
CRISPR/Cas9 Screening Highlights PFKFB3 Gene as a Major Contributor to 5-Fluorouracil Resistance in Esophageal Cancer

 , , and
, , and
Simple Summary
Abstract
1. Introduction
2. Results
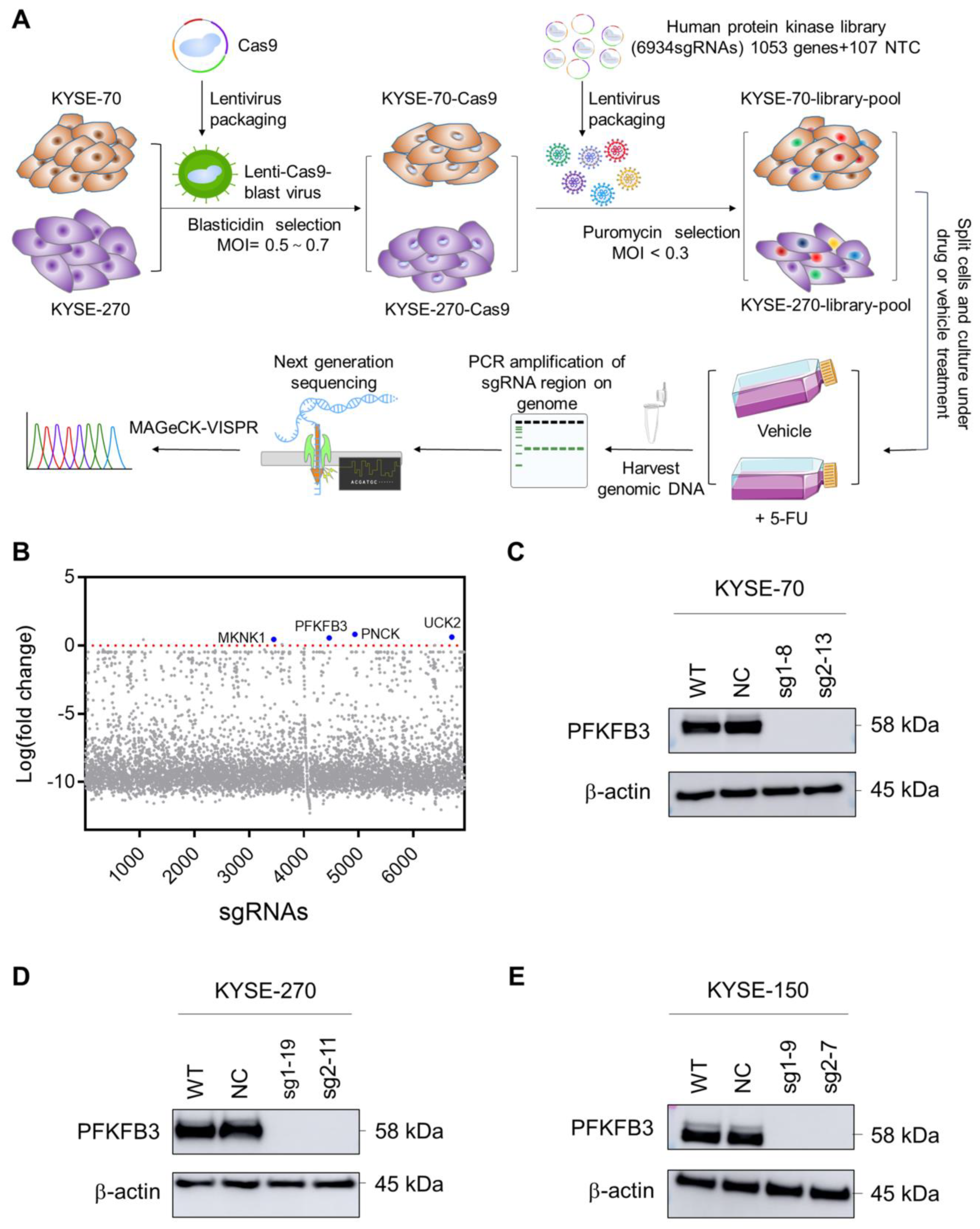
2.1. Human Protein Kinase Library Screening Identifies PFKFB3 as Target Gene Involved in Esophageal Cancer Chemoresistance
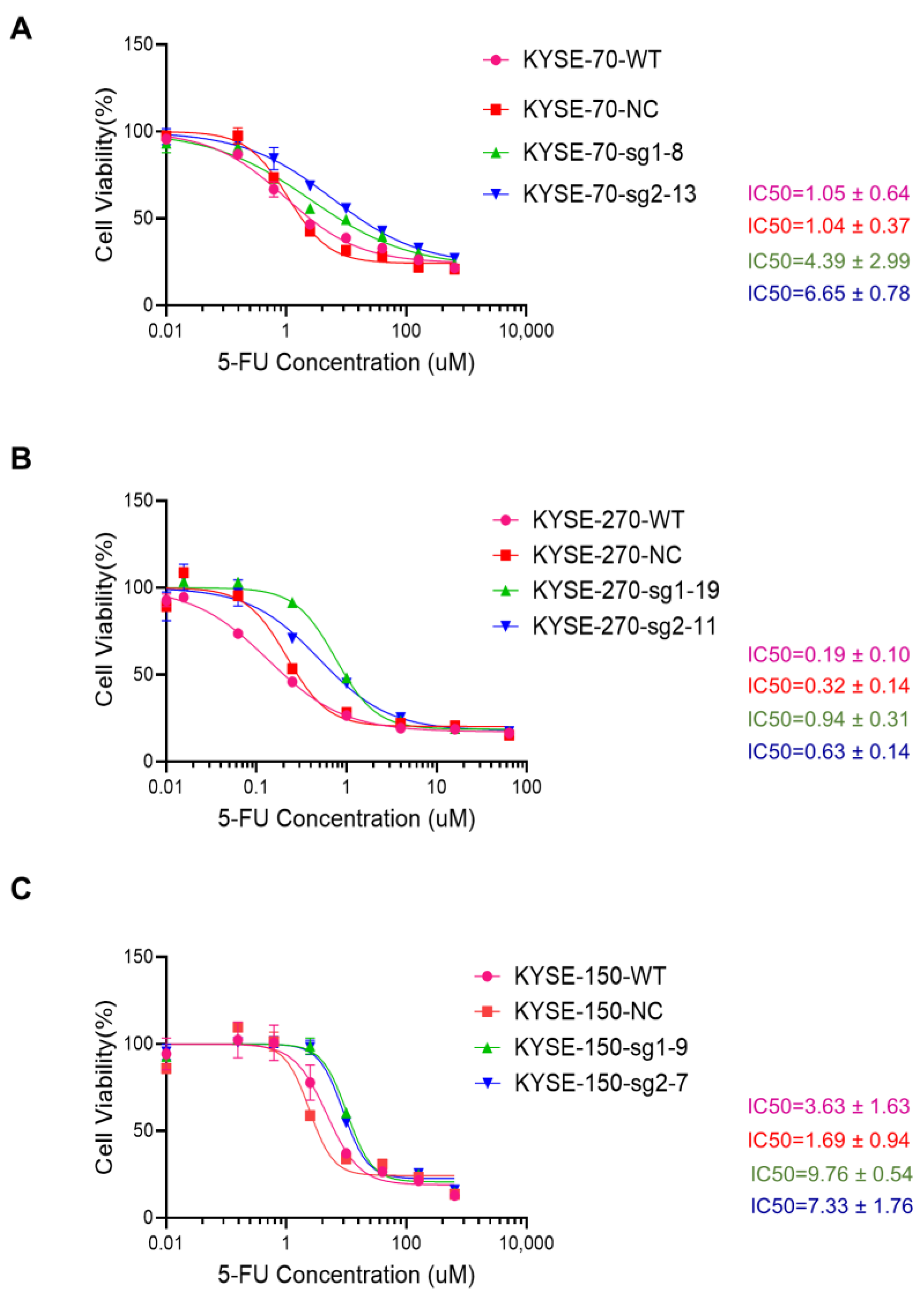
2.2. Knockout of PFKFB3 Enhances 5-FU Resistance in Human Esophageal Cancer Cell Lines
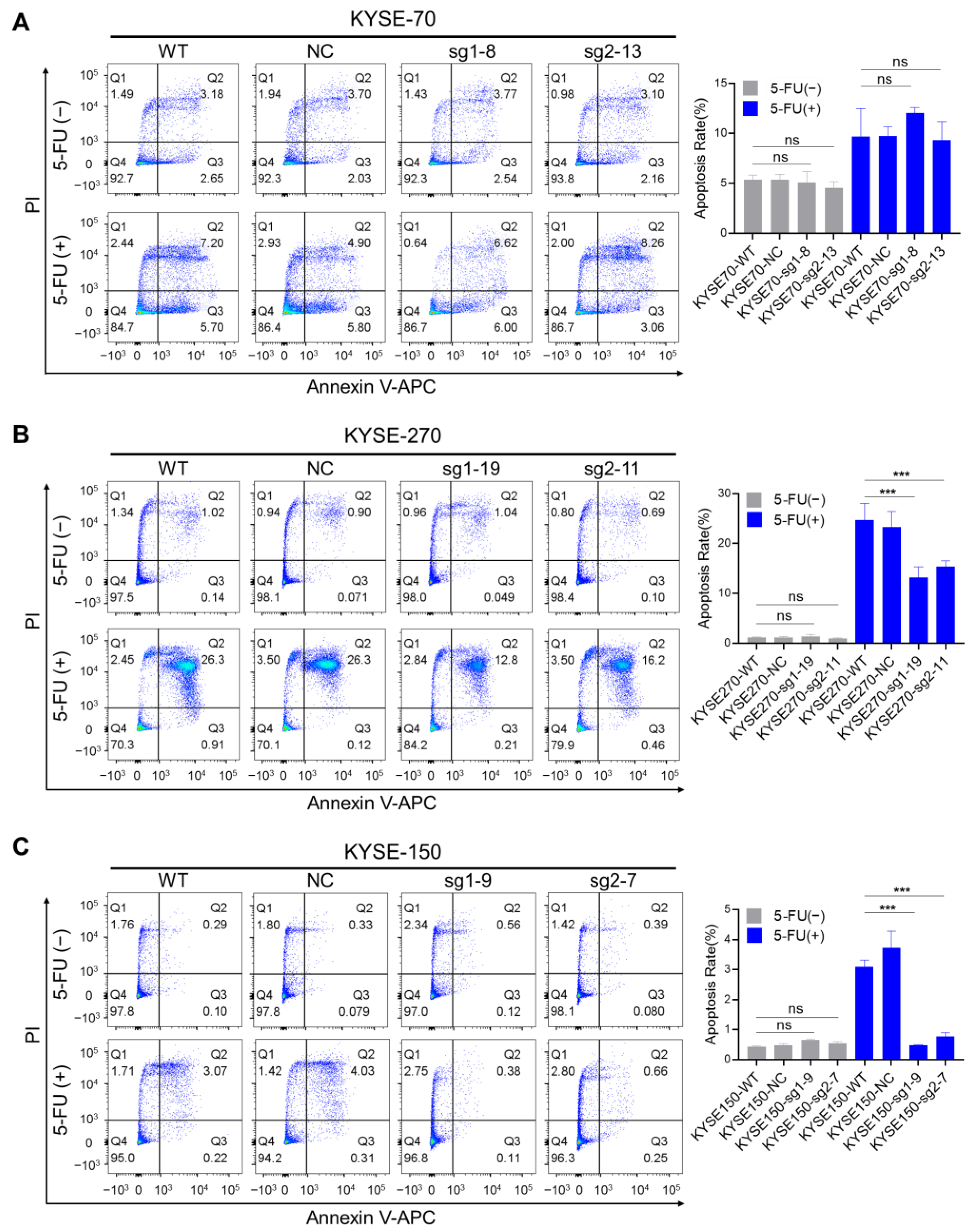
2.3. The Impact of PFKFB3 on Apoptosis in Human Esophageal Cancer Cell Lines
2.4. The Impact of PFKFB3 on Cell Cycle in Human Esophageal Cancer Cell Lines
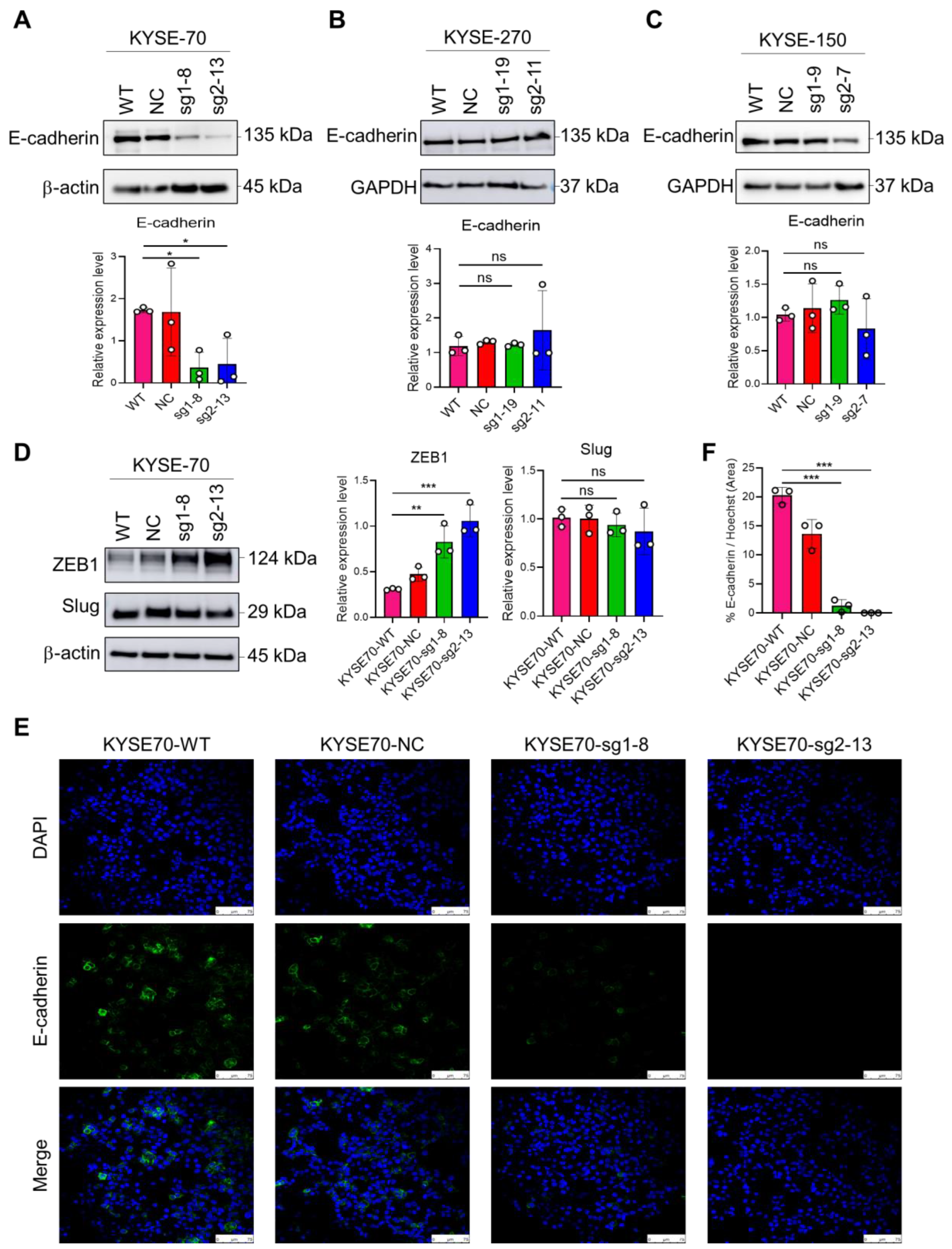
2.5. The Impact of PFKFB3 on EMT in Human Esophageal Cancer Cell Lines
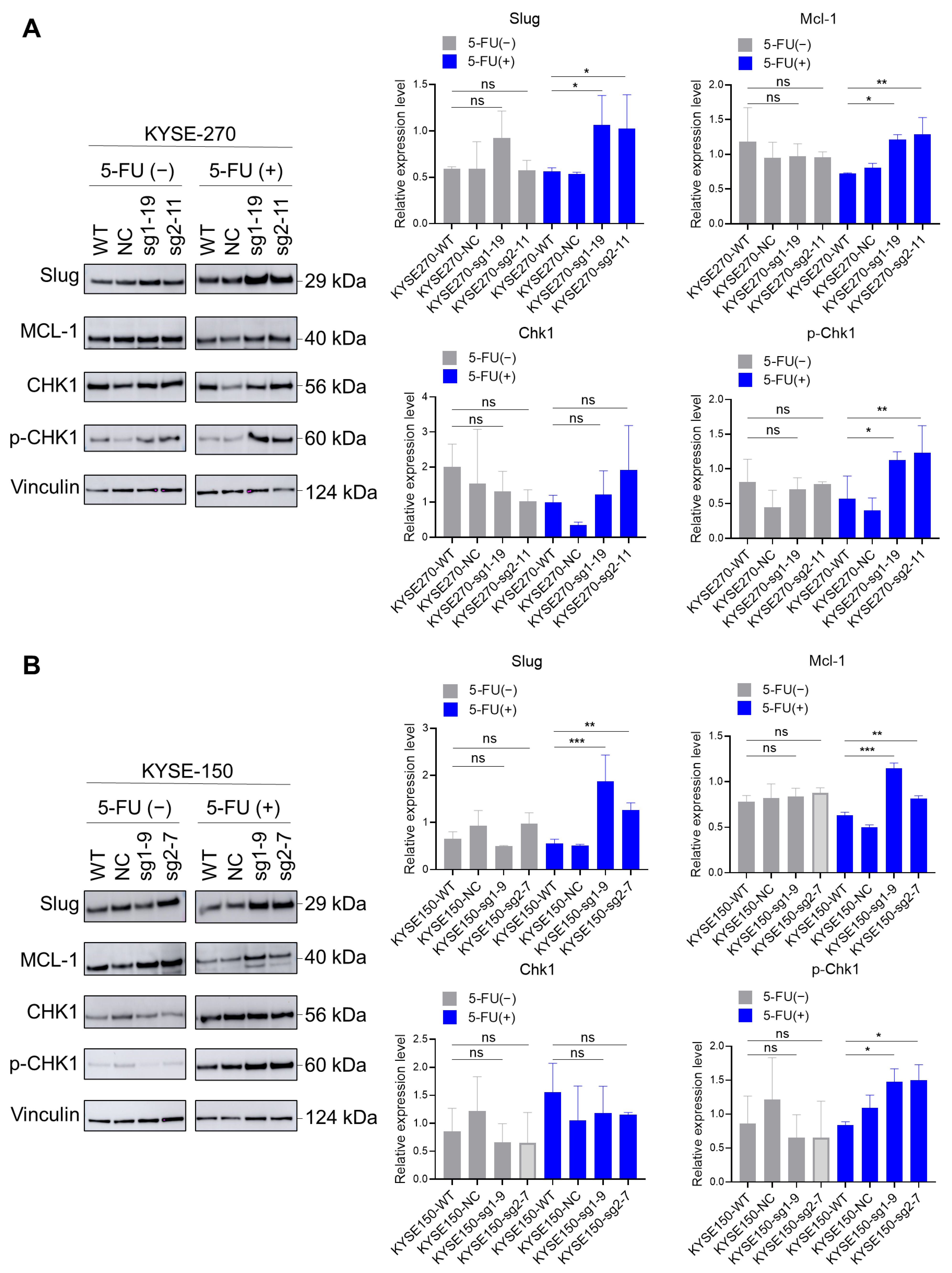
2.6. Loss of PFKFB3 Indirectly Regulates Phosphorylation of Checkpoint Kinase (Chk1) by Increasing the Expression of Slug and Mcl-1, Thereby Inhibiting 5-FU-Induced Apoptosis in KYSE-270 and KYSE-150 Cells
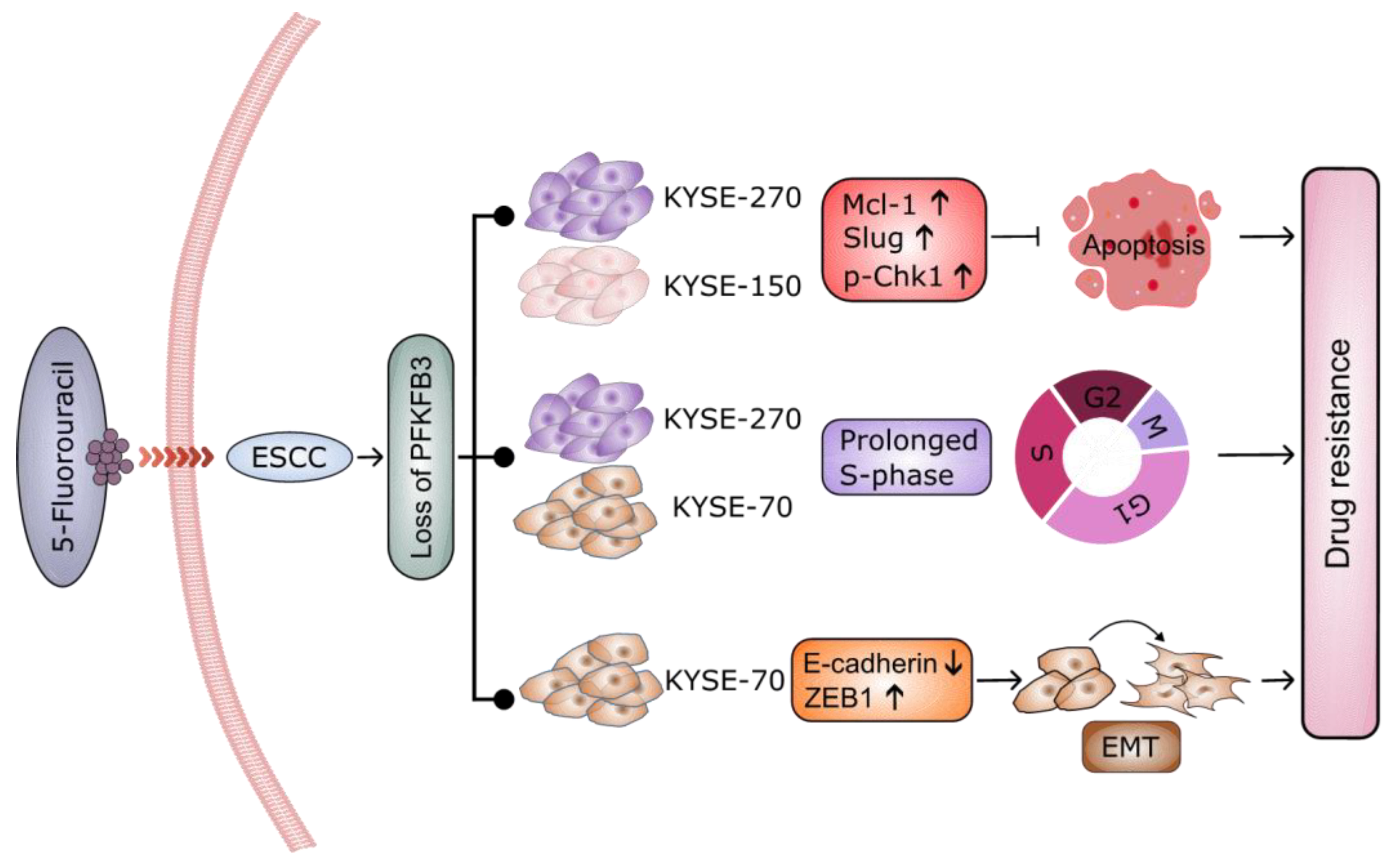
3. Discussion
4. Conclusions
5. Methods
5.1. Cell Culture
5.2. Production of Pooled sgRNA Library Lentivirus
5.3. Lenti-Library-Puro Transduction
5.4. CRISPR/Cas9 Screening with 5-FU
5.5. Isolation of Genomic DNA and Deep Sequencing
5.6. Detection of Cell Viability
5.7. Construction of Stable Knockout Cell Lines with CRISPR/Cas9
5.8. Western Blot
5.9. Eurofins Genomics DNA Sequencing
5.10. Apoptosis Assay
5.11. Cell Cycle Analysis
5.12. Immunofluorescence Staining
5.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Obermannova, R.; Alsina, M.; Cervantes, A.; Leong, T.; Lordick, F.; Nilsson, M.; van Grieken, N.C.T.; Vogel, A.; Smyth, E.C. Oesophageal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Q.; Ma, Y.L.; Qin, Q.; Wang, P.H.; Luo, Y.; Xu, P.F.; Cui, Y. Epidemiology of esophageal cancer in 2020 and projections to 2030 and 2040. Thorac. Cancer 2023, 14, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Lohan-Codeco, M.; Barambo-Wagner, M.L.; Nasciutti, L.E.; Ribeiro Pinto, L.F.; Meireles Da Costa, N.; Palumbo, A., Jr. Molecular mechanisms associated with chemoresistance in esophageal cancer. Cell. Mol. Life Sci. 2022, 79, 116. [Google Scholar] [CrossRef] [PubMed]
- Deboever, N.; Jones, C.M.; Yamashita, K.; Ajani, J.A.; Hofstetter, W.L. Advances in diagnosis and management of cancer of the esophagus. BMJ 2024, 385, e074962. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Abak, A.; Tondro Anamag, F.; Shoorei, H.; Fattahi, F.; Javadinia, S.A.; Basiri, A.; Taheri, M. 5-Fluorouracil: A Narrative Review on the Role of Regulatory Mechanisms in Driving Resistance to This Chemotherapeutic Agent. Front. Oncol. 2021, 11, 658636. [Google Scholar] [CrossRef]
- Sun, J.M.; Shen, L.; Shah, M.A.; Enzinger, P.; Adenis, A.; Doi, T.; Kojima, T.; Metges, J.P.; Li, Z.; Kim, S.B.; et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): A randomised, placebo-controlled, phase 3 study. Lancet 2021, 398, 759–771. [Google Scholar] [CrossRef]
- Okui, J.; Nagashima, K.; Matsuda, S.; Sato, Y.; Kawakubo, H.; Takeuchi, M.; Hirata, K.; Yamamoto, S.; Nomura, M.; Tsushima, T.; et al. Investigating the synergistic effects of immunochemotherapy in esophageal squamous cell carcinoma. Esophagus 2025, 22, 188–197. [Google Scholar] [CrossRef]
- Khademi, Z.; Mahmoudi, Z.; Sukhorukov, V.N.; Jamialahmadi, T.; Sahebkar, A. CRISPR/Cas9 Technology: A Novel Approach to Obesity Research. Curr. Pharm. Des. 2024, 30, 1791–1803. [Google Scholar] [CrossRef]
- Bock, C.; Datlinger, P.; Chardon, F.; Coelho, M.A.; Dong, M.B.; Lawson, K.A.; Lu, T.; Maroc, L.; Norman, T.M.; Song, B.; et al. High-content CRISPR screening. Nat. Rev. Methods Prim. 2022, 2, 9. [Google Scholar] [CrossRef]
- Wang, B.; Xu, Y.; Wan, A.H.; Wan, G.; Wang, Q.P. Integrating genome-wide CRISPR screens and in silico drug profiling for targeted antidote development. Nat. Protoc. 2024, 19, 2739–2770. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Ngan, K.C.; Lue, N.Z.; Lee, C.; Liau, B.B. CRISPR-Suppressor Scanning for Systematic Discovery of Drug-Resistance Mutations. Curr. Protoc. 2022, 2, e614. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Rajakulendran, N.; Angers, S. Identification of Drug Resistance Mechanisms Using Genome-Wide CRISPR-Cas9 Screens. Methods Mol. Biol. 2022, 2535, 141–156. [Google Scholar] [CrossRef]
- Selvakumar, S.C.; Preethi, K.A.; Ross, K.; Tusubira, D.; Khan, M.W.A.; Mani, P.; Rao, T.N.; Sekar, D. CRISPR/Cas9 and next generation sequencing in the personalized treatment of Cancer. Mol. Cancer 2022, 21, 83. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ramsay, L.; Ahanfeshar-Adams, M.; Lajoie, M.; Schadendorf, D.; Alain, T.; Watson, I.R. Mutations in the IFNgamma-JAK-STAT Pathway Causing Resistance to Immune Checkpoint Inhibitors in Melanoma Increase Sensitivity to Oncolytic Virus Treatment. Clin. Cancer Res. 2021, 27, 3432–3442. [Google Scholar] [CrossRef]
- Li, W.; Koster, J.; Xu, H.; Chen, C.H.; Xiao, T.; Liu, J.S.; Brown, M.; Liu, X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015, 16, 281. [Google Scholar] [CrossRef]
- Cui, Y.; Cheng, X.; Chen, Q.; Song, B.; Chiu, A.; Gao, Y.; Dawson, T.; Chao, L.; Zhang, W.; Li, D.; et al. CRISP-view: A database of functional genetic screens spanning multiple phenotypes. Nucleic Acids Res. 2021, 49, D848–D854. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J.; Jang, H. Anticancer drug resistance: An update and perspective. Drug Resist. Updat. 2021, 59, 100796. [Google Scholar] [CrossRef]
- Khan, S.U.; Fatima, K.; Aisha, S.; Malik, F. Unveiling the mechanisms and challenges of cancer drug resistance. Cell Commun. Signal. 2024, 22, 109. [Google Scholar] [CrossRef]
- Levatic, J.; Salvadores, M.; Fuster-Tormo, F.; Supek, F. Mutational signatures are markers of drug sensitivity of cancer cells. Nat. Commun. 2022, 13, 2926. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Robichaux, J.P.; Le, X.; Vijayan, R.S.K.; Hicks, J.K.; Heeke, S.; Elamin, Y.Y.; Lin, H.Y.; Udagawa, H.; Skoulidis, F.; Tran, H.; et al. Structure-based classification predicts drug response in EGFR-mutant NSCLC. Nature 2021, 597, 732–737. [Google Scholar] [CrossRef]
- Humeniuk, R.; Menon, L.G.; Mishra, P.J.; Gorlick, R.; Sowers, R.; Rode, W.; Pizzorno, G.; Cheng, Y.C.; Kemeny, N.; Bertino, J.R.; et al. Decreased levels of UMP kinase as a mechanism of fluoropyrimidine resistance. Mol. Cancer Ther. 2009, 8, 1037–1044. [Google Scholar] [CrossRef]
- Niu, Y.; Fan, X.; Wang, Y.; Lin, J.; Hua, L.; Li, X.; Qian, R.; Lu, C. Genome-wide CRISPR Screening Reveals Pyrimidine Metabolic Reprogramming in 5-FU Chronochemotherapy of Colorectal Cancer. Front. Oncol. 2022, 12, 949715. [Google Scholar] [CrossRef]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in cancer. Signal Transduct. Target. Ther. 2017, 2, 17044. [Google Scholar] [CrossRef]
- Kotowski, K.; Rosik, J.; Machaj, F.; Supplitt, S.; Wiczew, D.; Jablonska, K.; Wiechec, E.; Ghavami, S.; Dziegiel, P. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers 2021, 13, 909. [Google Scholar] [CrossRef]
- Deng, X.; Li, D.; Ke, X.; Wang, Q.; Yan, S.; Xue, Y.; Wang, Q.; Zheng, H. Mir-488 alleviates chemoresistance and glycolysis of colorectal cancer by targeting PFKFB3. J. Clin. Lab. Anal. 2021, 35, e23578. [Google Scholar] [CrossRef]
- Jones, B.C.; Pohlmann, P.R.; Clarke, R.; Sengupta, S. Treatment against glucose-dependent cancers through metabolic PFKFB3 targeting of glycolytic flux. Cancer Metastasis Rev. 2022, 41, 447–458. [Google Scholar] [CrossRef]
- Lypova, N.; Dougherty, S.M.; Lanceta, L.; Chesney, J.; Imbert-Fernandez, Y. PFKFB3 Inhibition Impairs Erlotinib-Induced Autophagy in NSCLCs. Cells 2021, 10, 1679. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Li, Q.; Li, S.; Ai, Z.; Yuan, D. The role of PFKFB3 in maintaining colorectal cancer cell proliferation and stemness. Mol. Biol. Rep. 2022, 49, 9877–9891. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Cheng, Y.; Li, H.; He, X.; Yu, S.; Ma, J.; Li, X.; Chen, J.; Xiao, H.; Guan, H.; et al. PFKFB3 facilitates cell proliferation and migration in anaplastic thyroid carcinoma via the WNT/beta-catenin signaling pathway. Endocrine 2024, 85, 737–750. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C. Glycolysis-induced drug resistance in tumors-A response to danger signals? Neoplasia 2021, 23, 234–245. [Google Scholar] [CrossRef]
- Sethy, C.; Kundu, C.N. 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer: Implication of DNA repair inhibition. Biomed. Pharmacother. 2021, 137, 111285. [Google Scholar] [CrossRef]
- Ohashi, S.; Kikuchi, O.; Nakai, Y.; Ida, T.; Saito, T.; Kondo, Y.; Yamamoto, Y.; Mitani, Y.; Nguyen Vu, T.H.; Fukuyama, K.; et al. Synthetic Lethality with Trifluridine/Tipiracil and Checkpoint Kinase 1 Inhibitor for Esophageal Squamous Cell Carcinoma. Mol. Cancer Ther. 2020, 19, 1363–1372. [Google Scholar] [CrossRef]
- Federica, G.; Michela, C.; Giovanna, D. Targeting the DNA damage response in cancer. MedComm 2024, 5, e788. [Google Scholar] [CrossRef]
- Drew, Y.; Zenke, F.T.; Curtin, N.J. DNA damage response inhibitors in cancer therapy: Lessons from the past, current status and future implications. Nat. Rev. Drug Discov. 2025, 24, 19–39. [Google Scholar] [CrossRef]
- Okita, N.; Minato, S.; Ohmi, E.; Tanuma, S.; Higami, Y. DNA damage-induced CHK1 autophosphorylation at Ser296 is regulated by an intramolecular mechanism. FEBS Lett. 2012, 586, 3974–3979. [Google Scholar] [CrossRef]
- Tantawy, S.I.; Timofeeva, N.; Sarkar, A.; Gandhi, V. Targeting MCL-1 protein to treat cancer: Opportunities and challenges. Front. Oncol. 2023, 13, 1226289. [Google Scholar] [CrossRef]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Yin, C.; Gong, P.; Zhang, Z.; Zhao, L.; Waxman, S.; Jing, Y. Artesunate improves venetoclax plus cytarabine AML cell targeting by regulating the Noxa/Bim/Mcl-1/p-Chk1 axis. Cell Death Dis. 2022, 13, 379. [Google Scholar] [CrossRef] [PubMed]
- Long, S.; Wang, J.; Weng, F.; Pei, Z.; Zhou, S.; Sun, G.; Xiang, D. ECM1 regulates the resistance of colorectal cancer to 5-FU treatment by modulating apoptotic cell death and epithelial-mesenchymal transition induction. Front. Pharmacol. 2022, 13, 1005915. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ma, Y.; Chen, S.; Guo, S.; Hu, J.; Yue, T.; Zhang, J.; Zhu, J.; Wang, P.; Chen, G.; et al. SPARC enhances 5-FU chemosensitivity in gastric cancer by modulating epithelial-mesenchymal transition and apoptosis. Biochem. Biophys. Res. Commun. 2021, 558, 134–140. [Google Scholar] [CrossRef]
- Tiu, Y.C.; Gong, L.; Zhang, Y.; Luo, J.; Yang, Y.; Tang, Y.; Lee, W.M.; Guan, X.Y. GLIPR1 promotes proliferation, metastasis and 5-fluorouracil resistance in hepatocellular carcinoma by activating the PI3K/PDK1/ROCK1 pathway. Cancer Gene Ther. 2022, 29, 1720–1730. [Google Scholar] [CrossRef]
- Matsuura, N.; Tanaka, K.; Yamasaki, M.; Yamashita, K.; Saito, T.; Makino, T.; Yamamoto, K.; Takahashi, T.; Kurokawa, Y.; Nakajima, K.; et al. NOTCH3 limits the epithelial-mesenchymal transition and predicts a favorable clinical outcome in esophageal cancer. Cancer Med. 2021, 10, 3986–3996. [Google Scholar] [CrossRef]
- Saitoh, M. Transcriptional regulation of EMT transcription factors in cancer. Semin. Cancer Biol. 2023, 97, 21–29. [Google Scholar] [CrossRef]
- Ang, H.L.; Mohan, C.D.; Shanmugam, M.K.; Leong, H.C.; Makvandi, P.; Rangappa, K.S.; Bishayee, A.; Kumar, A.P.; Sethi, G. Mechanism of epithelial-mesenchymal transition in cancer and its regulation by natural compounds. Med. Res. Rev. 2023, 43, 1141–1200. [Google Scholar] [CrossRef]
- Kim, S.; Yao, J.; Suyama, K.; Qian, X.; Qian, B.Z.; Bandyopadhyay, S.; Loudig, O.; De Leon-Rodriguez, C.; Zhou, Z.N.; Segall, J.; et al. Slug promotes survival during metastasis through suppression of Puma-mediated apoptosis. Cancer Res. 2014, 74, 3695–3706. [Google Scholar] [CrossRef]
- Assani, G.; Zhou, Y. Effect of modulation of epithelial-mesenchymal transition regulators Snail1 and Snail2 on cancer cell radiosensitivity by targeting of the cell cycle, cell apoptosis and cell migration/invasion. Oncol. Lett. 2019, 17, 23–30. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, S.; Jiao, X.; Wang, H.; Zhang, D.; Niu, Z.; Shen, Y.; Lv, L.; Zhou, Y. Slug regulates proliferation and invasiveness of esophageal adenocarcinoma cells in vitro and in vivo. Med. Oncol. 2011, 28, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.S.; Dutta, A.; Srinivasan, G.; Yuan, Y.; Zhou, D.; Shaheen, M.; Sadideen, D.T.; Kirby, A.; Williamson, E.A.; Gupta, Y.K.; et al. TATDN2 resolution of R-loops is required for survival of BRCA1-mutant cancer cells. Nucleic Acids Res. 2023, 51, 12224–12241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pei, J.; Durham, J.; Bos, T.; Cong, Q. Computed cancer interactome explains the effects of somatic mutations in cancers. Protein Sci. 2022, 31, e4479. [Google Scholar] [CrossRef]
- Hardison, K.L.; Hawk, T.M.; Bouley, R.A.; Petreaca, R.C. KAT5 histone acetyltransferase mutations in cancer cells. microPubl. Biol. 2022. [Google Scholar] [CrossRef]
- Ren, F.; Ding, X.; Zheng, M.; Korzinkin, M.; Cai, X.; Zhu, W.; Mantsyzov, A.; Aliper, A.; Aladinskiy, V.; Cao, Z.; et al. AlphaFold accelerates artificial intelligence powered drug discovery: Efficient discovery of a novel CDK20 small molecule inhibitor. Chem. Sci. 2023, 14, 1443–1452. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KO | Cells | Mutation Number | Mutation Type | Mutation Size |
|---|---|---|---|---|
| A. | KYSE-70-sg1-8 | 1 | Insertion | 2 bp |
| B. | KYSE-70-sg2-13 | 1 | Insertion | 1 bp |
| C. | KYSE-150-sg1-9 | 2 | Deletion Deletion | 43 bp 124 bp |
| D. | KYSE-150-sg2-7 | 1 | Insertion | 1 bp |
| E. | KYSE-270-sg1-19 | 1 | Deletion | 5 bp |
| F. | KYSE-270-sg2-11 | Insertion | 1 bp | |
| 2 | Deletion | 1 bp |
| Primer | Sequence (5′-3′) |
|---|---|
| Hu_PFKFB3_sg1_forward | CCAGCGCTAAGCAGTGTAGA |
| Hu_PFKFB3_sg1_reverse | CCAGTGGTCCTGTGGGTAAC |
| Hu_PFKFB3_sg2_forward | GAGCAAGTTCGTGGAGGAGCAGA |
| Hu_PFKFB3_sg2_reverse | TCCCCGGTGGGGTAGCGGTAATA |
| P5 | ACACTCTTTCCCTACACGACGCTCTTCCGAT CTNNNNNTCTTGTGGAAAGGACGAAACACCG |
| P7 | TCTACTATTCTTTCCCCTGCACTGT |
| sgRNA | Sequence (5′-3′) |
|---|---|
| Hu_PFKFB3_sg1_forward | CACCgGACATCTCTCAAGGCAGCTA |
| Hu_PFKFB3_sg1_reverse | AAACTAGCTGCCTTGAGAGATGTCC |
| Hu_PFKFB3_sg2_forward | CACCgGGCGCTCAATGAGATCGACG |
| Hu_PFKFB3_sg2_reverse | AAACCGTCGATCTCATTGAGCGCCC |
| Hu_None-targeting control_forward | CACCgGAAATGCTATGCTTCGGTTC |
| Hu_None-targeting control_reverse | AAACGAACCGAAGCATAGCATTTCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, F.; Yang, H.; Xu, P.; Zhang, S.; Britzen-Laurent, N.; Bao, L.-L.; Grützmann, R.; Krautz, C.; Pilarsky, C. CRISPR/Cas9 Screening Highlights PFKFB3 Gene as a Major Contributor to 5-Fluorouracil Resistance in Esophageal Cancer. Cancers 2025, 17, 1637. https://doi.org/10.3390/cancers17101637
Xue F, Yang H, Xu P, Zhang S, Britzen-Laurent N, Bao L-L, Grützmann R, Krautz C, Pilarsky C. CRISPR/Cas9 Screening Highlights PFKFB3 Gene as a Major Contributor to 5-Fluorouracil Resistance in Esophageal Cancer. Cancers. 2025; 17(10):1637. https://doi.org/10.3390/cancers17101637
Chicago/Turabian StyleXue, Feng, Hai Yang, Pengyan Xu, Shuman Zhang, Nathalie Britzen-Laurent, Li-Li Bao, Robert Grützmann, Christian Krautz, and Christian Pilarsky. 2025. "CRISPR/Cas9 Screening Highlights PFKFB3 Gene as a Major Contributor to 5-Fluorouracil Resistance in Esophageal Cancer" Cancers 17, no. 10: 1637. https://doi.org/10.3390/cancers17101637
APA StyleXue, F., Yang, H., Xu, P., Zhang, S., Britzen-Laurent, N., Bao, L.-L., Grützmann, R., Krautz, C., & Pilarsky, C. (2025). CRISPR/Cas9 Screening Highlights PFKFB3 Gene as a Major Contributor to 5-Fluorouracil Resistance in Esophageal Cancer. Cancers, 17(10), 1637. https://doi.org/10.3390/cancers17101637