrated humoral and cellular response is elicited in pancreatic cancer by alpha-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int. J. Cancer 2009, 125, 639–648. [Google Scholar] [CrossRef]
- Tjomsland, V.; Sandström, P.; Spångeus, A.; Messmer, D.; Emilsson, J.; Falkmer, U.; Falkmer, S.; Magnusson, K.-E.; Borch, K.; Larsson, M. Pancreatic adenocarcinoma exerts systemic effects on the peripheral blood myeloid and plasmacytoid dendritic cells: An indicator of disease severity? BMC Cancer 2010, 10, 87. [Google Scholar] [CrossRef]
- Lunardi, S.; Muschel, R.J.; Brunner, T.B. The stromal compartments in pancreatic cancer: Are there any therapeutic targets? Cancer Lett. 2014, 343, 147–155. [Google Scholar] [CrossRef]
- Plesca, I.; Benešová, I.; Beer, C.; Sommer, U.; Müller, L.; Wehner, R.; Heiduk, M.; Aust, D.; Baretton, G.; Bachmann, M.P.; et al. Clinical Significance of Tumor-Infiltrating Conventional and Plasmacytoid Dendritic Cells in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 1216. [Google Scholar] [CrossRef]
- Tjomsland, V.; Spångeus, A.; Sandström, P.; Borch, K.; Messmer, D.; Larsson, M. Semi mature blood dendritic cells exist in patients with ductal pancreatic adenocarcinoma owing to inflammatory factors released from the tumor. PLoS ONE 2010, 5, e13441. [Google Scholar] [CrossRef]
- Hiraoka, N.; Yamazaki-Itoh, R.; Ino, Y.; Mizuguchi, Y.; Yamada, T.; Hirohashi, S.; Kanai, Y. CXCL17 and ICAM2 are associated with a potential anti-tumor immune response in early intraepithelial stages of human pancreatic carcinogenesis. Gastroenterology 2011, 140, 310–321. [Google Scholar] [CrossRef]
- Elieh Ali Komi, D.; Wöhrl, S.; Bielory, L. Mast Cell Biology at Molecular Level: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2020, 58, 342–365. [Google Scholar] [CrossRef] [PubMed]
- Reber, L.L.; Sibilano, R.; Mukai, K.; Galli, S.J. Potential effector and immunoregulatory functions of mast cells in mucosal immunity. Mucosal Immunol. 2015, 8, 444–463. [Google Scholar] [CrossRef]
- Frossi, B.; Mion, F.; Sibilano, R.; Danelli, L.; Pucillo, C.E.M. Is it time for a new classification of mast cells? What do we know about mast cell heterogeneity? Immunol. Rev. 2018, 282, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Gri, G.; Frossi, B.; D’Inca, F.; Danelli, L.; Betto, E.; Mion, F.; Sibilano, R.; Pucillo, C. Mast cell: An emerging partner in immune interaction. Front. Immunol. 2012, 3, 120. [Google Scholar] [CrossRef]
- Tsai, M.; Valent, P.; Galli, S.J. KIT as a master regulator of the mast cell lineage. J. Allergy Clin. Immunol. 2022, 149, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Vukman, K.V.; Försönits, A.; Oszvald, Á.; Tóth, E.Á.; Buzás, E.I. Mast cell secretome: Soluble and vesicular components. Semin. Cell Dev. Biol. 2017, 67, 65–73. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Conti, P. Mast cells: The Jekyll and Hyde of tumor growth. Trends Immunol. 2004, 25, 235–241. [Google Scholar] [CrossRef]
- Ribatti, D.; Crivellato, E. The controversial role of mast cells in tumor growth. Int. Rev. Cell Mol. Biol. 2009, 275, 89–131. [Google Scholar] [CrossRef] [PubMed]
- Aponte-López, A.; Muñoz-Cruz, S. Mast Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1273, 159–173. [Google Scholar] [CrossRef]
- Wang, B.; Li, L.; Liao, Y.; Li, J.; Yu, X.; Zhang, Y.; Xu, J.; Rao, H.; Chen, S.; Zhang, L.; et al. Mast cells expressing interleukin 17 in the muscularis propria predict a favorable prognosis in esophageal squamous cell carcinoma. Cancer Immunol. Immunother. 2013, 62, 1575–1585. [Google Scholar] [CrossRef]
- Chan, J.K.; Magistris, A.; Loizzi, V.; Lin, F.; Rutgers, J.; Osann, K.; DiSaia, P.J.; Samoszuk, M. Mast cell density, angiogenesis, blood clotting, and prognosis in women with advanced ovarian cancer. Gynecol. Oncol. 2005, 99, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Hedström, G.; Berglund, M.; Molin, D.; Fischer, M.; Nilsson, G.; Thunberg, U.; Book, M.; Sundström, C.; Rosenquist, R.; Roos, G.; et al. Mast cell infiltration is a favourable prognostic factor in diffuse large B-cell lymphoma. Br. J. Haematol. 2007, 138, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Pittoni, P.; Tripodo, C.; Piconese, S.; Mauri, G.; Parenza, M.; Rigoni, A.; Sangaletti, S.; Colombo, M.P. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res. 2011, 71, 5987–5997. [Google Scholar] [CrossRef]
- Ribatti, D.; Ennas, M.G.; Vacca, A.; Ferreli, F.; Nico, B.; Orru, S.; Sirigu, P. Tumor vascularity and tryptase-positive mast cells correlate with a poor prognosis in melanoma. Eur. J. Clin. Investig. 2003, 33, 420–425. [Google Scholar] [CrossRef]
- Somasundaram, R.; Connelly, T.; Choi, R.; Choi, H.; Samarkina, A.; Li, L.; Gregorio, E.; Chen, Y.; Thakur, R.; Abdel-Mohsen, M.; et al. Tumor-infiltrating mast cells are associated with resistance to anti-PD-1 therapy. Nat. Commun. 2021, 12, 346. [Google Scholar] [CrossRef]
- Reddy, S.M.; Reuben, A.; Barua, S.; Jiang, H.; Zhang, S.; Wang, L.; Gopalakrishnan, V.; Hudgens, C.W.; Tetzlaff, M.T.; Reuben, J.M.; et al. Poor Response to Neoadjuvant Chemotherapy Correlates with Mast Cell Infiltration in Inflammatory Breast Cancer. Cancer Immunol. Res. 2019, 7, 1025–1035. [Google Scholar] [CrossRef]
- Rajput, A.B.; Turbin, D.A.; Cheang, M.C.; Voduc, D.K.; Leung, S.; Gelmon, K.A.; Gilks, C.B.; Huntsman, D.G. Stromal mast cells in invasive breast cancer are a marker of favourable prognosis: A study of 4,444 cases. Breast Cancer Res. Treat. 2008, 107, 249–257. [Google Scholar] [CrossRef]
- Sammarco, G.; Varricchi, G.; Ferraro, V.; Ammendola, M.; de Fazio, M.; Altomare, D.F.; Luposella, M.; Maltese, L.; Currò, G.; Marone, G.; et al. Mast Cells, Angiogenesis and Lymphangiogenesis in Human Gastric Cancer. Int. J. Mol. Sci. 2019, 20, 2106. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Kinuta, M.; Tateishi, H.; Nakano, Y.; Matsui, S.; Monden, T.; Okamura, J.; Sakai, M.; Okamoto, S. Mast cell infiltration around gastric cancer cells correlates with tumor angiogenesis and metastasis. Gastric Cancer 1999, 2, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, S.; Gao, Q. An integrated overview of the immunosuppression features in the tumor microenvironment of pancreatic cancer. Front. Immunol. 2023, 14, 1258538. [Google Scholar] [CrossRef]
- Cai, S.-W.; Yang, S.-Z.; Gao, J.; Pan, K.; Chen, J.-Y.; Wang, Y.-L.; Wei, L.-X.; Dong, J.-H. Prognostic significance of mast cell count following curative resection for pancreatic ductal adenocarcinoma. Surgery 2011, 149, 576–584. [Google Scholar] [CrossRef]
- Chang, D.Z.; Ma, Y.; Ji, B.; Wang, H.; Deng, D.; Liu, Y.; Abbruzzese, J.L.; Liu, Y.; Logsdon, C.D.; Hwu, P. Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2011, 17, 7015–7023. [Google Scholar] [CrossRef] [PubMed]
- Strouch, M.J.; Cheon, E.C.; Salabat, M.R.; Krantz, S.B.; Gounaris, E.; Melstrom, L.G.; Dangi-Garimella, S.; Wang, E.; Munshi, H.G.; Khazaie, K.; et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin. Cancer Res. 2010, 16, 2257–2265. [Google Scholar] [CrossRef]
- Tóth, T.; Tóth-Jakatics, R.; Jimi, S.; Takebayashi, S. Increased density of interstitial mast cells in amyloid A renal amyloidosis. Mod. Pathol. 2000, 13, 1020–1028. [Google Scholar] [CrossRef]
- Sawatsubashi, M.; Yamada, T.; Fukushima, N.; Mizokami, H.; Tokunaga, O.; Shin, T. Association of vascular endothelial growth factor and mast cells with angiogenesis in laryngeal squamous cell carcinoma. Virchows Arch. 2000, 436, 243–248. [Google Scholar] [CrossRef]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef]
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Swigart, L.B.; Evan, G.I. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar] [CrossRef]
- Norrby, K. Mast cells and angiogenesis. J. Pathol. Microbiol. Immunol. 2002, 110, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Hiromatsu, Y.; Toda, S. Mast cells and angiogenesis. Microsc. Res. Tech. 2003, 60, 64–69. [Google Scholar] [CrossRef] [PubMed]
- McHale, C.; Mohammed, Z.; Gomez, G. Human Skin-Derived Mast Cells Spontaneously Secrete Several Angiogenesis-Related Factors. Front. Immunol. 2019, 10, 1445. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.; Tamma, R.; Brunetti, O.; Pisconti, S.; Argentiero, A.; Silvestris, N.; Ribatti, D. Mast cells and angiogenesis in pancreatic ductal adenocarcinoma. Clin. Exp. Med. 2018, 18, 319–323. [Google Scholar] [CrossRef]
- Guo, X.; Zhai, L.; Xue, R.; Shi, J.; Zeng, Q.; Gao, C. Mast Cell Tryptase Contributes to Pancreatic Cancer Growth through Promoting Angiogenesis via Activation of Angiopoietin-1. Int. J. Mol. Sci. 2016, 17, 834. [Google Scholar] [CrossRef]
- Wroblewski, M.; Bauer, R.; Cubas Córdova, M.; Udonta, F.; Ben-Batalla, I.; Legler, K.; Hauser, C.; Egberts, J.; Janning, M.; Velthaus, J.; et al. Mast cells decrease efficacy of anti-angiogenic therapy by secreting matrix-degrading granzyme B. Nat. Commun. 2017, 8, 269. [Google Scholar] [CrossRef]
- Ma, Y.; Hwang, R.F.; Logsdon, C.D.; Ullrich, S.E. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 2013, 73, 3927–3937. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, C.; Jiang, K.; Werner, J.; Bazhin, A.V.; D’Haese, J.G. The Role of Stellate Cells in Pancreatic Ductal Adenocarcinoma: Targeting Perspectives. Front. Oncol. 2020, 10, 621937. [Google Scholar] [CrossRef]
- Lichterman, J.N.; Reddy, S.M. Mast Cells: A New Frontier for Cancer Immunotherapy. Cells 2021, 10, 1270. [Google Scholar] [CrossRef]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef]
- Deplanque, G.; Demarchi, M.; Hebbar, M.; Flynn, P.; Melichar, B.; Atkins, J.; Nowara, E.; Moyé, L.; Piquemal, D.; Ritter, D.; et al. A randomized, placebo-controlled phase III trial of masitinib plus gemcitabine in the treatment of advanced pancreatic cancer. Ann. Oncol. 2015, 26, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Mitry, E.; Hammel, P.; Deplanque, G.; Mornex, F.; Levy, P.; Seitz, J.-F.; Moussy, A.; Kinet, J.-P.; Hermine, O.; Rougier, P.; et al. Safety and activity of masitinib in combination with gemcitabine in patients with advanced pancreatic cancer. Cancer Chemother. Pharmacol. 2010, 66, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Dancey, J.T.; Deubelbeiss, K.A.; Harker, L.A.; Finch, C.A. Neutrophil kinetics in man. J. Clin. Investig. 1976, 58, 705–715. [Google Scholar] [CrossRef]
- Ho-Tin-Noé, B.; Boulaftali, Y.; Camerer, E. Platelets and vascular integrity: How platelets prevent bleeding in inflammation. Blood 2018, 131, 277–288. [Google Scholar] [CrossRef]
- Thanabalasuriar, A.; Scott, B.N.V.; Peiseler, M.; Willson, M.E.; Zeng, Z.; Warrener, P.; Keller, A.E.; Surewaard, B.G.J.; Dozier, E.A.; Korhonen, J.T.; et al. Neutrophil Extracellular Traps Confine Pseudomonas aeruginosa Ocular Biofilms and Restrict Brain Invasion. Cell Host Microbe 2019, 25, 526–536.e4. [Google Scholar] [CrossRef] [PubMed]
- Drummond, R.A.; Lionakis, M.S. Measuring In Vivo Neutrophil Trafficking Responses During Fungal Infection Using Mixed Bone Marrow Chimeras. Methods Mol. Biol. 2021, 2260, 179–196. [Google Scholar] [CrossRef]
- Iversen, M.B.; Reinert, L.S.; Thomsen, M.K.; Bagdonaite, I.; Nandakumar, R.; Cheshenko, N.; Prabakaran, T.; Vakhrushev, S.Y.; Krzyzowska, M.; Kratholm, S.K.; et al. An innate antiviral pathway acting before interferons at epithelial surfaces. Nat. Immunol. 2016, 17, 150–158. [Google Scholar] [CrossRef]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Sandoval, D.; Gukovskaya, A.; Reavey, P.; Gukovsky, S.; Sisk, A.; Braquet, P.; Pandol, S.J.; Poucell-Hatton, S. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology 1996, 111, 1081–1091. [Google Scholar] [CrossRef]
- Felix, K.; Gaida, M.M. Neutrophil-Derived Proteases in the Microenvironment of Pancreatic Cancer -Active Players in Tumor Progression. Int. J. Biol. Sci. 2016, 12, 302–313. [Google Scholar] [CrossRef]
- Gaida, M.M.; Steffen, T.G.; Günther, F.; Tschaharganeh, D.F.; Felix, K.; Bergmann, F.; Schirmacher, P.; Hänsch, G.M. Polymorphonuclear neutrophils promote dyshesion of tumor cells and elastase-mediated degradation of E-cadherin in pancreatic tumors. Eur. J. Immunol. 2012, 42, 3369–3380. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Adrover, J.M.; McDowell, S.A.C.; He, X.-Y.; Quail, D.F.; Egeblad, M. NETworking with cancer: The bidirectional interplay between cancer and neutrophil extracellular traps. Cancer Cell 2023, 41, 505–526. [Google Scholar] [CrossRef]
- Jung, H.S.; Gu, J.; Kim, J.-E.; Nam, Y.; Song, J.W.; Kim, H.K. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS ONE 2019, 14, e0216055. [Google Scholar] [CrossRef]
- Zhang, Y.; Chandra, V.; Riquelme Sanchez, E.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17–induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217, e20190354. [Google Scholar] [CrossRef] [PubMed]
- Schoeps, B.; Eckfeld, C.; Prokopchuk, O.; Böttcher, J.; Häußler, D.; Steiger, K.; Demir, I.E.; Knolle, P.; Soehnlein, O.; Jenne, D.E.; et al. TIMP1 Triggers Neutrophil Extracellular Trap Formation in Pancreatic Cancer. Cancer Res. 2021, 81, 3568–3579. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Yin, H.; Li, H.; Yu, X.-J.; Xu, H.-X.; Liu, L. Neutrophil extracellular DNA traps promote pancreatic cancer cells migration and invasion by activating EGFR/ERK pathway. J. Cell. Mol. Med. 2021, 25, 5443–5456. [Google Scholar] [CrossRef]
- Munir, H.; Jones, J.O.; Janowitz, T.; Hoffmann, M.; Euler, M.; Martins, C.P.; Welsh, S.J.; Shields, J.D. Stromal-driven and Amyloid β-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat. Commun. 2021, 12, 683. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Frere, C.; Bournet, B.; Gourgou, S.; Fraisse, J.; Canivet, C.; Connors, J.M.; Buscail, L.; Farge, D. Incidence of Venous Thromboembolism in Patients With Newly Diagnosed Pancreatic Cancer and Factors Associated With Outcomes. Gastroenterology 2020, 158, 1346–1358.e4. [Google Scholar] [CrossRef] [PubMed]
- Lasser, S.A.; Ozbay Kurt, F.G.; Arkhypov, I.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat. Rev. Clin. Oncol. 2024, 21, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Marigo, I.; Bosio, E.; Solito, S.; Mesa, C.; Fernandez, A.; Dolcetti, L.; Ugel, S.; Sonda, N.; Bicciato, S.; Falisi, E.; et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010, 32, 790–802. [Google Scholar] [CrossRef]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef]
- Menjivar, R.E.; Nwosu, Z.C.; Du, W.; Donahue, K.L.; Hong, H.S.; Espinoza, C.; Brown, K.; Velez-Delgado, A.; Yan, W.; Lima, F.; et al. Arginase 1 is a key driver of immune suppression in pancreatic cancer. eLife 2023, 12, e80721. [Google Scholar] [CrossRef]
- Panni, R.Z.; Sanford, D.E.; Belt, B.A.; Mitchem, J.B.; Worley, L.A.; Goetz, B.D.; Mukherjee, P.; Wang-Gillam, A.; Link, D.C.; DeNardo, D.G.; et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol. Immunother. 2014, 63, 513–528. [Google Scholar] [CrossRef]
- Porembka, M.R.; Mitchem, J.B.; Belt, B.A.; Hsieh, C.-S.; Lee, H.-M.; Herndon, J.; Gillanders, W.E.; Linehan, D.C.; Goedegebuure, P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 2012, 61, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Sanford, D.E.; Porembka, M.R.; Panni, R.Z.; Mitchem, J.B.; Belt, B.A.; Plambeck-Suess, S.M.; Lin, G.; DeNardo, D.G.; Fields, R.C.; Hawkins, W.G.; et al. A Study of Zoledronic Acid as Neo-Adjuvant, Perioperative Therapy in Patients with Resectable Pancreatic Ductal Adenocarcinoma. J. Cancer Ther. 2013, 4, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef]
- Siret, C.; Collignon, A.; Silvy, F.; Robert, S.; Cheyrol, T.; André, P.; Rigot, V.; Iovanna, J.; van de Pavert, S.; Lombardo, D.; et al. Deciphering the Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells in Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2019, 10, 3070. [Google Scholar] [CrossRef]
- Sarhan, D.; Eisinger, S.; He, F.; Bergsland, M.; Pelicano, C.; Driescher, C.; Westberg, K.; Benitez, I.I.; Humoud, R.; Palano, G.; et al. Targeting myeloid suppressive cells revives cytotoxic anti-tumor responses in pancreatic cancer. iScience 2022, 25, 105317. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.-G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef]
- Suzuki, E.; Kapoor, V.; Jassar, A.S.; Kaiser, L.R.; Albelda, S.M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef]
- Sun, R.; Sun, Y.; Wu, C.; Liu, Y.; Zhou, M.; Dong, Y.; Du, G.; Luo, H.; Shi, B.; Jiang, H.; et al. CXCR4-modified CAR-T cells suppresses MDSCs recruitment via STAT3/NF-κB/SDF-1α axis to enhance efficacy against pancreatic cancer. Mol. Ther. 2023, 31, 3193–3209. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef]
- Takeuchi, S.; Baghdadi, M.; Tsuchikawa, T.; Wada, H.; Nakamura, T.; Abe, H.; Nakanishi, S.; Usui, Y.; Higuchi, K.; Takahashi, M.; et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res. 2015, 75, 2629–2640. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M. Differentiation and proliferation of hematopoietic stem cells. Blood 1993, 81, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Gardner, F.H.; Cohen, P. Platelet Life Span. Transfusion 1966, 6, 23–31. [Google Scholar] [CrossRef]
- Yun, S.-H.; Sim, E.-H.; Goh, R.-Y.; Park, J.-I.; Han, J.-Y. Platelet Activation: The Mechanisms and Potential Biomarkers. BioMed Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef]
- Bottsford-Miller, J.; Choi, H.-J.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Haemmerle, M.; Nick, A.M.; Pradeep, S.; Zand, B.; Previs, R.A.; et al. Differential platelet levels affect response to taxane-based therapy in ovarian cancer. Clin. Cancer Res. 2015, 21, 602–610. [Google Scholar] [CrossRef]
- Haemmerle, M.; Bottsford-Miller, J.; Pradeep, S.; Taylor, M.L.; Choi, H.-J.; Hansen, J.M.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Nick, A.M.; et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J. Clin. Investig. 2016, 126, 1885–1896. [Google Scholar] [CrossRef]
- Haemmerle, M.; Taylor, M.L.; Gutschner, T.; Pradeep, S.; Cho, M.S.; Sheng, J.; Lyons, Y.M.; Nagaraja, A.S.; Dood, R.L.; Wen, Y.; et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun. 2017, 8, 310. [Google Scholar] [CrossRef]
- Ali, R.A.; Wuescher, L.M.; Worth, R.G. Platelets: Essential components of the immune system. Curr. Trends Immunol. 2015, 16, 65–78. [Google Scholar]
- Nicolai, L.; Gaertner, F.; Massberg, S. Platelets in Host Defense: Experimental and Clinical Insights. Trends Immunol. 2019, 40, 922–938. [Google Scholar] [CrossRef]
- Dib, P.R.B.; Quirino-Teixeira, A.C.; Merij, L.B.; Pinheiro, M.B.M.; Rozini, S.V.; Andrade, F.B.; Hottz, E.D. Innate immune receptors in platelets and platelet-leukocyte interactions. J. Leukoc. Biol. 2020, 108, 1157–1182. [Google Scholar] [CrossRef]
- Chapman, L.M.; Aggrey, A.A.; Field, D.J.; Srivastava, K.; Ture, S.; Yui, K.; Topham, D.J.; Baldwin, W.M.; Morrell, C.N. Platelets present antigen in the context of MHC class I. J. Immunol. 2012, 189, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Pitchford, S.C.; Momi, S.; Giannini, S.; Casali, L.; Spina, D.; Page, C.P.; Gresele, P. Platelet P-selectin is required for pulmonary eosinophil and lymphocyte recruitment in a murine model of allergic inflammation. Blood 2005, 105, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Pitchford, S.C.; Yano, H.; Lever, R.; Riffo-Vasquez, Y.; Ciferri, S.; Rose, M.J.; Giannini, S.; Momi, S.; Spina, D.; O’connor, B.; et al. Platelets are essential for leukocyte recruitment in allergic inflammation. J. Allergy Clin. Immunol. 2003, 112, 109–118. [Google Scholar] [CrossRef]
- de Stoppelaar, S.F.; van’t Veer, C.; Claushuis, T.A.M.; Albersen, B.J.A.; Roelofs, J.J.T.H.; van der Poll, T. Thrombocytopenia impairs host defense in gram-negative pneumonia-derived sepsis in mice. Blood 2014, 124, 3781–3790. [Google Scholar] [CrossRef]
- van den Boogaard, F.E.; Schouten, M.; de Stoppelaar, S.F.; Roelofs, J.J.T.H.; Brands, X.; Schultz, M.J.; van’t Veer, C.; van der Poll, T. Thrombocytopenia impairs host defense during murine Streptococcus pneumoniae pneumonia. Crit. Care Med. 2015, 43, e75–e83. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Männel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar]
- Maurer, S.; Kropp, K.N.; Klein, G.; Steinle, A.; Haen, S.P.; Walz, J.S.; Hinterleitner, C.; Märklin, M.; Kopp, H.-G.; Salih, H.R. Platelet-mediated shedding of NKG2D ligands impairs NK cell immune-surveillance of tumor cells. Oncoimmunology 2018, 7, e1364827. [Google Scholar] [CrossRef]
- Clar, K.L.; Hinterleitner, C.; Schneider, P.; Salih, H.R.; Maurer, S. Inhibition of NK Reactivity Against Solid Tumors by Platelet-Derived RANKL. Cancers 2019, 11, 277. [Google Scholar] [CrossRef]
- Kissel, K.; Berber, S.; Nockher, A.; Santoso, S.; Bein, G.; Hackstein, H. Human platelets target dendritic cell differentiation and production of proinflammatory cytokines. Transfusion 2006, 46, 818–827. [Google Scholar] [CrossRef]
- Langer, H.F.; Daub, K.; Braun, G.; Schönberger, T.; May, A.E.; Schaller, M.; Stein, G.M.; Stellos, K.; Bueltmann, A.; Siegel-Axel, D.; et al. Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1463–1470. [Google Scholar] [CrossRef]
- Carestia, A.; Mena, H.A.; Olexen, C.M.; Ortiz Wilczyñski, J.M.; Negrotto, S.; Errasti, A.E.; Gómez, R.M.; Jenne, C.N.; Carrera Silva, E.A.; Schattner, M. Platelets Promote Macrophage Polarization toward Pro-inflammatory Phenotype and Increase Survival of Septic Mice. Cell Rep. 2019, 28, 896–908.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, J.; Liu, Z.; Wu, T.; Li, S.; Zhang, Y.; Yin, X.; Yang, G.; Zhang, G. Tumor cell-induced platelet aggregation accelerates hematogenous metastasis of malignant melanoma by triggering macrophage recruitment. J. Exp. Clin. Cancer Res. 2023, 42, 277. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Shaked, I.; Little, K.M.; Ley, K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J. Immunol. 2010, 184, 4810–4818. [Google Scholar] [CrossRef] [PubMed]
- Lucotti, S.; Ogitani, Y.; Kenific, C.M.; Geri, J.; Kim, Y.H.; Gu, J.; Balaji, U.; Bojmar, L.; Shaashua, L.; Song, Y.; et al. Extracellular vesicles from the lung pro-thrombotic niche drive cancer-associated thrombosis and metastasis via integrin beta 2. Cell 2025, 188, 1642–1661.e24. [Google Scholar] [CrossRef]
- Santoso, S.; Sachs, U.J.H.; Kroll, H.; Linder, M.; Ruf, A.; Preissner, K.T.; Chavakis, T. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J. Exp. Med. 2002, 196, 679–691. [Google Scholar] [CrossRef]
- Haselmayer, P.; Grosse-Hovest, L.; von Landenberg, P.; Schild, H.; Radsak, M.P. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood 2007, 110, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Zhang, S.; Chew, M.; Ersson, A.; Jeppsson, B.; Thorlacius, H. Platelet-derived CD40L (CD154) mediates neutrophil upregulation of Mac-1 and recruitment in septic lung injury. Ann. Surg. 2009, 250, 783–790. [Google Scholar] [CrossRef]
- Zuchtriegel, G.; Uhl, B.; Puhr-Westerheide, D.; Pörnbacher, M.; Lauber, K.; Krombach, F.; Reichel, C.A. Platelets Guide Leukocytes to Their Sites of Extravasation. PLoS Biol. 2016, 14, e1002459. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- von Brühl, M.-L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Rosell, A.; Martinod, K.; Mackman, N.; Thålin, C. Neutrophil extracellular traps and cancer-associated thrombosis. Thromb. Res. 2022, 213 (Suppl. 1), S35–S41. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.; Soundararajan, R.; Vasaikar, S.; Yang, F.; Allton, K.L.; Tian, L.; den Hollander, P.; Isgandarova, S.; Haemmerle, M.; Mino, B.; et al. CD8+ T cells inhibit metastasis and CXCL4 regulates its function. Br. J. Cancer 2021, 125, 176–189. [Google Scholar] [CrossRef]
- Servais, L.; Wéra, O.; Dibato Epoh, J.; Delierneux, C.; Bouznad, N.; Rahmouni, S.; Mazzucchelli, G.; Baiwir, D.; Delvenne, P.; Lancellotti, P.; et al. Platelets contribute to the initiation of colitis-associated cancer by promoting immunosuppression. J. Thromb. Haemost. 2018, 16, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Karhausen, J.; Choi, H.W.; Maddipati, K.R.; Mathew, J.P.; Ma, Q.; Boulaftali, Y.; Lee, R.H.; Bergmeier, W.; Abraham, S.N. Platelets trigger perivascular mast cell degranulation to cause inflammatory responses and tissue injury. Sci. Adv. 2020, 6, eaay6314. [Google Scholar] [CrossRef]
- de Giovanni, M.; Dang, E.V.; Chen, K.Y.; An, J.; Madhani, H.D.; Cyster, J.G. Platelets and mast cells promote pathogenic eosinophil recruitment during invasive fungal infection via the 5-HIAA-GPR35 ligand-receptor system. Immunity 2023, 56, 1548–1560.e5. [Google Scholar] [CrossRef]
- Xu, H.; He, A.; Liu, A.; Tong, W.; Cao, D. Evaluation of the prognostic role of platelet-lymphocyte ratio in cancer patients treated with immune checkpoint inhibitors: A systematic review and meta-analysis. Int. Immunopharmacol. 2019, 77, 105957. [Google Scholar] [CrossRef]
- Cho, M.S.; Lee, H.; Gonzalez-Delgado, R.; Li, D.; Sasano, T.; Carlos-Alcalde, W.; Ma, Q.; Liu, J.; Sood, A.K.; Afshar-Kharghan, V. Platelets Increase the Expression of PD-L1 in Ovarian Cancer. Cancers 2022, 14, 2498. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Malloy, M.W.; Roweth, H.G.; McAllister, S.S.; Italiano, J.E.; Battinelli, E.M. Platelets upregulate tumor cell programmed death ligand 1 in an epidermal growth factor receptor-dependent manner in vitro. Blood Adv. 2022, 6, 5668–5675. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.-W.; Li, Y.; Yang, Y.; Yang, H.-K.; Dong, J.-M.; Xiao, Z.-H.; He, X.; Guo, J.-H.; Wang, R.-Q.; Dai, B.; et al. Tumor immunotherapy resistance: Revealing the mechanism of PD-1/PD-L1-mediated tumor immune escape. Biomed. Pharmacother. 2024, 171, 116203. [Google Scholar] [CrossRef]
- Zaslavsky, A.B.; Adams, M.P.; Cao, X.; Maj, T.; Choi, J.E.; Stangl-Kremser, J.; Patel, S.; Putelo, A.; Lee, S.K.; Nallandhighal, S.; et al. Platelet PD-L1 suppresses anti-cancer immune cell activity in PD-L1 negative tumors. Sci. Rep. 2020, 10, 19296. [Google Scholar] [CrossRef] [PubMed]
- Hinterleitner, C.; Strähle, J.; Malenke, E.; Hinterleitner, M.; Henning, M.; Seehawer, M.; Bilich, T.; Heitmann, J.; Lutz, M.; Mattern, S.; et al. Platelet PD-L1 reflects collective intratumoral PD-L1 expression and predicts immunotherapy response in non-small cell lung cancer. Nat. Commun. 2021, 12, 7005. [Google Scholar] [CrossRef]
- Lutz, M.S.; Klimovich, B.; Maurer, S.; Heitmann, J.S.; Märklin, M.; Zekri, L.; Jung, G.; Salih, H.R.; Hinterleitner, C. Platelets subvert antitumor efficacy of T cell-recruiting bispecific antibodies. J. Immunother. Cancer 2022, 10, e003655. [Google Scholar] [CrossRef]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; Paulos, C.M.; Rubinstein, M.P.; Garrett-Mayer, E.; Hennig, M.; et al. Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci. Immunol. 2017, 2, eaai7911. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Yang, J.; Yamashita-Kanemaru, Y.; Morris, B.I.; Contursi, A.; Trajkovski, D.; Xu, J.; Patrascan, I.; Benson, J.; Evans, A.C.; Conti, A.G.; et al. Aspirin prevents metastasis by limiting platelet TXA2 suppression of T cell immunity. Nature 2025, 640, 1052–1061. [Google Scholar] [CrossRef]
- Liu, X.; Song, J.; Zhang, H.; Liu, X.; Zuo, F.; Zhao, Y.; Zhao, Y.; Yin, X.; Guo, X.; Wu, X.; et al. Immune checkpoint HLA-E:CD94-NKG2A mediates evasion of circulating tumor cells from NK cell surveillance. Cancer Cell 2023, 41, 272–287.e9. [Google Scholar] [CrossRef]
- Heinmöller, E.; Schropp, T.; Kisker, O.; Simon, B.; Seitz, R.; Weinel, R.J. Tumor cell-induced platelet aggregation in vitro by human pancreatic cancer cell lines. Scand. J. Gastroenterol. 1995, 30, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-G.; Geddings, J.E.; Aleman, M.M.; Cardenas, J.C.; Chantrathammachart, P.; Williams, J.C.; Kirchhofer, D.; Bogdanov, V.Y.; Bach, R.R.; Rak, J.; et al. Tumor-derived tissue factor activates coagulation and enhances thrombosis in a mouse xenograft model of human pancreatic cancer. Blood 2012, 119, 5543–5552. [Google Scholar] [CrossRef]
- Hisada, Y.; Ay, C.; Auriemma, A.C.; Cooley, B.C.; Mackman, N. Human pancreatic tumors grown in mice release tissue factor-positive microvesicles that increase venous clot size. J. Thromb. Haemost. 2017, 15, 2208–2217. [Google Scholar] [CrossRef]
- Ansari, D.; Ansari, D.; Andersson, R.; Andrén-Sandberg, Å. Pancreatic cancer and thromboembolic disease, 150 years after Trousseau. Hepatobiliary Surg. Nutr. 2015, 4, 325–335. [Google Scholar] [CrossRef]
- Brown, K.M.; Domin, C.; Aranha, G.V.; Yong, S.; Shoup, M. Increased preoperative platelet count is associated with decreased survival after resection for adenocarcinoma of the pancreas. Am. J. Surg. 2005, 189, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Chadha, A.S.; Kocak-Uzel, E.; Das, P.; Minsky, B.D.; Delclos, M.E.; Mahmood, U.; Guha, S.; Ahmad, M.; Varadhachary, G.R.; Javle, M.; et al. Paraneoplastic thrombocytosis independently predicts poor prognosis in patients with locally advanced pancreatic cancer. Acta Oncol. 2015, 54, 971–978. [Google Scholar] [CrossRef]
- Iodice, S.; Gandini, S.; Löhr, M.; Lowenfels, A.B.; Maisonneuve, P. Venous thromboembolic events and organ-specific occult cancers: A review and meta-analysis. J. Thromb. Haemost. 2008, 6, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Tian, C.; Wang, K.; Zhang, R.-J.; Zou, S.-B. Preoperative platelet lymphocyte ratio as independent predictors of prognosis in pancreatic cancer: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0178762. [Google Scholar] [CrossRef]
- Shirai, Y.; Shiba, H.; Haruki, K.; Horiuchi, T.; Saito, N.; Fujiwara, Y.; Sakamoto, T.; Uwagawa, T.; Yanaga, K. Preoperative Platelet–to–Albumin Ratio Predicts Prognosis of Patients with Pancreatic Ductal Adenocarcinoma After Pancreatic Resection. Anticancer. Res. 2017, 37, 787–794. [Google Scholar] [CrossRef]
- Best, M.G.; Sol, N.; Kooi, I.; Tannous, J.; Westerman, B.A.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Koster, J.; et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell 2015, 28, 666–676. [Google Scholar] [CrossRef]
- Assoian, R.K.; Komoriya, A.; Meyers, C.A.; Miller, D.M.; Sporn, M.B. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 1983, 258, 7155–7160. [Google Scholar] [CrossRef]
- Tindall, R.R.; Bailey-Lundberg, J.M.; Cao, Y.; Ko, T.C. The TGF-β superfamily as potential therapeutic targets in pancreatic cancer. Front. Oncol. 2024, 14, 1362247. [Google Scholar] [CrossRef] [PubMed]
- Shek, F.W.-T.; Benyon, R.C.; Walker, F.M.; McCrudden, P.R.; Pender, S.L.F.; Williams, E.J.; Johnson, P.A.; Johnson, C.D.; Bateman, A.C.; Fine, D.R.; et al. Expression of Transforming Growth Factor-β1 by Pancreatic Stellate Cells and Its Implications for Matrix Secretion and Turnover in Chronic Pancreatitis. Am. J. Pathol. 2002, 160, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Chuvin, N.; Vincent, D.F.; Pommier, R.M.; Alcaraz, L.B.; Gout, J.; Caligaris, C.; Yacoub, K.; Cardot, V.; Roger, E.; Kaniewski, B.; et al. Acinar-to-Ductal Metaplasia Induced by Transforming Growth Factor Beta Facilitates KRAS G12D -driven Pancreatic Tumorigenesis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, S.; Tolley, N.D.; Dixon, D.A.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Weyrich, A.S. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J. Cell Biol. 2001, 154, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Pennings, G.J.; Reddel, C.J.; Traini, M.; Lam, M.; Kockx, M.; Chen, V.M.; Kritharides, L. Rapid Release of Interleukin-1β from Human Platelets Is Independent of NLRP3 and Caspase. Thromb. Haemost. 2022, 122, 517–528. [Google Scholar] [CrossRef]
- Takahashi, R.; Macchini, M.; Sunagawa, M.; Jiang, Z.; Tanaka, T.; Valenti, G.; Renz, B.W.; White, R.A.; Hayakawa, Y.; Westphalen, C.B.; et al. Interleukin-1β-induced pancreatitis promotes pancreatic ductal adenocarcinoma via B lymphocyte-mediated immune suppression. Gut 2021, 70, 330–341. [Google Scholar] [CrossRef]
- Marrache, F.; Tu, S.P.; Bhagat, G.; Pendyala, S.; Osterreicher, C.H.; Gordon, S.; Ramanathan, V.; Penz-Osterreicher, M.; Betz, K.S.; Song, Z.; et al. Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis. Gastroenterology 2008, 135, 1277–1287. [Google Scholar] [CrossRef]
- Li, H.; Jiang, W.; Zhang, S.-R.; Li, P.-C.; Li, T.-J.; Jin, W.; Xu, H.-X.; Yu, X.-J.; Liu, L. The platelet pannexin 1-IL-1β axis orchestrates pancreatic ductal adenocarcinoma invasion and metastasis. Oncogene 2023, 42, 1453–1465. [Google Scholar] [CrossRef]
- Zhao, J.-G.; Li, Y.-J.; Wu, Y.; Zhang, K.; Peng, L.-J.; Chen, H. Revealing platelet-related subtypes and prognostic signature in pancreatic adenocarcinoma. BMC Med. Genom. 2023, 16, 106. [Google Scholar] [CrossRef]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Buckland, G.R.; Wilding, S.A.; McDonnell, D.; Hamady, Z.Z.R. The role of aspirin in the prevention of pancreatic cancer: A nested case-control study in the UK Biobank. Pancreatology 2024, 24, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Gao, M.; Lu, L.; Gui, R.; Fu, Y. Doxorubicin-Loaded Platelet Decoys for Enhanced Chemoimmunotherapy Against Triple-Negative Breast Cancer in Mice Model. Int. J. Nanomed. 2023, 18, 3577–3593. [Google Scholar] [CrossRef] [PubMed]
- Mai, X.; Zhang, Y.; Fan, H.; Song, W.; Chang, Y.; Chen, B.; Shi, J.; Xin, X.; Teng, Z.; Sun, J.; et al. Integration of immunogenic activation and immunosuppressive reversion using mitochondrial-respiration-inhibited platelet-mimicking nanoparticles. Biomaterials 2020, 232, 119699. [Google Scholar] [CrossRef]
- Chen, C.; Chen, N.; Qi, Y.; Lyu, M.; Wu, C.; Xie, C.; Yu, H. Copper-Based Single-Atom Nanozyme System Mimicking Platelet Cells for Enhancing the Outcome of Radioimmunotherapy. Int. J. Nanomed. 2024, 19, 403–414. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Yao, Y.; Wang, S.; Zhang, Y.; Dotti, G.; Yu, J.; Gu, Z. T cell-mimicking platelet-drug conjugates. Matter 2023, 6, 2340–2355. [Google Scholar] [CrossRef]
- Kött, J.; Zell, T.; Zimmermann, N.; Rünger, A.; Smit, D.J.; Abeck, F.; Geidel, G.; Hansen-Abeck, I.; Heidrich, I.; Weichenthal, M.; et al. Improved survival of advanced melanoma patients receiving immunotherapy with concomitant antithrombotic therapy—A multicenter study on 2419 patients from the prospective skin cancer registry ADOReg. Eur. J. Cancer 2024, 214, 115159. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Gupta, Y.H.; Khanom, A.; Acton, S.E. Control of Dendritic Cell Function Within the Tumour Microenvironment. Front. Immunol. 2022, 13, 733800. [Google Scholar] [CrossRef]
- Crouse, J.; Xu, H.C.; Lang, P.A.; Oxenius, A. NK cells regulating T cell responses: Mechanisms and outcome. Trends Immunol. 2015, 36, 49–58. [Google Scholar] [CrossRef]
- Krieger, T.G.; Le Blanc, S.; Jabs, J.; Ten, F.W.; Ishaque, N.; Jechow, K.; Debnath, O.; Leonhardt, C.-S.; Giri, A.; Eils, R.; et al. Single-cell analysis of patient-derived PDAC organoids reveals cell state heterogeneity and a conserved developmental hierarchy. Nat. Commun. 2021, 12, 5826. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Noel, P.; Borazanci, E.H.; Lee, J.; Amini, A.; Han, I.W.; Heo, J.S.; Jameson, G.S.; Fraser, C.; Steinbach, M.; et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 2020, 12, 80. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.-F.; Chen, C.-Y.; Zhou, J.-Y.; Chen, Y.-S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.-S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.G.; Carpenter, E.S.; Kemp, S.B.; Sirihorachai, V.R.; The, S.; Delrosario, L.; Lazarus, J.; Amir, E.-A.D.; Gunchick, V.; Espinoza, C.; et al. Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat. Cancer 2020, 1, 1097–1112. [Google Scholar] [CrossRef]
- Liudahl, S.M.; Betts, C.B.; Sivagnanam, S.; Morales-Oyarvide, V.; Da Silva, A.; Yuan, C.; Hwang, S.; Grossblatt-Wait, A.; Leis, K.R.; Larson, W.; et al. Leukocyte Heterogeneity in Pancreatic Ductal Adenocarcinoma: Phenotypic and Spatial Features Associated with Clinical Outcome. Cancer Discov. 2021, 11, 2014–2031. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Grünwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e18. [Google Scholar] [CrossRef]
- Pei, G.; Min, J.; Rajapakshe, K.I.; Branchi, V.; Liu, Y.; Selvanesan, B.C.; Thege, F.; Sadeghian, D.; Zhang, D.; Cho, K.S.; et al. Spatial mapping of transcriptomic plasticity in metastatic pancreatic cancer. Nature 2025. [Google Scholar] [CrossRef]
- Zhou, D.C.; Jayasinghe, R.G.; Chen, S.; Herndon, J.M.; Iglesia, M.D.; Navale, P.; Wendl, M.C.; Caravan, W.; Sato, K.; Storrs, E.; et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat. Genet. 2022, 54, 1390–1405. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NK Cells | |||
| Compound | Clinical Study Phase | Trial Number | Combination Drugs/Treatment |
| CAR-NK Cells (CL-NK-001) | Phase 1 | NCT06816823 | - |
| Allogeneic Magicell-NK infusion | Phases 1/2 | NCT06730009 | SLOG chemotherapy |
| CAR-T/CAR-NK cells | Phase 1 | NCT06572956 | - |
| Chemotherapy Sequential NKG2D CAR-NK Cell | Phase 1 | NCT06503497 | Second-line chemotherapy (not specified) |
| Intratumoral Injection and intravenous infusion of NKG2D CAR-NK cells | Phase 1 | NCT06478459 | - |
| CAR-NK cells (CB CAR-NK182) targeting Claudin18.2 | Phase 1 | NCT06464965 | - |
| TROP2-CAR/IL15-transduced CB-NK cells | Phases 1/2 | NCT05922930 | Cyclophosphamide, Fludarabine |
| NK cell infusion | Phases 1/2 | NCT02718859 | Irreversible electroporation (IRE) |
| Cytokine-induced Killer Cells | Phases 1/2 | NCT01868490 | - |
| Dendritic cell-activated Cytokine-induced killer treatment (DC-CIK) | Phases 1/2 | NCT01781520 | S-1 |
| Interleukin-2 | Phase 2 | NCT05810792 | Histamine Dihydrochloride (HDC) |
| FT500 | Phase 1 | NCT03841110 | Cyclophosphamide, Fludarabine, Nivolumab, Pembrolizumab, Atezolizumab, IL-2 |
| FATE-NK100 | Phase 1 | NCT03319459 | Cetuximab, Trastuzumab |
| Macrophages | |||
| Compound | Preclinical/clinical study | Trial Number | Combination drug |
| BMS-813160 (CCR2/CCR5 antagonist) | Phases 1/2 | NCT03184870 | Nivolumab, nab-paclitaxel, gemcitabine, 5-FU, leucovorin, irinotecan |
| BMS-813160 | Phases 1/2 | NCT03767582 | Nivolumab, GVAX |
| TAK-500 (CCR2 and STING agonist) | Phase 1 | NCT05070247 | Pembrolizumab |
| PLX3397 (CSF1R inhibitors) | Phase 1 | NCT02777710 | Durvalumab |
| LY3022855 (anti-CSF1R antibody) | Phase 1 | NCT03153410 | Cyclophosphamide, GVAX, pembrolizumab |
| Cabiralizumab (anti-CSF1R antibody) | Phase 1 | NCT02526017 | Nivolumab |
| Lurbinectedin | Phase 2 | NCT05229588 | - |
| BI 754,091 (anti-CD47/SIRPα antibody) | Phase 1 | NCT04752215 | Ezabenlimab |
| CP-870, 893 (CD40 agonist antibody) | Phase 1 | NCT01456585 | Gemcitabine |
| APX005M (CD40 agonist antibody) | Phase 1 | NCT02600949 | Pembrolizumab, sotigalimab |
| APX005M | Phases 1/2 | NCT05419479 | Zimberelimab, domvanalimab |
| RO7009789 (CD40 agonist antibody) | Phase 1 | NCT02588443 | Nab-paclitaxel, gemcitabine |
| Gedatolisib (PI3K/mTOR inhibitor) | Phase 1 | NCT03065062 | Palbociclib |
| Metformin | Phase 1 | NCT02336087 | Gemcitabine, paclitaxel albumin-stabilized nanoparticle formulation |
| Metformin | Phase 2 | NCT01666730 | Oxaliplatin, leucovorin calcium, fluorouracil |
| Metformin | Phase 2 | NCT04033107 | Vitamin C |
| CT-0508 (CAR macrophages) | Phase 1 | NCT04660929 | Pembrolizumab |
| Dendritic cells | |||
| Compound | Preclinical/clinical study | Trial Number | Combination drug |
| Metformin | Phase 2 | PREOPANC trial EudraCT: 2012-003181-40 | - |
| Toripalimab | Phases 1b/2 | ChiCTR2000032293 | chemotherapy |
| Ibrutinib | Phase 1/2 | NCT02562898 | Paclitaxel, gemcitabine |
| Wilms’ tumor 1 (WT1) peptides | Phase 2 | jRCTc030190195 | multiagent chemotherapy |
| Mast cells | |||
| Compound | Preclinical/clinical study | Trial Number | Combination drug |
| Masitinib (TKI targeting KIT) | Phase 3 | NCT00789633 | Gemcitabine |
| Neutrophils | |||
| Compound | Preclinical/clinical study | Trial Number | Combination drug |
| Nadunolimab (IL1RAP inhibition) | Phases 1/2 | NCT03267316 | Cisplatin, gemcitabine, nab-paclitaxel, carboplatin, pemetrexed |
| MDSCs | |||
| Compound | Preclinical/clinical study | Trial Number | Combination drug |
| Entinostat | Phase 2 | NCT03250273 | Nivolumab |
| SX-682 (CXCR1/2 Inhibitor) | Phase 1 | NCT04477343 | Nivolumab |
| Platelets | |||
| Compound | Preclinical/clinical study | Trial Number | Combination drug |
| Avatrombopag | Phase 1 | NCT06182072 | Gemcitabine, nab-Paclitaxel |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blümke, J.; Schameitat, M.; Verma, A.; Limbecker, C.; Arlt, E.; Kessler, S.M.; Kielstein, H.; Krug, S.; Bazwinsky-Wutschke, I.; Haemmerle, M. Innate Immunity and Platelets: Unveiling Their Role in Chronic Pancreatitis and Pancreatic Cancer. Cancers 2025, 17, 1689. https://doi.org/10.3390/cancers17101689
Blümke J, Schameitat M, Verma A, Limbecker C, Arlt E, Kessler SM, Kielstein H, Krug S, Bazwinsky-Wutschke I, Haemmerle M. Innate Immunity and Platelets: Unveiling Their Role in Chronic Pancreatitis and Pancreatic Cancer. Cancers. 2025; 17(10):1689. https://doi.org/10.3390/cancers17101689
Chicago/Turabian StyleBlümke, Juliane, Moritz Schameitat, Atul Verma, Celina Limbecker, Elise Arlt, Sonja M. Kessler, Heike Kielstein, Sebastian Krug, Ivonne Bazwinsky-Wutschke, and Monika Haemmerle. 2025. "Innate Immunity and Platelets: Unveiling Their Role in Chronic Pancreatitis and Pancreatic Cancer" Cancers 17, no. 10: 1689. https://doi.org/10.3390/cancers17101689
APA StyleBlümke, J., Schameitat, M., Verma, A., Limbecker, C., Arlt, E., Kessler, S. M., Kielstein, H., Krug, S., Bazwinsky-Wutschke, I., & Haemmerle, M. (2025). Innate Immunity and Platelets: Unveiling Their Role in Chronic Pancreatitis and Pancreatic Cancer. Cancers, 17(10), 1689. https://doi.org/10.3390/cancers17101689