
The Role of the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma: Recent Advancements and Emerging Therapeutic Strategies

 , , , ,
, , , ,  and
and
Simple Summary
Abstract
1. Introduction
2. The Structure of the Microenvironment
2.1. Pancreatic Stellate Cells (PSCs)
2.2. Cancer-Associated Fibroblasts (CAFs)
How Cancer-Associated Fibroblasts Impact Tumor Behavior
2.3. Extracellular Matrix
2.4. Regulatory Lymphocytes (Treg Lymphocytes)
2.5. Myeloid-Derived Suppressor Cells (MDSCs)
2.6. Tumor-Associated Macrophages (TAMs)
2.7. Angiogenesis
3. Stromal Cell Relations and the Influence of Connective Tissue on PDAC
Epithelium–Stroma Relations: How Independent Is Stroma from Epithelium?
4. Neuronal PDAC Infiltration and Contribution to Metastasis
5. Bacterial and Fungal Influence on PDAC Behavior and Treatment Response
6. Repressing Immune Response via Microenvironmental Components
7. Clinical Trials
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| Treg | Regulatory T lymphocytes |
| CD4 | Cluster of Differentiation 4 |
| CD25 | Cluster of Differentiation 25 |
| FOXP3 | Forkhead box P3 |
| TGFβ | Transforming Growth Factor Beta |
| IL-10 | Interleukin 10 |
| IL-1 | Interleukin 1 |
| MDSC | Myeloid-Derived Suppressor Cells |
| M-MDSC | Monocyte-like Myeloid-Derived Suppressor Cells |
| PMN-MDSC | Polymorphonuclear Myeloid-Derived Suppressor Cells |
| CD8+ | Cytotoxic T lymphocyte |
| NK | Natural Killer cells |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| IL-13 | Interleukin 13 |
| HIFs | Hypoxia-Inducible Factors |
| TAMs | Tumor-Associated Macrophages |
| TNFα | Tumor Necrosis Factor Alpha |
| HIF2 | Hypoxia-Inducible Factor 2 |
| CAFs | Cancer-Associated Fibroblasts |
| myCAF | Myofibroblastic Cancer-Associated Fibroblasts |
| iCAF | inflammatory Cancer-Associated Fibroblasts |
| apCAF | Antigen-presenting Cancer-Associated Fibroblasts |
| ECM | Extracellular Matrix |
| CK-19 | Cytokeratin-19 |
| SOX9 | SRY-Box Transcription Factor 9 |
| FOLFIRINOX | Fluorouracil, Leucovorin, Irinotecan, Oxaliplatin |
| GnP | Gemcitabine and Nab-Paclitaxel |
| R-PDAC | Resectable Pancreatic Ductal Adenocarcinoma |
| BR-PDAC | Borderline Resectable Pancreatic Ductal Adenocarcinoma |
| MSI-high | Microsatellite Instability-high |
| PD-L1 | Programmed Death-Ligand 1 |
| MEK1/2 | Mitogen-Activated Protein Kinase ½ |
| CAR-T | Chimeric Antigen Receptor T-cells |
| CEA | Carcinoembryonic Antigen |
| PSCA | Prostate Stem Cell Antigen |
| MSLN | Mesothelin |
| ICIs | Immune Checkpoint Inhibitors |
| CXCL12 | C-X-C Motif Chemokine Ligand 12 |
| CCL2 | C-C motif chemokine 2 |
| NGF | Nerve Growth Factor |
| GDNF | Glial Cell-Derived Neurotrophic Factor |
| HGF | Hepatocyte Growth Factor |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| β2 | Beta-2 Adrenergic Receptor |
| TME | Tumor Microenvironment |
| OS | Overall Survival |
| DFS | Disease-Free Survival |
| PAR-2 | Protease-Activated Receptor 2 |
| ICPs | Immune Checkpoints |
| TANs | Tumor-Associated Neutrophils |
| FAP | Fibroblast Activation Protein |
| ERK | Extracellular Signal-Regulated Kinase |
| TILs | Tumor-Infiltrating Lymphocytes |
| PC | Pancreatic Cancer |
| CK-19 | Cytokeratin-19 |
| SEER | Surveillance, Epidemiology, and End Results Programme |
| PSC | Pancreatic Stellate Cells |
| PDGF | Platelet-derived growth factor |
| FGF | fibroblast growth factor |
| CTGF | Connective tissue growth factor |
| αSMA | α-smooth muscle actin |
| IGF1 | Insulin-like growth factor 1 |
| VEGF | Vascular endothelial growth factor |
| FSP1 | Fibroblasts-specific protein 1 |
| ETM | Endothelial-to-mesenchymal transition |
| SDF-1 | Stromal-derived factor-1 |
| ROS | Reactive Oxygen Species |
| NOXs | NADPH oxidases |
| ER | Endoplasmatic Reticulum |
| siRNAs | Small interfering RNAs |
| SMA+ | Alpha-smooth muscle actin |
| SHH, | Sonic Hedgehog Homolog |
| Ptch | Patched transmembrane protein |
| SMO | Smoothened transmembrane protein |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| TIMP | Matrix-metalloproteinases inhibitors |
| PDX | Patient-dervied xenografts |
| RA | Retinoid acid |
| ATRA | All-trans-retinoid-acid |
| HMGA1 | High mobility group A1 |
| ADM | Acinar-to-ductal metaplasia |
| GFRA1 | GDNF family receptor alpha 1 |
| PNI | Perineural invasion |
| L1CAM | L1 cell adhesion molecule |
| CSF1 | Colony stimulating factor 1 |
| ACh | Acetylcholine |
| AChR | Acetylcholine receptor |
| PAR-2 | Protease-activated receptor 2 |
| LTS | Long-time survivors |
| STS | Short-time survivors |
| 3-IAA | Indole-3-acetic acid |
| GP | Gammaproteobacterium |
| NSCLC | Non-small cell lung cancer |
| RCC | Renal cell cancer |
| TSAs | Tumor-specific antigens |
| GVAX | Pancreatic cancer cell line-based vaccine |
| BIRC5 | Baculoviral IAP Repeat Containing 5 |
| CCND1 | Cyclin D1 |
| CDH1 | Cadherin 1 |
| CD74 | Cluster of Differentiation 74 |
| COL6A3 | Collagen type VI alpha three chain |
| CTHRC1 | Collagen triple helix repeat containing 1 |
| IL-13RA2 | Interleukin 13 receptor subunit alpha 2 |
| LRP1 | Low-density lipoprotein receptor-related protein 1 |
| MAP1LC3A/B | Microtubule-associated protein one light chain three alpha/beta |
| MDK | Midkine |
| MYBL2 | MYB proto-oncogene like 2 |
| MYC | MYC proto-oncogene |
| PODXL | Podocalyxin-like protein |
| PTN | Pleiotrophin |
| SDC2 | Syndecan 2 |
| SDC3 | Syndecan 3 |
| SNCG | Synuclein gamma |
| TIMP2 | Tissue inhibitor of metalloproteinases 2 |
| VGF | VGF nerve growth factor |
| WASL | WASP-like actin nucleation-promoting factor |
| FAS | FAS cell surface death receptor |
| POP1 | Processing of precursor 1, ribonuclease P/MRP subunit |
| CADM4 | Cell adhesion molecule 4 |
| HPGD | 15-hydroxyprostaglandin dehydrogenase |
| CD11b | Cluster of differentiation 11b |
| VEGFR1 | Vascular endothelial growth factor receptor 1 |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| VEGFR3 | Vascular endothelial growth factor receptor 3 |
| CSF-1R | Colony-stimulating-factor receptor 1 |
| TLR3 | Toll-like receptor 3 |
| PARP1 | Poly(ADP-ribose) polymerase 1 |
| PARP2 | Poly(ADP-ribose) polymerase 2 |
| LRRC15 | Leucine-rich repeat containing 15 protein |
| IGF1R | Insulin-like growth factor 1 receptor |
| HA | Hyaluronic acid |
| LIF | Leukemia inhibitory factor |
| FOLFIRINOX | 5-Fluorouracil, Irinotecan, Oxaliplatin, Leucovorin |
References
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, D.; Wei, D. Pancreatic Acinar-to-Ductal Metaplasia and Pancreatic Cancer. Methods Mol. Biol. 2019, 1882, 299–308. [Google Scholar] [CrossRef]
- Grant, T.J.; Hua, K.; Singh, A. Molecular Pathogenesis of Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Beatty, G.L. Tumor Microenvironment in Pancreatic Cancer Pathogenesis and Therapeutic Resistance. Annu. Rev. Pathol. 2023, 18, 123–148. [Google Scholar] [CrossRef]
- National Cancer Institute. SEER Cancer Statistics; National Cancer Institute: Bethesda, MD, USA, 2025.
- Polish National Cancer Registry. Polish National Cancer Registry Statistics; Polish National Cancer Registry: Warszawa, Poland, 2025. [Google Scholar]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Qin, Q.; Yu, R.; Eriksson, J.E.; Tsai, H.-I.; Zhu, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma therapy: Challenges and opportunities. Cancer Lett. 2024, 591, 216859. [Google Scholar] [CrossRef] [PubMed]
- Kwaśniewska, D.; Fudalej, M.; Nurzyński, P.; Badowska-Kozakiewicz, A.; Czerw, A.; Cipora, E.; Sygit, K.; Bandurska, E.; Deptała, A. How A Patient with Resectable or Borderline Resectable Pancreatic Cancer should Be Treated-A Comprehensive Review. Cancers 2023, 15, 4275. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2018, 24, 1344–1354. [Google Scholar] [CrossRef]
- Veenstra, V.L.; Garcia-Garijo, A.; van Laarhoven, H.W.; Bijlsma, M.F. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers 2018, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Mareel, M. Role of tissue stroma in cancer cell invasion. J. Pathol. 2003, 200, 429–447. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7, S44–S47. [Google Scholar] [CrossRef]
- Jang, J.E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef]
- Sperb, N.; Tsesmelis, M.; Wirth, T. Crosstalk between Tumor and Stromal Cells in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 5486. [Google Scholar] [CrossRef]
- Dougan, S.K. The Pancreatic Cancer Microenvironment. Cancer J. 2017, 23, 321–325. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: Implications for pancreatic fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef]
- Jin, G.; Hong, W.; Guo, Y.; Bai, Y.; Chen, B. Molecular Mechanism of Pancreatic Stellate Cells Activation in Chronic Pancreatitis and Pancreatic Cancer. J. Cancer 2020, 11, 1505–1515. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Mardhian, D.F.; van Baarlen, J.; Östman, A.; Prakash, J. Integrin α11 in pancreatic stellate cells regulates tumor stroma interaction in pancreatic cancer. FASEB J. 2019, 33, 6609–6621. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Wagner, E.F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 2016, 30, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Mantoni, T.S.; Lunardi, S.; Al-Assar, O.; Masamune, A.; Brunner, T.B. Pancreatic stellate cells radioprotect pancreatic cancer cells through β1-integrin signaling. Cancer Res. 2011, 71, 3453–3458. [Google Scholar] [CrossRef]
- Cabrera, M.C.; Tilahun, E.; Nakles, R.; Diaz-Cruz, E.S.; Charabaty, A.; Suy, S.; Jackson, P.; Ley, L.; Slack, R.; Jha, R.; et al. Human Pancreatic Cancer-Associated Stellate Cells Remain Activated after in vivo Chemoradiation. Front. Oncol. 2014, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Mękal, D.; Sobocki, J.; Badowska-Kozakiewicz, A.; Sygit, K.; Cipora, E.; Bandurska, E.; Czerw, A.; Deptała, A. Evaluation of Nutritional Status and the Impact of Nutritional Treatment in Patients with Pancreatic Cancer. Cancers 2023, 15, 3816. [Google Scholar] [CrossRef]
- Vaish, U.; Jain, T.; Are, A.C.; Dudeja, V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. Int. J. Mol. Sci. 2021, 22, 13408. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Anderberg, C.; Pietras, K. On the origin of cancer-associated fibroblasts. Cell Cycle 2009, 8, 1461–1462. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Rimal, R.; Desai, P.; Daware, R.; Hosseinnejad, A.; Prakash, J.; Lammers, T.; Singh, S. Cancer-associated fibroblasts: Origin, function, imaging, and therapeutic targeting. Adv. Drug Deliv. Rev. 2022, 189, 114504. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Flaberg, E.; Markasz, L.; Petranyi, G.; Stuber, G.; Dicso, F.; Alchihabi, N.; Oláh, È.; Csízy, I.; Józsa, T.; Andrén, O.; et al. High-throughput live-cell imaging reveals differential inhibition of tumor cell proliferation by human fibroblasts. Int. J. Cancer 2011, 128, 2793–2802. [Google Scholar] [CrossRef]
- Mahajan, U.M.; Langhoff, E.; Goni, E.; Costello, E.; Greenhalf, W.; Halloran, C.; Ormanns, S.; Kruger, S.; Boeck, S.; Ribback, S.; et al. Immune Cell and Stromal Signature Associated With Progression-Free Survival of Patients with Resected Pancreatic Ductal Adenocarcinoma. Gastroenterology 2018, 155, 1625–1639.e1622. [Google Scholar] [CrossRef]
- Guo, J.; Zeng, H.; Chen, Y. Emerging Nano Drug Delivery Systems Targeting Cancer-Associated Fibroblasts for Improved Antitumor Effect and Tumor Drug Penetration. Mol. Pharm. 2020, 17, 1028–1048. [Google Scholar] [CrossRef]
- Sharbeen, G.; McCarroll, J.A.; Akerman, A.; Kopecky, C.; Youkhana, J.; Kokkinos, J.; Holst, J.; Boyer, C.; Erkan, M.; Goldstein, D.; et al. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma Determine Response to SLC7A11 Inhibition. Cancer Res. 2021, 81, 3461–3479. [Google Scholar] [CrossRef]
- Sato, H.; Shiiya, A.; Kimata, M.; Maebara, K.; Tamba, M.; Sakakura, Y.; Makino, N.; Sugiyama, F.; Yagami, K.; Moriguchi, T.; et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 2005, 280, 37423–37429. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jiang, J.; Lei, Y.; Zhou, S.; Wei, Y.; Huang, C. Targeting Metabolic-Redox Circuits for Cancer Therapy. Trends Biochem. Sci. 2019, 44, 401–414. [Google Scholar] [CrossRef]
- Weber, C.K.; Liptay, S.; Wirth, T.; Adler, G.; Schmid, R.M. Suppression of NF-kappaB activity by sulfasalazine is mediated by direct inhibition of IkappaB kinases alpha and beta. Gastroenterology 2000, 119, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.; Ling, V.; Low, C.; Wang, Y.Z.; Gout, P.W. Potential use of the anti-inflammatory drug, sulfasalazine, for targeted therapy of pancreatic cancer. Curr. Oncol. 2010, 17, 9–16. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, S.; Tavormina, J.; Tampe, D.; Zeisberg, M.; Wang, H.; Mahadevan, K.K.; Wu, C.J.; Sugimoto, H.; Chang, C.C.; et al. Oncogenic collagen I homotrimers from cancer cells bind to α3β1 integrin and impact tumor microbiome and immunity to promote pancreatic cancer. Cancer Cell 2022, 40, 818–834.e819. [Google Scholar] [CrossRef] [PubMed]
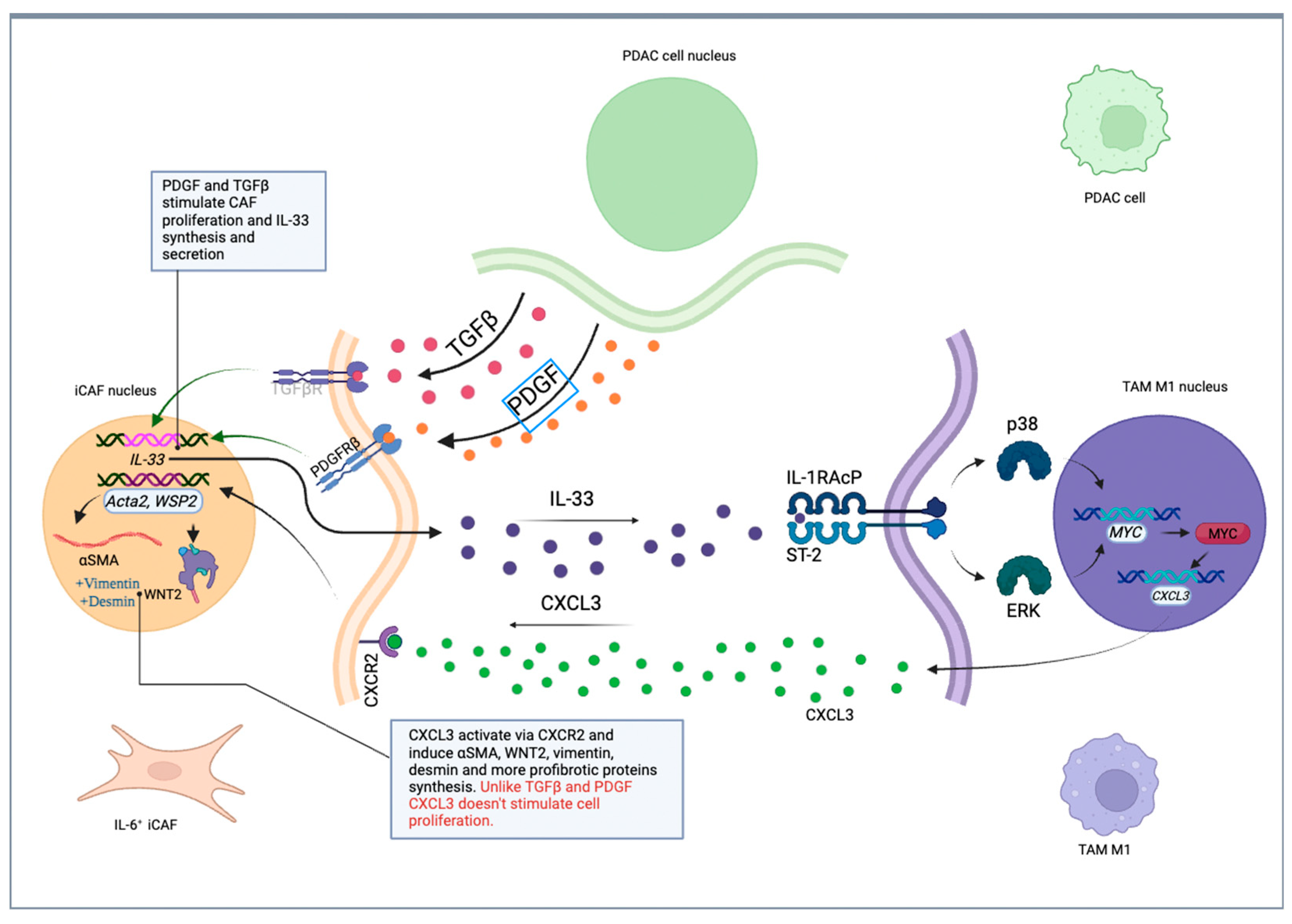
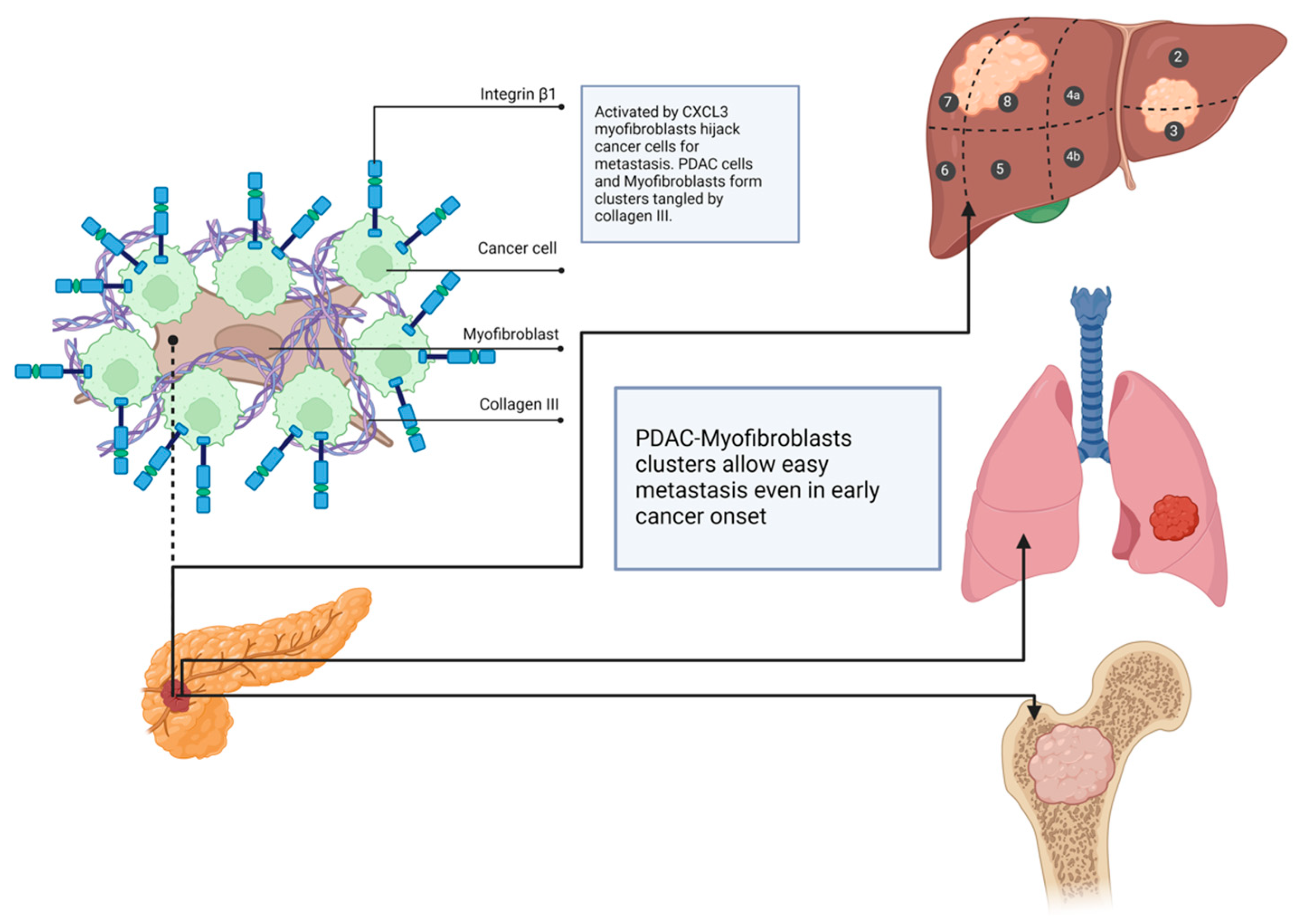
- Sun, X.; He, X.; Zhang, Y.; Hosaka, K.; Andersson, P.; Wu, J.; Wu, J.; Jing, X.; Du, Q.; Hui, X.; et al. Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut 2022, 71, 129–147. [Google Scholar] [CrossRef]
- Maurer, C.; Holmstrom, S.R.; He, J.; Laise, P.; Su, T.; Ahmed, A.; Hibshoosh, H.; Chabot, J.A.; Oberstein, P.E.; Sepulveda, A.R.; et al. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 2019, 68, 1034–1043. [Google Scholar] [CrossRef]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e546. [Google Scholar] [CrossRef]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Kayed, H.; Kleeff, J.; Osman, T.; Keleg, S.; Büchler, M.W.; Friess, H. Hedgehog signaling in the normal and diseased pancreas. Pancreas 2006, 32, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Ishii, G.; Sangai, T.; Ito, T.; Hasebe, T.; Endoh, Y.; Sasaki, H.; Harigaya, K.; Ochiai, A. In vivo and in vitro characterization of human fibroblasts recruited selectively into human cancer stroma. Int. J. Cancer 2005, 117, 212–220. [Google Scholar] [CrossRef]
- Yoo, Y.A.; Kang, M.H.; Kim, J.S.; Oh, S.C. Sonic hedgehog signaling promotes motility and invasiveness of gastric cancer cells through TGF-beta-mediated activation of the ALK5-Smad 3 pathway. Carcinogenesis 2008, 29, 480–490. [Google Scholar] [CrossRef]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.N.; Berthezène, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Ohlund, D.; Lundin, C.; Ardnor, B.; Oman, M.; Naredi, P.; Sund, M. Type IV collagen is a tumour stroma-derived biomarker for pancreas cancer. Br. J. Cancer 2009, 101, 91–97. [Google Scholar] [CrossRef]
- Knapinska, A.M.; Estrada, C.A.; Fields, G.B. The Roles of Matrix Metalloproteinases in Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2017, 148, 339–354. [Google Scholar] [CrossRef]
- Ottaviano, A.J.; Sun, L.; Ananthanarayanan, V.; Munshi, H.G. Extracellular matrix-mediated membrane-type 1 matrix metalloproteinase expression in pancreatic ductal cells is regulated by transforming growth factor-beta1. Cancer Res. 2006, 66, 7032–7040. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Ng, S.; Barrett, M.T.; Hostetter, G.; Von Hoff, D.D.; Han, H. Inhibition of ROCK1 kinase modulates both tumor cells and stromal fibroblasts in pancreatic cancer. PLoS ONE 2017, 12, e0183871. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [PubMed]
- Grage-Griebenow, E.; Schäfer, H.; Sebens, S. The fatal alliance of cancer and T cells: How pancreatic tumor cells gather immunosuppressive T cells. Oncoimmunology 2014, 3, e29382. [Google Scholar] [CrossRef] [PubMed]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, T.; Ijsselsteijn, M.; Oosting, J.; Ruano, D.; van der Ploeg, M.; Dijk, F.; Bonsing, B.; Fariña, A.; Morreau, H.; Vahrmeijer, A.; et al. A Paradoxical Role for Regulatory T Cells in the Tumor Microenvironment of Pancreatic Cancer. Cancers 2022, 14, 3862. [Google Scholar] [CrossRef]
- Karakhanova, S.; Link, J.; Heinrich, M.; Shevchenko, I.; Yang, Y.; Hassenpflug, M.; Bunge, H.; von Ahn, K.; Brecht, R.; Mathes, A.; et al. Characterization of myeloid leukocytes and soluble mediators in pancreatic cancer: Importance of myeloid-derived suppressor cells. Oncoimmunology 2015, 4, e998519. [Google Scholar] [CrossRef]
- Pergamo, M.; Miller, G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017, 24, 100–105. [Google Scholar] [CrossRef]
- Metzger, P.; Kirchleitner, S.V.; Kluge, M.; Koenig, L.M.; Hörth, C.; Rambuscheck, C.A.; Böhmer, D.; Ahlfeld, J.; Kobold, S.; Friedel, C.C.; et al. Immunostimulatory RNA leads to functional reprogramming of myeloid-derived suppressor cells in pancreatic cancer. J. Immunother. Cancer 2019, 7, 288. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rébé, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef]
- Liu, R.; Li, J.; Liu, L.; Wang, W.; Jia, J. Tumor-associated macrophages (TAMs): Constructing an immunosuppressive microenvironment bridge for pancreatic ductal adenocarcinoma (PDAC). Cancer Pathog. Ther. 2024, 2, E01–E50. [Google Scholar] [CrossRef]
- Chen, S.J.; Lian, G.D.; Li, J.J.; Zhang, Q.B.; Zeng, L.J.; Yang, K.G.; Huang, C.M.; Li, Y.Q.; Chen, Y.T.; Huang, K.H. Tumor-driven like macrophages induced by conditioned media from pancreatic ductal adenocarcinoma promote tumor metastasis via secreting IL-8. Cancer Med. 2018, 7, 5679–5690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Pan, X.; Fujiwara, K.; Jurcak, N.; Muth, S.; Zhou, J.; Xiao, Q.; Li, A.; Che, X.; Li, Z.; et al. Pancreatic cancer cells render tumor-associated macrophages metabolically reprogrammed by a GARP and DNA methylation-mediated mechanism. Signal Transduct. Target. Ther. 2021, 6, 366. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Wang, J.; Zhu, C.; Xu, J.; Chen, P.; Jiang, X.; Chen, Y.; Jiang, J.; Sun, C. Exosome-derived FGD5-AS1 promotes tumor-associated macrophage M2 polarization-mediated pancreatic cancer cell proliferation and metastasis. Cancer Lett. 2022, 548, 215751. [Google Scholar] [CrossRef]
- Kratochvill, F.; Neale, G.; Haverkamp, J.M.; Van de Velde, L.A.; Smith, A.M.; Kawauchi, D.; McEvoy, J.; Roussel, M.F.; Dyer, M.A.; Qualls, J.E.; et al. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep. 2015, 12, 1902–1914. [Google Scholar] [CrossRef]
- Xiong, C.; Zhu, Y.; Xue, M.; Jiang, Y.; Zhong, Y.; Jiang, L.; Shi, M.; Chen, H. Tumor-associated macrophages promote pancreatic ductal adenocarcinoma progression by inducing epithelial-to-mesenchymal transition. Aging 2021, 13, 3386–3404. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Di Maggio, F.; Arumugam, P.; Delvecchio, F.R.; Batista, S.; Lechertier, T.; Hodivala-Dilke, K.; Kocher, H.M. Pancreatic stellate cells regulate blood vessel density in the stroma of pancreatic ductal adenocarcinoma. Pancreatology 2016, 16, 995–1004. [Google Scholar] [CrossRef]
- Fernández-Cortés, M.; Delgado-Bellido, D.; Oliver, F.J. Vasculogenic Mimicry: Become an Endothelial Cell “But Not So Much”. Front. Oncol. 2019, 9, 803. [Google Scholar] [CrossRef]
- Benjakul, N.; Prakobphol, N.; Tangshewinsirikul, C.; Dulyaphat, W.; Svasti, J.; Charngkaew, K.; Kangsamaksin, T. Notch signaling regulates vasculogenic mimicry and promotes cell morphogenesis and the epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma. PLoS ONE 2022, 17, e0279001. [Google Scholar] [CrossRef]
- Hawighorst, T.; Velasco, P.; Streit, M.; Hong, Y.K.; Kyriakides, T.R.; Brown, L.F.; Bornstein, P.; Detmar, M. Thrombospondin-2 plays a protective role in multistep carcinogenesis: A novel host anti-tumor defense mechanism. EMBO J. 2001, 20, 2631–2640. [Google Scholar] [CrossRef] [PubMed]
- Bausch, D.; Fritz, S.; Bolm, L.; Wellner, U.F.; Fernandez-Del-Castillo, C.; Warshaw, A.L.; Thayer, S.P.; Liss, A.S. Hedgehog signaling promotes angiogenesis directly and indirectly in pancreatic cancer. Angiogenesis 2020, 23, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: A possible control point in tumor growth. Ann. Intern. Med. 1975, 82, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef]
- Nicolle, R.; Blum, Y.; Marisa, L.; Loncle, C.; Gayet, O.; Moutardier, V.; Turrini, O.; Giovannini, M.; Bian, B.; Bigonnet, M.; et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Rep. 2017, 21, 2458–2470. [Google Scholar] [CrossRef]
- Sun, L.; Zheng, M.; Gao, Y.; Brigstock, D.R.; Gao, R. Retinoic acid signaling pathway in pancreatic stellate cells: Insight into the anti-fibrotic effect and mechanism. Eur. J. Pharmacol. 2024, 967, 176374. [Google Scholar] [CrossRef]
- Guan, J.; Zhang, H.; Wen, Z.; Gu, Y.; Cheng, Y.; Sun, Y.; Zhang, T.; Jia, C.; Lu, Z.; Chen, J. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett. 2014, 345, 132–139. [Google Scholar] [CrossRef]
- Go, Y.H.; Choi, W.H.; Bae, W.J.; Jung, S.I.; Cho, C.H.; Lee, S.A.; Park, J.S.; Ahn, J.M.; Kim, S.W.; Lee, K.J.; et al. Modeling Pancreatic Cancer with Patient-Derived Organoids Integrating Cancer-Associated Fibroblasts. Cancers 2022, 14, 2077. [Google Scholar] [CrossRef] [PubMed]
- Chia, L.; Wang, B.; Kim, J.H.; Luo, L.Z.; Shuai, S.; Herrera, I.; Chen, S.Y.; Li, L.; Xian, L.; Huso, T.; et al. HMGA1 induces FGF19 to drive pancreatic carcinogenesis and stroma formation. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Parte, S.; Kaur, A.B.; Nimmakayala, R.K.; Ogunleye, A.O.; Chirravuri, R.; Vengoji, R.; Leon, F.; Nallasamy, P.; Rauth, S.; Alsafwani, Z.W.; et al. Cancer-Associated Fibroblast Induces Acinar-to-Ductal Cell Transdifferentiation and Pancreatic Cancer Initiation Via LAMA5/ITGA4 Axis. Gastroenterology 2024, 166, 842–858.e845. [Google Scholar] [CrossRef]
- Picard, F.S.R.; Lutz, V.; Brichkina, A.; Neuhaus, F.; Ruckenbrod, T.; Hupfer, A.; Raifer, H.; Klein, M.; Bopp, T.; Pfefferle, P.I.; et al. IL-17A-producing CD8(+) T cells promote PDAC via induction of inflammatory cancer-associated fibroblasts. Gut 2023, 72, 1510–1522. [Google Scholar] [CrossRef] [PubMed]
- Salvioli, B.; Bovara, M.; Barbara, G.; De Ponti, F.; Stanghellini, V.; Tonini, M.; Guerrini, S.; Cremon, C.; Degli Esposti, M.; Koumandou, M.; et al. Neurology and neuropathology of the pancreatic innervation. JOP 2002, 3, 26–33. [Google Scholar] [PubMed]
- Demir, I.E.; Friess, H.; Ceyhan, G.O. Neural plasticity in pancreatitis and pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 649–659. [Google Scholar] [CrossRef]
- Boilly, B.; Faulkner, S.; Jobling, P.; Hondermarck, H. Nerve Dependence: From Regeneration to Cancer. Cancer Cell 2017, 31, 342–354. [Google Scholar] [CrossRef]
- Dai, H.; Li, R.; Wheeler, T.; Ozen, M.; Ittmann, M.; Anderson, M.; Wang, Y.; Rowley, D.; Younes, M.; Ayala, G.E. Enhanced survival in perineural invasion of pancreatic cancer: An in vitro approach. Hum. Pathol. 2007, 38, 299–307. [Google Scholar] [CrossRef]
- Gil, Z.; Cavel, O.; Kelly, K.; Brader, P.; Rein, A.; Gao, S.P.; Carlson, D.L.; Shah, J.P.; Fong, Y.; Wong, R.J. Paracrine regulation of pancreatic cancer cell invasion by peripheral nerves. J. Natl. Cancer Inst. 2010, 102, 107–118. [Google Scholar] [CrossRef]
- Liebl, F.; Demir, I.E.; Mayer, K.; Schuster, T.; D’Haese, J.G.; Becker, K.; Langer, R.; Bergmann, F.; Wang, K.; Rosenberg, R.; et al. The impact of neural invasion severity in gastrointestinal malignancies: A clinicopathological study. Ann. Surg. 2014, 260, 900–907; discussion 907–908. [Google Scholar] [CrossRef]
- Li, J.; Kang, R.; Tang, D. Cellular and molecular mechanisms of perineural invasion of pancreatic ductal adenocarcinoma. Cancer Commun. 2021, 41, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Ceyhan, G.O.; Demir, I.E.; Altintas, B.; Rauch, U.; Thiel, G.; Müller, M.W.; Giese, N.A.; Friess, H.; Schäfer, K.H. Neural invasion in pancreatic cancer: A mutual tropism between neurons and cancer cells. Biochem. Biophys. Res. Commun. 2008, 374, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, K.; Rao, S.; Friess, H.; Weiss, J.; Büchler, M.W.; Korc, M. Reverse transcription-PCR analysis of laser-captured cells points to potential paracrine and autocrine actions of neurotrophins in pancreatic cancer. Clin. Cancer Res. 2003, 9, 5127–5136. [Google Scholar] [PubMed]
- Banh, R.S.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.W.; Walters, B.; Kuljanin, M.; Gikandi, A.; Wang, H.; Mancias, J.D.; Schneider, R.J.; et al. Neurons Release Serine to Support mRNA Translation in Pancreatic Cancer. Cell 2020, 183, 1202–1218.e1225. [Google Scholar] [CrossRef]
- Saloman, J.L.; Singhi, A.D.; Hartman, D.J.; Normolle, D.P.; Albers, K.M.; Davis, B.M. Systemic Depletion of Nerve Growth Factor Inhibits Disease Progression in a Genetically Engineered Model of Pancreatic Ductal Adenocarcinoma. Pancreas 2018, 47, 856–863. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Zeng, Q.; Cheng, Y.; Zhu, Q.; Yu, Z.; Wu, X.; Huang, K.; Zhou, M.; Han, S.; Zhang, Q. The relationship between overexpression of glial cell-derived neurotrophic factor and its RET receptor with progression and prognosis of human pancreatic cancer. J. Int. Med. Res. 2008, 36, 656–664. [Google Scholar] [CrossRef]
- Lian, E.Y.; Hyndman, B.D.; Moodley, S.; Maritan, S.M.; Mulligan, L.M. RET isoforms contribute differentially to invasive processes in pancreatic ductal adenocarcinoma. Oncogene 2020, 39, 6493–6510. [Google Scholar] [CrossRef]
- Marchesi, F.; Piemonti, L.; Fedele, G.; Destro, A.; Roncalli, M.; Albarello, L.; Doglioni, C.; Anselmo, A.; Doni, A.; Bianchi, P.; et al. The chemokine receptor CX3CR1 is involved in the neural tropism and malignant behavior of pancreatic ductal adenocarcinoma. Cancer Res. 2008, 68, 9060–9069. [Google Scholar] [CrossRef]
- Nan, L.; Qin, T.; Xiao, Y.; Qian, W.; Li, J.; Wang, Z.; Ma, J.; Ma, Q.; Wu, Z. Pancreatic Stellate Cells Facilitate Perineural Invasion of Pancreatic Cancer via HGF/c-Met Pathway. Cell Transplant. 2019, 28, 1289–1298. [Google Scholar] [CrossRef]
- Guo, K.; Ma, Q.; Li, J.; Wang, Z.; Shan, T.; Li, W.; Xu, Q.; Xie, K. Interaction of the sympathetic nerve with pancreatic cancer cells promotes perineural invasion through the activation of STAT3 signaling. Mol. Cancer Ther. 2013, 12, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell 2018, 33, 75–90.e77. [Google Scholar] [CrossRef] [PubMed]
- Swanson, B.J.; McDermott, K.M.; Singh, P.K.; Eggers, J.P.; Crocker, P.R.; Hollingsworth, M.A. MUC1 is a counter-receptor for myelin-associated glycoprotein (Siglec-4a) and their interaction contributes to adhesion in pancreatic cancer perineural invasion. Cancer Res. 2007, 67, 10222–10229. [Google Scholar] [CrossRef] [PubMed]
- Deborde, S.; Omelchenko, T.; Lyubchik, A.; Zhou, Y.; He, S.; McNamara, W.F. Schwann Cells Promote Cancer Cell Invasion. Cancer Discov. 2016, 6, 473. [Google Scholar] [CrossRef]
- Deborde, S.; Omelchenko, T.; Lyubchik, A.; Zhou, Y.; He, S.; McNamara, W.F.; Chernichenko, N.; Lee, S.Y.; Barajas, F.; Chen, C.H.; et al. Schwann cells induce cancer cell dispersion and invasion. J. Clin. Investig. 2016, 126, 1538–1554. [Google Scholar] [CrossRef]
- Na’ara, S.; Amit, M.; Gil, Z. L1CAM induces perineural invasion of pancreas cancer cells by upregulation of metalloproteinase expression. Oncogene 2019, 38, 596–608. [Google Scholar] [CrossRef]
- Roger, E.; Martel, S.; Bertrand-Chapel, A.; Depollier, A.; Chuvin, N.; Pommier, R.M.; Yacoub, K.; Caligaris, C.; Cardot-Ruffino, V.; Chauvet, V.; et al. Schwann cells support oncogenic potential of pancreatic cancer cells through TGFβ signaling. Cell Death Dis. 2019, 10, 886. [Google Scholar] [CrossRef]
- Bakst, R.L.; Xiong, H.; Chen, C.-H.; Deborde, S.; Lyubchik, A.; Zhou, Y.; He, S.; McNamara, W.; Lee, S.-Y.; Olson, O.C.; et al. Inflammatory Monocytes Promote Perineural Invasion via CCL2-Mediated Recruitment and Cathepsin B Expression. Cancer Res. 2017, 77, 6400–6414. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef]
- Azad, A.; Yin Lim, S.; D’Costa, Z.; Jones, K.; Diana, A.; Sansom, O.J.; Kruger, P.; Liu, S.; McKenna, W.G.; Dushek, O.; et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol. Med. 2017, 9, 167–180. [Google Scholar] [CrossRef]
- Furuhashi, S.; Sakaguchi, T.; Murakami, T.; Fukushima, M.; Morita, Y.; Ikegami, K.; Kikuchi, H.; Setou, M.; Takeuchi, H. Tenascin C in the Tumor-Nerve Microenvironment Enhances Perineural Invasion and Correlates With Locoregional Recurrence in Pancreatic Ductal Adenocarcinoma. Pancreas 2020, 49, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Z.; Ma, Q.; Xu, Q.; Liu, H.; Duan, W.; Lei, J.; Ma, J.; Wang, X.; Lv, S.; et al. Sonic hedgehog paracrine signaling activates stromal cells to promote perineural invasion in pancreatic cancer. Clin. Cancer Res. 2014, 20, 4326–4338. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Guo, Y.; Liang, J.; Chen, S.; Peng, P.; Zhang, Q.; Su, H.; Chen, Y.; Huang, K. Perineural Invasion and TAMs in Pancreatic Ductal Adenocarcinomas: Review of the Original Pathology Reports Using Immunohistochemical Enhancement and Relationships with Clinicopathological Features. J. Cancer 2014, 5, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Cavel, O.; Shomron, O.; Shabtay, A.; Vital, J.; Trejo-Leider, L.; Weizman, N.; Krelin, Y.; Fong, Y.; Wong, R.J.; Amit, M.; et al. Endoneurial macrophages induce perineural invasion of pancreatic cancer cells by secretion of GDNF and activation of RET tyrosine kinase receptor. Cancer Res. 2012, 72, 5733–5743. [Google Scholar] [CrossRef]
- Yang, M.W.; Tao, L.Y.; Jiang, Y.S.; Yang, J.Y.; Huo, Y.M.; Liu, D.J.; Li, J.; Fu, X.L.; He, R.; Lin, C.; et al. Perineural Invasion Reprograms the Immune Microenvironment through Cholinergic Signaling in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2020, 80, 1991–2003. [Google Scholar] [CrossRef]
- Partecke, L.I.; Käding, A.; Trung, D.N.; Diedrich, S.; Sendler, M.; Weiss, F.; Kühn, J.P.; Mayerle, J.; Beyer, K.; von Bernstorff, W.; et al. Subdiaphragmatic vagotomy promotes tumor growth and reduces survival via TNFα in a murine pancreatic cancer model. Oncotarget 2017, 8, 22501–22512. [Google Scholar] [CrossRef]
- Koide, N.; Yamada, T.; Shibata, R.; Mori, T.; Fukuma, M.; Yamazaki, K.; Aiura, K.; Shimazu, M.; Hirohashi, S.; Nimura, Y.; et al. Establishment of perineural invasion models and analysis of gene expression revealed an invariant chain (CD74) as a possible molecule involved in perineural invasion in pancreatic cancer. Clin. Cancer Res. 2006, 12, 2419–2426. [Google Scholar] [CrossRef]
- Torer, N.; Kayaselcuk, F.; Nursal, T.Z.; Yildirim, S.; Tarim, A.; Nòyan, T.; Karakayali, H. Adhesion molecules as prognostic markers in pancreatic adenocarcinoma. J. Surg. Oncol. 2007, 96, 419–423. [Google Scholar] [CrossRef]
- Lebe, B.; Sağol, O.; Ulukuş, C.; Coker, A.; Karademir, S.; Astarcioglu, H.; Küpelioğlu, A.; Astarcioğlu, I.; Obuz, F. The importance of cyclin D1 and Ki67 expression on the biological behavior of pancreatic adenocarcinomas. Pathol. Res. Pract. 2004, 200, 389–396. [Google Scholar] [CrossRef]
- Bianchini, M.; Giambelluca, M.; Scavuzzo, M.C.; Di Franco, G.; Guadagni, S.; Palmeri, M.; Furbetta, N.; Gianardi, D.; Costa, A.; Gentiluomo, M.; et al. In Pancreatic Adenocarcinoma Alpha-Synuclein Increases and Marks Peri-Neural Infiltration. Int. J. Mol. Sci. 2022, 23, 3775. [Google Scholar] [CrossRef]
- Jurcak, N.R.; Rucki, A.A.; Muth, S.; Thompson, E.; Sharma, R.; Ding, D.; Zhu, Q.; Eshleman, J.R.; Anders, R.A.; Jaffee, E.M.; et al. Axon Guidance Molecules Promote Perineural Invasion and Metastasis of Orthotopic Pancreatic Tumors in Mice. Gastroenterology 2019, 157, 838–850.e836. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, D.; Hirth, M.; Gandla, J.; Kuner, R. A mouse model for pain and neuroplastic changes associated with pancreatic ductal adenocarcinoma. Pain 2017, 158, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Demir, I.E.; Schorn, S.; Schremmer-Danninger, E.; Wang, K.; Kehl, T.; Giese, N.A.; Algül, H.; Friess, H.; Ceyhan, G.O. Perineural Mast Cells Are Specifically Enriched in Pancreatic Neuritis and Neuropathic Pain in Pancreatic Cancer and Chronic Pancreatitis. PLoS ONE 2013, 8, e60529. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, X.; Song, Q.; Liu, L.; Forbes, E.; Tian, W.; Zhang, Z.; Kang, Y.; Wang, H.; Fleming, J.B.; et al. IGFBP2 promotes tumor progression by inducing alternative polarization of macrophages in pancreatic ductal adenocarcinoma through the STAT3 pathway. Cancer Lett. 2021, 500, 132–146. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e326. [Google Scholar] [CrossRef]
- Sperti, C.; Pasquali, C.; Piccoli, A.; Pedrazzoli, S. Survival after resection for ductal adenocarcinoma of the pancreas. Br. J. Surg. 1996, 83, 625–631. [Google Scholar] [CrossRef]
- Chen, J.W.; Bhandari, M.; Astill, D.S.; Wilson, T.G.; Kow, L.; Brooke-Smith, M.; Toouli, J.; Padbury, R.T. Predicting patient survival after pancreaticoduodenectomy for malignancy: Histopathological criteria based on perineural infiltration and lymphovascular invasion. HPB 2010, 12, 101–108. [Google Scholar] [CrossRef]
- Crippa, S.; Pergolini, I.; Javed, A.A.; Honselmann, K.C.; Weiss, M.J.; Di Salvo, F.; Burkhart, R.; Zamboni, G.; Belfiori, G.; Ferrone, C.R.; et al. Implications of Perineural Invasion on Disease Recurrence and Survival After Pancreatectomy for Pancreatic Head Ductal Adenocarcinoma. Ann. Surg. 2022, 276, 378–385. [Google Scholar] [CrossRef]
- Li, B.; Wang, Y.; Jiang, H.; Li, B.; Shi, X.; Gao, S.; Ni, C.; Zhang, Z.; Guo, S.; Xu, J.; et al. Pros and Cons: High Proportion of Stromal Component Indicates Better Prognosis in Patients With Pancreatic Ductal Adenocarcinoma-A Research Based on the Evaluation of Whole-Mount Histological Slides. Front. Oncol. 2020, 10, 1472. [Google Scholar] [CrossRef]
- Kondo, N.; Murakami, Y.; Uemura, K.; Hashimoto, Y.; Nakagawa, N.; Sasaki, H.; Sueda, T. An Increased Number of Perineural Invasions Is Independently Associated With Poor Survival of Patients With Resectable Pancreatic Ductal Adenocarcinoma. Pancreas 2015, 44, 1345–1351. [Google Scholar] [CrossRef]
- Zhou, W.; Jin, W.; Wang, D.; Lu, C.; Xu, X.; Zhang, R.; Kuang, T.; Zhou, Y.; Wu, W.; Jin, D.; et al. Laparoscopic versus open pancreaticoduodenectomy for pancreatic ductal adenocarcinoma: A propensity score matching analysis. Cancer Commun. 2019, 39, 66. [Google Scholar] [CrossRef] [PubMed]
- McCracken, M.N.; Cha, A.C.; Weissman, I.L. Molecular Pathways: Activating T Cells after Cancer Cell Phagocytosis from Blockade of CD47 “Don’t Eat Me” Signals. Clin. Cancer Res. 2015, 21, 3597–3601. [Google Scholar] [CrossRef] [PubMed]
- Michaels, A.D.; Newhook, T.E.; Adair, S.J.; Morioka, S.; Goudreau, B.J.; Nagdas, S.; Mullen, M.G.; Persily, J.B.; Bullock, T.N.J.; Slingluff, C.L., Jr.; et al. CD47 Blockade as an Adjuvant Immunotherapy for Resectable Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Kadera, B.E.; Sunjaya, D.B.; Isacoff, W.H.; Li, L.; Hines, O.J.; Tomlinson, J.S.; Dawson, D.W.; Rochefort, M.M.; Donald, G.W.; Clerkin, B.M.; et al. Locally Advanced Pancreatic Cancer: Association Between Prolonged Preoperative Treatment and Lymph-Node Negativity and Overall Survival. JAMA Surg. 2014, 149, 145–153. [Google Scholar] [CrossRef]
- Zhu, J.; Miao, X.-R.; Tao, K.-M.; Zhu, H.; Liu, Z.-Y.; Yu, D.-W.; Chen, Q.-B.; Qiu, H.-B.; Lu, Z.-J. Trypsin-protease activated receptor-2 signaling contributes to pancreatic cancer pain. Oncotarget 2017, 8, 61810. [Google Scholar] [CrossRef]
- Karamitopoulou, E.; Shoni, M.; Theoharides, T.C. Increased number of non-degranulated mast cells in pancreatic ductal adenocarcinoma but not in acute pancreatitis. Int. J. Immunopathol. Pharmacol. 2014, 27, 213–220. [Google Scholar] [CrossRef]
- Demir, I.E.; Kujundzic, K.; Pfitzinger, P.L.; Saricaoglu, Ö.C.; Teller, S.; Kehl, T.; Reyes, C.M.; Ertl, L.S.; Miao, Z.; Schall, T.J.; et al. Early pancreatic cancer lesions suppress pain through CXCL12-mediated chemoattraction of Schwann cells. Proc. Natl. Acad. Sci. USA 2017, 114, E85–E94. [Google Scholar] [CrossRef]
- Demir, I.E.; Tieftrunk, E.; Schorn, S.; Saricaoglu, Ö.C.; Pfitzinger, P.L.; Teller, S.; Wang, K.; Waldbaur, C.; Kurkowski, M.U.; Wörmann, S.M.; et al. Activated Schwann cells in pancreatic cancer are linked to analgesia via suppression of spinal astroglia and microglia. Gut 2016, 65, 1001–1014. [Google Scholar] [CrossRef]
- Chatterjee, D.; Katz, M.H.; Rashid, A.; Wang, H.; Iuga, A.C.; Varadhachary, G.R.; Wolff, R.A.; Lee, J.E.; Pisters, P.W.; Crane, C.H.; et al. Perineural and intraneural invasion in posttherapy pancreaticoduodenectomy specimens predicts poor prognosis in patients with pancreatic ductal adenocarcinoma. Am. J. Surg. Pathol. 2012, 36, 409–417. [Google Scholar] [CrossRef]
- Bakst, R.L.; Lee, N.; He, S.; Chernichenko, N.; Chen, C.H.; Linkov, G.; Le, H.C.; Koutcher, J.; Vakiani, E.; Wong, R.J. Radiation impairs perineural invasion by modulating the nerve microenvironment. PLoS ONE 2012, 7, e39925. [Google Scholar] [CrossRef]
- Lu, Z.; Dong, T.H.; Si, P.R.; Shen, W.; Bi, Y.L.; Min, M.; Chen, X.; Liu, Y. Continuous Low-dose-rate Irradiation of Iodine-125 Seeds Inhibiting Perineural Invasion in Pancreatic Cancer. Chin. Med. J. 2016, 129, 2460–2468. [Google Scholar] [CrossRef] [PubMed]
- Koujima, T.; Tazawa, H.; Ieda, T.; Araki, H.; Fushimi, T.; Shoji, R.; Kuroda, S.; Kikuchi, S.; Yoshida, R.; Umeda, Y.; et al. Oncolytic Virus-Mediated Targeting of the ERK Signaling Pathway Inhibits Invasive Propensity in Human Pancreatic Cancer. Mol. Ther. Oncolytics 2020, 17, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, A.; et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 2019, 178, 795–806.e712. [Google Scholar] [CrossRef] [PubMed]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Kim, Y.I.; Park, J.E.; Brand, D.D.; Fitzpatrick, E.A.; Yi, A.K. Protein kinase D1 is essential for the proinflammatory response induced by hypersensitivity pneumonitis-causing thermophilic actinomycetes Saccharopolyspora rectivirgula. J. Immunol. 2010, 184, 3145–3156. [Google Scholar] [CrossRef]
- Hezaveh, K.; Shinde, R.S.; Klötgen, A.; Halaby, M.J.; Lamorte, S.; Ciudad, M.T.; Quevedo, R.; Neufeld, L.; Liu, Z.Q.; Jin, R.; et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity 2022, 55, 324–340.e328. [Google Scholar] [CrossRef]
- Beatty, G.L.; Delman, D.; Yu, J.; Liu, M.; Li, J.H.; Zhang, L.; Lee, J.W.; Chang, R.B.; Bahary, N.; Kennedy, E.P.; et al. Treatment Response in First-Line Metastatic Pancreatic Ductal Adenocarcinoma Is Stratified By a Composite Index of Tumor Proliferation and CD8 T-Cell Infiltration. Clin. Cancer Res. 2023, 29, 3514–3525. [Google Scholar] [CrossRef]
- Tintelnot, J.; Xu, Y.; Lesker, T.R.; Schönlein, M.; Konczalla, L.; Giannou, A.D.; Pelczar, P.; Kylies, D.; Puelles, V.G.; Bielecka, A.A.; et al. Microbiota-derived 3-IAA influences chemotherapy efficacy in pancreatic cancer. Nature 2023, 615, 168–174. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Pan, Y.; Lu, F.; Fei, Q.; Yu, X.; Xiong, P.; Yu, X.; Dang, Y.; Hou, Z.; Lin, W.; Lin, X.; et al. Single-cell RNA sequencing reveals compartmental remodeling of tumor-infiltrating immune cells induced by anti-CD47 targeting in pancreatic cancer. J. Hematol. Oncol. 2019, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Watanabe, T.; Oga, Y.; Matsumoto, S.; Kimura, N.; Nagamori, M.; Tanaka, H.; Shibuya, K.; Yoshioka, I.; Fujii, T. A case of MSI-high pancreatic body-tail cancer successfully treated with radical resection after pembrolizumab. Clin. J. Gastroenterol. 2025, 18, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Fudalej, M.; Kwaśniewska, D.; Nurzyński, P.; Badowska-Kozakiewicz, A.; Mękal, D.; Czerw, A.; Sygit, K.; Deptała, A. New Treatment Options in Metastatic Pancreatic Cancer. Cancers 2023, 15, 2327. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, P.M.; Rendo, M.J.; Uy, M.D.; Adams, A.M.; O’Shea, A.E.; Nelson, D.W.; Fenderson, J.L.; Cebe, K.M.; Krell, R.W.; Clifton, G.T.; et al. Near Complete Pathologic Response to PD-1 Inhibitor and Radiotherapy in a Patient with Locally Advanced Pancreatic Ductal Adenocarcinoma. Onco Targets Ther. 2021, 14, 3537–3544. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Fan, J.Q.; Wang, M.F.; Chen, H.L.; Shang, D.; Das, J.K.; Song, J. Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma. Mol. Cancer 2020, 19, 32. [Google Scholar] [CrossRef]
- Poschke, I.; Faryna, M.; Bergmann, F.; Flossdorf, M.; Lauenstein, C.; Hermes, J.; Hinz, U.; Hank, T.; Ehrenberg, R.; Volkmar, M.; et al. Identification of a tumor-reactive T-cell repertoire in the immune infiltrate of patients with resectable pancreatic ductal adenocarcinoma. Oncoimmunology 2016, 5, e1240859. [Google Scholar] [CrossRef]
- Stromnes, I.M.; Hulbert, A.; Pierce, R.H.; Greenberg, P.D.; Hingorani, S.R. T-cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2017, 5, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Tassi, E.; Gavazzi, F.; Albarello, L.; Senyukov, V.; Longhi, R.; Dellabona, P.; Doglioni, C.; Braga, M.; Di Carlo, V.; Protti, M.P. Carcinoembryonic antigen-specific but not antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients. J. Immunol. 2008, 181, 6595–6603. [Google Scholar] [CrossRef]
- Turk, J.L.; Parker, D. Effect of cyclophosphamide on immunological control mechanisms. Immunol. Rev. 1982, 65, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Madondo, M.T.; Quinn, M.; Plebanski, M. Low dose cyclophosphamide: Mechanisms of T cell modulation. Cancer Treat. Rev. 2016, 42, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Heylmann, D.; Bauer, M.; Becker, H.; van Gool, S.; Bacher, N.; Steinbrink, K.; Kaina, B. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: Implications for the immune response. PLoS ONE 2013, 8, e83384. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef]
- Timmer, F.E.F.; Geboers, B.; Nieuwenhuizen, S.; Dijkstra, M.; Schouten, E.A.C.; Puijk, R.S.; de Vries, J.J.J.; van den Tol, M.P.; Bruynzeel, A.M.E.; Streppel, M.M.; et al. Pancreatic Cancer and Immunotherapy: A Clinical Overview. Cancers 2021, 13, 4138. [Google Scholar] [CrossRef]
- Haen, S.P.; Löffler, M.W.; Rammensee, H.G.; Brossart, P. Towards new horizons: Characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020, 17, 595–610. [Google Scholar] [CrossRef]
- Zuo, C.; Baer, J.M.; Knolhoff, B.L.; Belle, J.I.; Liu, X.; Alarcon De La Lastra, A.; Fu, C.; Hogg, G.D.; Kingston, N.L.; Breden, M.A.; et al. Stromal and therapy-induced macrophage proliferation promotes PDAC progression and susceptibility to innate immunotherapy. J. Exp. Med. 2023, 220, e20212062. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Felix, K.; Gaida, M.M. Neutrophil-Derived Proteases in the Microenvironment of Pancreatic Cancer -Active Players in Tumor Progression. Int. J. Biol. Sci. 2016, 12, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef]
- Ray, U.; Pathoulas, C.L.; Thirusangu, P.; Purcell, J.W.; Kannan, N.; Shridhar, V. Exploiting LRRC15 as a Novel Therapeutic Target in Cancer. Cancer Res. 2022, 82, 1675–1681. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Williams, A.; Schreiber, J.; Hohmann, N.; Pruefer, U.; Krauss, J.; Jäger, D.; Frömming, A.; Beyer, D.; Eulberg, D.; et al. Combined inhibition of CXCL12 and PD-1 in MSS colorectal and pancreatic cancer: Modulation of the microenvironment and clinical effects. J. Immunother. Cancer 2021, 9, e002505. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Angevin, E.; Tabernero, J.; Elez, E.; Cohen, S.J.; Bahleda, R.; van Laethem, J.L.; Ottensmeier, C.; Lopez-Martin, J.A.; Clive, S.; Joly, F.; et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2192–2204. [Google Scholar] [CrossRef]
- Abdel-Wahab, R.; Varadhachary, G.R.; Bhosale, P.R.; Wang, X.; Fogelman, D.R.; Shroff, R.T.; Overman, M.J.; Wolff, R.A.; Javle, M. Randomized, phase I/II study of gemcitabine plus IGF-1R antagonist (MK-0646) versus gemcitabine plus erlotinib with and without MK-0646 for advanced pancreatic adenocarcinoma. J. Hematol. Oncol. 2018, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Kundranda, M.; Gracian, A.C.; Zafar, S.F.; Meiri, E.; Bendell, J.; Algül, H.; Rivera, F.; Ahn, E.R.; Watkins, D.; Pelzer, U.; et al. Randomized, double-blind, placebo-controlled phase II study of istiratumab (MM-141) plus nab-paclitaxel and gemcitabine versus nab-paclitaxel and gemcitabine in front-line metastatic pancreatic cancer (CARRIE). Ann. Oncol. 2020, 31, 79–87. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Azevedo, S.; Okusaka, T.; Van Laethem, J.L.; Lipton, L.R.; Riess, H.; Szczylik, C.; Moore, M.J.; Peeters, M.; Bodoky, G.; et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: The GAMMA trial. Ann. Oncol. 2015, 26, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [PubMed]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br. J. Cancer 2020, 122, 498–505. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Carr, R.M.; Duma, N.; McCleary-Wheeler, A.L.; Almada, L.L.; Marks, D.L.; Graham, R.P.; Smyrk, T.C.; Lowe, V.; Borad, M.J.; Kim, G.; et al. Targeting of the Hedgehog/GLI and mTOR pathways in advanced pancreatic cancer, a phase 1 trial of Vismodegib and Sirolimus combination. Pancreatology 2020, 20, 1115–1122. [Google Scholar] [CrossRef]
- Pijnappel, E.N.; Wassenaar, N.P.M.; Gurney-Champion, O.J.; Klaassen, R.; van der Lee, K.; Pleunis-van Empel, M.C.H.; Richel, D.J.; Legdeur, M.C.; Nederveen, A.J.; van Laarhoven, H.W.M.; et al. Phase I/II Study of LDE225 in Combination with Gemcitabine and Nab-Paclitaxel in Patients with Metastatic Pancreatic Cancer. Cancers 2021, 13, 4869. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; Saddala, M.S.; Foote, J.B.; Khaliq, A.M.; Masood, A.; Golivi, Y.; Bandi, D.S.R.; Sarvesh, S.; Reddy, S.P.; Switchenko, J.; et al. Mechanism of enhancing chemotherapy efficacy in pancreatic ductal adenocarcinoma with paricalcitol and hydroxychloroquine. Cell Rep. Med. 2025, 6, 101881. [Google Scholar] [CrossRef]
- Chung, V.; Alistar, A.; Becerra, C.; Kasi, A.; Borazanci, E.; Jameson, G.S.; Roe, D.J.; Wertheim, B.C.; Cridebring, D.; Truitt, M.; et al. Pembrolizumab ± paricalcitol in metastatic pancreatic cancer postmaximal cytoreduction. Oncologist 2025, 30, oyae323. [Google Scholar] [CrossRef]
- Grierson, P.M.; Suresh, R.; Tan, B.; Pedersen, K.S.; Amin, M.; Park, H.; Trikalinos, N.A.; Liu, J.; Boice, N.; Brown, A.; et al. A Pilot Study of Paricalcitol plus Nanoliposomal Irinotecan and 5-FU/LV in Advanced Pancreatic Cancer Patients after Progression on Gemcitabine-Based Therapy. Clin. Cancer Res. 2023, 29, 4733–4739. [Google Scholar] [CrossRef] [PubMed]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; deSouza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat. Commun. 2020, 11, 4841. [Google Scholar] [CrossRef] [PubMed]
- Kocher, H.M.; Sasieni, P.; Corrie, P.; McNamara, M.G.; Sarker, D.; Froeling, F.E.M.; Christie, A.; Gillmore, R.; Khan, K.; Propper, D. Study protocol: Multi-centre, randomised controlled clinical trial exploring stromal targeting in locally advanced pancreatic cancer; STARPAC2. BMC Cancer 2025, 25, 106. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Oh, D.Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef] [PubMed]
- Gardner, F.P.; Wainberg, Z.A.; Fountzilas, C.; Bahary, N.; Womack, M.S.; Macarulla, T.; Garrido-Laguna, I.; Peterson, P.M.; Borazanci, E.; Johnson, M.; et al. Results of a Randomized, Double-Blind, Placebo-Controlled, Phase 1b/2 Trial of Nabpaclitaxel + Gemcitabine ± Olaratumab in Treatment-Naïve Participants with Metastatic Pancreatic Cancer. Cancers 2024, 16, 1323. [Google Scholar] [CrossRef]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef]
- Mayer, P.; Jiang, Y.; Kuder, T.A.; Bergmann, F.; Khristenko, E.; Steinle, V.; Kaiser, J.; Hackert, T.; Kauczor, H.U.; Klauß, M.; et al. Diffusion Kurtosis Imaging-A Superior Approach to Assess Tumor-Stroma Ratio in Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 1656. [Google Scholar] [CrossRef]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Type of Cells | Cytokines and Chemokines |
|---|---|
| Cancer cells | TGF-β FGF PDGF IGF-1 IL-1β IL-6 |
| Acinar cells | TGF-β CTGF IL-1β TNF-α |
| PSC | TGF-β CTGF IL-10 PDGF IL-6 IGF-1 CXCL12 |
| Inflammatory cells | IL-6 IL-10 TGF-β IL-6 IL-1β TNF-α IGF-1 |
| Platelets | PDGF TGF-β |
| Expression | |||
|---|---|---|---|
| Upregulated | Downregulated | ||
| Correlation with PNI | Positive | BIRC5, CCND1, CDH1, CD74, COL6A3, CTHRC1, CXCR4, IL-13RA2, LRP1, L1CAM, MAP1LC3A/B, MDK, MYBL2, MYC, PODXL, PTN, SDC2, SDC3, SNCG, TIMP2, VGF, WASL | |
| Negative | FAS, POP1 | CADM4, HPGD | |
| Component of TME | Description | Impact on Tumor Behavior | Impact on Metastasis | Impact on Treatment Response |
|---|---|---|---|---|
| PSCs | Quiescent cells in the exocrine pancreas; upon activation, they express markers like α-SMA and desmin and produce ECM components such as collagen and fibronectin. | Promote cancer cell proliferation and migration. Inhibit apoptosis. | Contribute to ECM remodeling, aiding tumor invasion and metastasis. | Contribute to chemotherapy and radiotherapy resistance. |
| CAFs | Heterogeneous cells responsible for ECM deposition, arise from fibroblasts, mesenchymal stem cells, or PSCs, and express markers like FAP, α-SMA, and vimentin. | Promote tumor growth, neovascularization, and invasion through the secretion of growth factors. | Enhance metastasis Via ECM remodeling and secretion of pro-invasive cytokines. | Suppress anti-tumor immune responses, affecting immunotherapy efficacy. |
| Tregs | CD4+CD25+FOXP3+ cells that suppress immune responses and maintain self-tolerance. | Impair anti-tumor immune responses, promoting tumor immune evasion. | Facilitate metastasis by suppressing immune responses to circulating tumor cells. | Correlate with poorer prognosis; suppress immune checkpoint responses. |
| MDSCs | Immature myeloid cells that are activated by cytokines like GM-CSF and IL-13. | Suppress anti-tumor immunity by activating Tregs and inhibiting CD8+ T cells. | Promote immune tolerance, aiding tumor cell survival in distant sites. | Limit the effectiveness of immunotherapies by suppressing T cell function. |
| TAMs | Monocytes that differentiate into M1 or M2 macrophages; M1 promotes inflammation, but M2 is more pro-tumor. | M1 promotes inflammation, M2 promotes tissue remodeling, immune suppression, and tumor progression. | M2 phenotype enhances metastasis by facilitating ECM remodeling and immune evasion. | M2 macrophages are linked to poor prognosis, suppressing response to treatment. |
| VM | Cancer cells form vessel-like structures independent of endothelial cells and angiogenesis. | Provides an alternative tumor blood supply, facilitating nutrient and oxygen supply to the tumor. | Complicates tumor vascular architecture and enhances metastasis by forming new blood pathways. | Linked to poor prognosis and chemotherapy resistance. |
| Clinical Trial | Status | Tested Drug | Target | Additional Interventions | Short Description, Objective of the Study, Outcome, If Available |
|---|---|---|---|---|---|
| NCT06904378 | Phase 1 (Finished)/ Phase 2 (Not yet recruiting) | Ontegimod | CD11b | Nab-Paclitaxel, Gemcitabine | An open-label phase I/II clinical trial of Ontegimod with gemcitabine and nab-paclitaxel in unresectable PDAC prior to future studies incorporating anti-PD1 checkpoint immunotherapy. Results are not available yet. |
| NCT06825546 | Phase 2 (Recruiting) | Icaritin | TNF-α, IL-6, PD-L1 | Nab-Paclitaxel, Gemcitabine | Regulating the tumor immune microenvironment by reducing the secretion of TNFa and IL-6 and inhibiting PD-L1 expression through decreasing MDSC proportion. Results are not available yet. |
| NCT06639724 | Phase 1 (Recruiting) | Fostamatinib | Syk kinase | Nab-Paclitaxel, Gemcitabine | Fostamatinib is a Syk kinase inhibitor currently FDA-approved for chronic idiopathic thrombocytopenia purpura, but it has not been studied in PDAC. The investigators hypothesize that Syk inhibition reprograms macrophages to an immunostimulatory phenotype in the TME. Thus, Syk inhibition with fostamatinib in combination with chemotherapy could improve outcomes for patients with PDAC while having a favorable safety profile. Results are not available yet. |
| NCT06492915 | Phase 2 (Recruiting) | Chiauranib | VEGFR2, VEGFR1, VEGFR3, PDGFRa and c-Kit, Aurora B kinase, CSF-1R | Nab-Paclitaxel, Gemcitabine | Chiauranib, which simultaneously targets VEGFR/Aurora B/CSF-1R, several key kinases involved in tumor angiogenesis, tumor cell mitosis, and chronic inflammatory microenvironment. Results are not available yet. |
| NCT06145074 | Phase 2 (Recruiting) | Propanolol | β-adrenergic receptors | Assessment of density and subtypes of tumor-infiltrating lymphocytes, desmoplasia, adrenergic receptor expression, and spatial distribution of immune cells in the TME. Results are not available yet. | |
| NCT05927142 | Phase 1 (Recruiting), Phase 2 (Recruiting) | Rintatolimod | TLR3 | Durvalumab | Immunotherapy effectiveness is improved by the agonist effect of Rintatolimod, which enhances dendritic cell maturation. Results are not available yet. |
| NCT05546853 | Phase 1 (Active, not recruiting) | NP137 | Netrin-1 | FOLFIRINOX | Inhibiting Netrin-1 impedes EMT, thereby reducing tumor progression and metastasis. Results are not available yet. |
| NCT04493060 | Phase 2 (Active, not recruiting) | Niraparib | PARP1, PARP2 | Dostarlimab | This phase II trial studies how well niraparib and dostarlimab work in treating patients with germline or somatic BRCA1/2 and PALB2 mutated pancreatic cancer that has spread to other places in the body (metastatic). Results are not available yet. |
| NCT03727880 | Phase 2 (Active, not recruiting) | Defacitinib | FAK kinase | Pembrolizumab | Evaluating if reprograming the TME by targeting FAK following chemotherapy can potentiate anti-PD-1 antibody. Results are not available yet. |
| NCT02651727 | Phase 1 (Terminated) | Defacitinib, | FAK kinase (VS-4718) | Nab-Paclitaxel, Gemcitabine | The company’s decision to de-prioritize 4718 development. |
| NCT03085914 | Phase 1 (Completed), Phase 2 (Completed) | Epacadostat, | Indoleamine 2,3-dioxygenase 1 | Nab-Paclitaxel, Gemcitabine, FOLFIRINOX, Pembrolizumab, Pemetrexed, Cyclophosphamide, Carboplatin, Cisplatin, | Additional cohorts (i.e., the mandatory biopsy cohorts) were designed to evaluate changes in the TME in participants with any advanced or metastatic solid tumor who had progressed on previous therapy with a PD-1 or a PD-L1 inhibitor. Results are not available yet. |
| NCT02600949 | Phase 1 (Recruiting) | Synthetic Tumoor-Associated Peptide Vaccine Therapy, | Personalized peptide vaccine targeting antigenes of PDAC tumor | Imiquimod, Pembrolizumab, Sotigalimab | A personalized peptide vaccine is developed from a patient’s tumor cells and blood to be used as a biological therapy. Biological therapies, such as personalized peptide vaccines, may attack tumor cells and stop them from growing or kill them. Results are not available yet. |
| NCT02565758 | Phase 1 (Completed) | ABBV-085 | LRRC15 | Targeting leucine-rich repeat-containing protein 15 (LRRC15) using specific antibody-drug conjugates (ABBV-085) has the potential to improve the outcome of patients with LRRC15+ cancers of mesenchymal origin or stromal desmoplasia [188]. | |
| NCT03932565 | Phase 1 (Status Unknown) | CAR-T therapy | FAP/Nectin-4 | Targeting Nectin-4 transmembrane protein, which is highly expressed on the surface of breast cancer, bladder cancer, non-small lung cancer, and pancreatic cancer [189]. | |
| NCT03168139 | Phase 1/Phase 2 (Completed) | NOX-A12, | CXCL12 | Pembrolizumab | Olaptesed pegol (NOX-A12) targets a key chemokine in tumor TME—CXCL12, which is involved in the homeostasis of blood and immune cells. The hypothesis is that inactivation of CXCL12 by NOX-A12 makes pancreatic tumors more susceptible to immunotherapy [190]. |
| NCT02765165 | Phase 1/Phase 2 (Terminated) | USL311, | CXCR4 | Lomustine | Terminated because of business reasons not related to safety. |
| NCT03277209 | Phase 1 (Terminated) | Plerixafor, | CXCR4 | Cemiplimab, Lomustine | Terminated due to slow accrual. |
| NCT02907099 | Phase 2 (Completed) | BL-8040 (motixafortide) | CXCR4 | Blocking CXCR4 with BL-8040 may stop the growth of tumor cells by blocking some of the enzymes needed for cell growth [191]. | |
| NCT00841191 | Phase 1/Phase 2 (Completed) | Siltuximab | IL-6 | IL-6 is one of the main agents responsible for inflammation. Blocking IL-6 with siltuximab was tolerated, but without clinical activity in solid tumors, including ovarian and KRAS-mutant cancers [192]. | |
| NCT00769483 | Phase 1/Phase 2 (Completed) | MK-0646, | IGF1R | Gemcitabine, Erlotinib | MK-0646 and Ganitumab (AMG 479) are monoclonal antibodies, while Istiratumab is a bispecific antibody, with all three targeting IGF1R (with additional blockade of ErbB3 by Istiratumab), preventing its binding with IGF1 and IGF2 ligands and inhibiting downstream signaling pathways, such as Pl3K/Akt and MAPK, which are involved in promoting tumor cell proliferation and survival and resistance to apoptosis. All three trials did not show prolonged PFS or OS compared to standard chemotherapy regimens [193,194,195]. |
| NCT02399137 | Phase 2 (Completed) | MM-141 (istiratumab), | Nab-Paclitaxel, Gemcitabine | ||
| NCT01231347 | Phase 3 (Completed) | AMG 479 (ganitumab), | Gemcitabine | ||
| NCT01383538 | Phase 1 (Completed) | IPI-926 | Hedgehog | FOLFIRINOX | IPI-926 is an oral SHH pathway inhibitor. The initial response rate was high, and patients receiving IPI-926 maintenance showed further declines in CA19-9 levels even after FOLFIRINOX discontinuation. However, the trial was closed early, as a separate phase II trial of IPI-926 + gemcitabine indicated the detrimental effects of this combination [196]. |
| NCT01537107 | Phase 1 (Completed) | Vismodegib | Sirolimus | Vismodegib is an SHH pathway inhibitor. SHH signaling is predominantly active in stromal cells rather than tumor cells, leading to desmoplasia and creating a dense TME. By inhibiting this pathway, vismodegib was supposed to remodel TME and enhance chemotherapy efficacy. However, all three studies showed that adding vismodegib did not enhance efficacy compared to standard chemotherapy regimens [197,198,199]. | |
| NCT01088815 | Phase 2 (Completed) | Vismodegib | Nab-Paclitaxel, Gemcitabine | ||
| NCT01195415 | Phase 2 (Completed) | Vismodegib, | Gemcitabine hydrochloride | ||
| NCT01485744 | Phase 1 (Completed) | LDE225 | FOLFIRINOX | LDE225, in combination with gemcitabine and nab-paclitaxel, was well-tolerated in patients with metastatic PDAC and has a promising efficacy after prior treatment with FOLFIRINOX. Quantitative MRI suggested that LDE225 causes increased tumor diffusion and works particularly well in patients with poor baseline tumor perfusion [200]. | |
| NCT02358161 | Phase 1/Phase 2 (Completed) | LDE225 | Nab-Paclitaxel, Gemcitabine | ||
| NCT03472833 | Phase 3 (Terminated) | High dose vitamin D | Vitamin D metabolism | Slow recruitment and patients lost to follow-up due to the COVID-19 pandemic. | |
| NCT04524702 | Phase 2 (Active, not recruiting) | Paricalcitol | Nab-Paclitaxel, Gemcitabine, Hydroxychloroquine | In preclinical models, the combination of paricalcitol and hydroxychloroquine has been shown to remodel TME by reducing stromal activation, decreasing cancer-associated fibroblasts, and enhancing immune cell infiltration. However, the efficacy of this regimen in the clinical trial was hard to establish due to the early termination of the trial due to COVID-19 and lower-than-expected enrolment. No published data on OS or PFS outcomes from this are available [201]. | |
| NCT04617067 | Phase 2 (Completed) | Nab-Paclitaxel, Gemcitabine | Results are not available at the moment. | ||
| NCT03520790 | Phase 1 (Terminated) | Phase II was not pursued due to futility based on the NAPOLI-3 therapeutic clinical trial results. | |||
| NCT03519308 | Early Phase 1 (Terminated | Nab-Paclitaxel, Gemcitabine, Nivolumab | The accrual goal was unmet, and the drug manufacturer pulled support. | ||
| NCT03331562 | Phase 2 (Completed) | Pembrolizumab | Paricalcitol did not improve pembrolizumab’s efficacy, likely related to its short half-life of only 5–7 h [202]. | ||
| NCT03883919 | Phase 1 (Completed) | 5-Fluorouracil, Liposomal Irinotecan, Leucovorin | The study showed increased tumor vascularity, potentially enhancing treatment efficacy. However, more insight is needed [203]. | ||
| NCT03307148 | Phase 1 (Completed) | ATRA | Vitamin A metabolism | Nab-Paclitaxel, Gemcitabine | The study showed that the combination of ATRA and Nab-Paclitaxel + Gemcitabine was safe and tolerable, establishing the recommended phase 2 dose. Pharmacodynamic studies indicated stromal modulation consistent with the proposed mechanism of action [204]. |
| NCT04241276 | Phase 2 (Active, not recruiting) | No clinical outcomes such as OS or PFS are reported at this stage [205]. | |||
| NCT04390763 | Phase 2 (Terminated) | NIS793 | TGF-β | Nab-Paclitaxel, Gemcitabine, FOLFIRINOX, Spartalizumab | The study was terminated early following the NIS793 treatment halt and urgent safety measure issued in July 2023, as the continued evaluation of Standard of Care alone will not support the original purpose of this phase 2 clinical trial. |
| NCT04935359 | Phase 3 (Completed) | Nab-Paclitaxel, Gemcitabine | Results are not available at the moment. | ||
| NCT05417386 | Phase 1 (Terminated) | FOLFIRINOX | NIS793 is no longer being developed. | ||
| NCT02734160 | Phase 1 (Completed) | Galunisertib | Durvalumab, Gemcitabine | Median OS was 5.72 months, and PFS was 1.87 months. The study concluded that, while the combination was tolerable, its clinical efficacy was modest, indicating that future research might focus on earlier lines of treatment or patient selection based on predictive biomarkers for TGF-β inhibition [206]. | |
| NCT04327986 | Phase 1/Phase 2 (Terminated) | M7824 | Gemcitabine | The study closed to accrual due to the worsening risk-benefit ratio for participants receiving bintrafusp alfa (M7824). | |
| NCT03451773 | Phase 1/Phase 2 (Terminated) | The study was closed after one treatment-related death. | |||
| NCT03086369 | Phase 1/Phase 2 (Completed) | Olaratumab | PDGF-α | Nab-Paclitaxel, Gemcitabine | The primary endpoint, OS, was not met: median OS was 9.1 months in the olaratumab arm versus 10.8 months in the placebo arm. Similarly, median PFS was 5.5 months with olaratumab compared to 6.4 months with placebo [207]. |
| NCT02550327 | Early Phase 1 (Completed) | Anakinra | IL-1 | Nab-Paclitaxel, Gemcitabine, Cisplatin | Results are not available at the moment. |
| NCT04999969 | Phase 2 (Active, not recruiting) | AZD0171 | LIF | Nab-Paclitaxel, Gemcitabine, Durvalumab | Results are not available at the moment. |
| NCT02119663 | Phase 3 (Terminated) | Ruxolitinib | JAK | Capecitabine | The safety committee found no safety issues, but recommended halting the study based on a lack of efficacy in a similar trial. The sponsor terminated the trial. |
| NCT02117479 | Phase 3 (Terminated) | The study was terminated early based on the results of the planned interim analysis. | |||
| NCT03563248 | Phase 2 (Active, not recruiting) | Losartan | Collagen and HA degradation | FOLFIRINOX | Results are not available at the moment. |
| NCT01821729 | Phase 2 (Unknown status) | In this single-arm phase 2 trial of 49 patients, the R0 resection rate was 61% among all eligible participants, with a PFS of 17.5 months and OS of 31.4 months, which suggests that this total neoadjuvant approach may offer improved respectability and survival benefits in this patient population [208]. | |||
| NCT02715804 | Phase 3 (Terminated) | PEGPH20, nab-paclitaxel, gemcitabine | Hyaluronan acid | Sponsor decision. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glapiński, F.; Zając, W.; Fudalej, M.; Deptała, A.; Czerw, A.; Sygit, K.; Kozłowski, R.; Badowska-Kozakiewicz, A. The Role of the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma: Recent Advancements and Emerging Therapeutic Strategies. Cancers 2025, 17, 1599. https://doi.org/10.3390/cancers17101599
Glapiński F, Zając W, Fudalej M, Deptała A, Czerw A, Sygit K, Kozłowski R, Badowska-Kozakiewicz A. The Role of the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma: Recent Advancements and Emerging Therapeutic Strategies. Cancers. 2025; 17(10):1599. https://doi.org/10.3390/cancers17101599
Chicago/Turabian StyleGlapiński, Franciszek, Weronika Zając, Marta Fudalej, Andrzej Deptała, Aleksandra Czerw, Katarzyna Sygit, Remigiusz Kozłowski, and Anna Badowska-Kozakiewicz. 2025. "The Role of the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma: Recent Advancements and Emerging Therapeutic Strategies" Cancers 17, no. 10: 1599. https://doi.org/10.3390/cancers17101599
APA StyleGlapiński, F., Zając, W., Fudalej, M., Deptała, A., Czerw, A., Sygit, K., Kozłowski, R., & Badowska-Kozakiewicz, A. (2025). The Role of the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma: Recent Advancements and Emerging Therapeutic Strategies. Cancers, 17(10), 1599. https://doi.org/10.3390/cancers17101599