
The Causes and Consequences of DNA Damage and Chromosomal Instability Induced by Human Papillomavirus
 , and
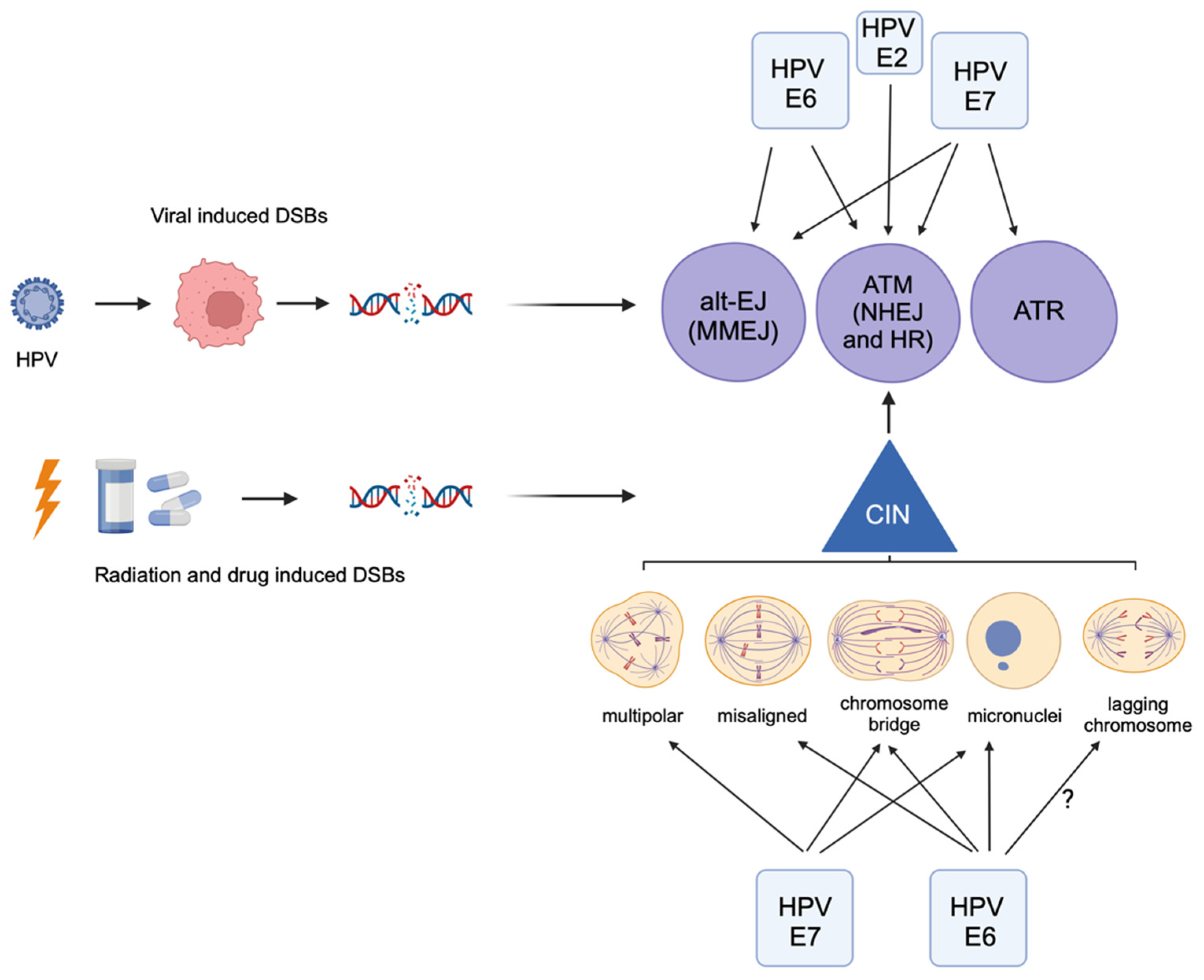
, and 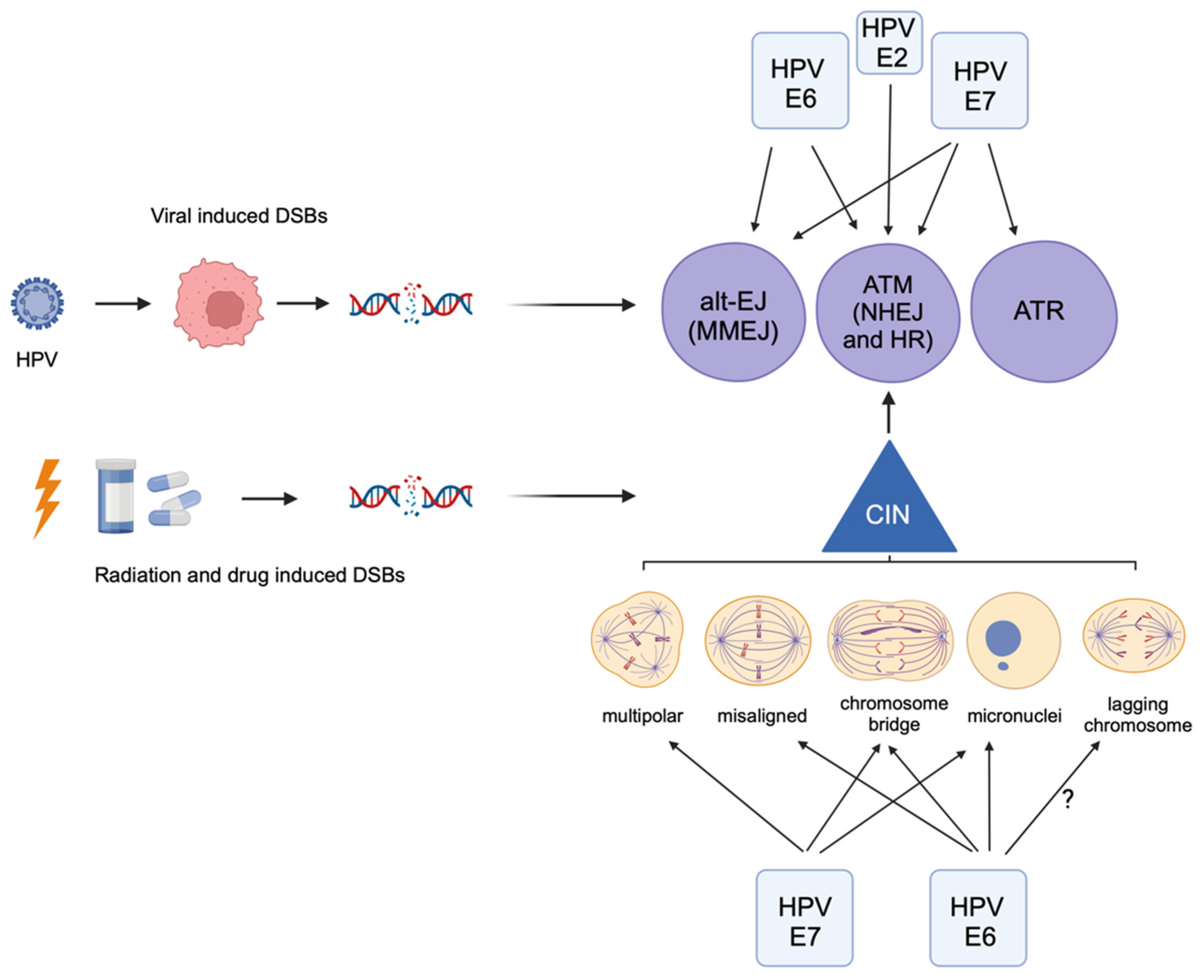{kind=link}
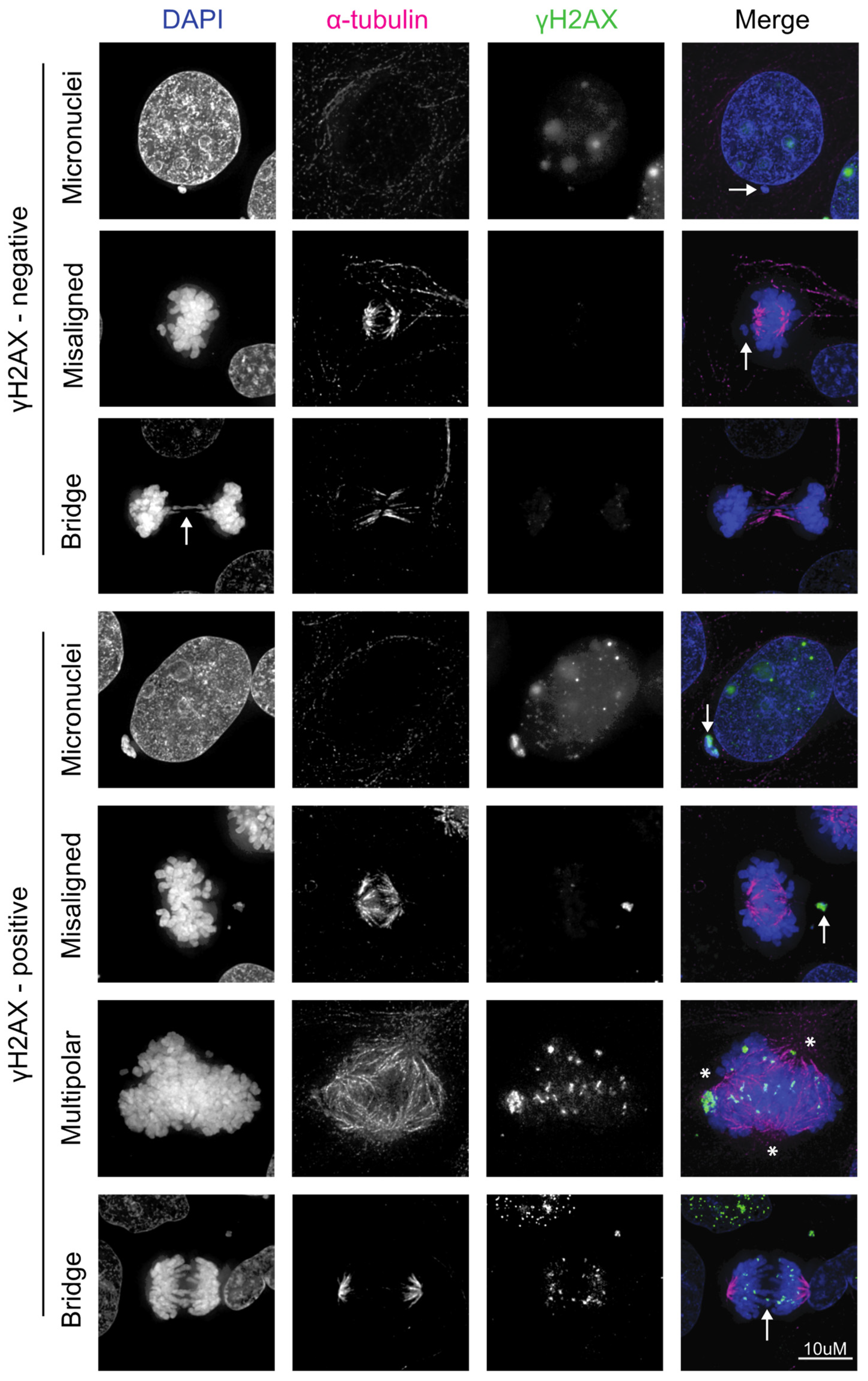
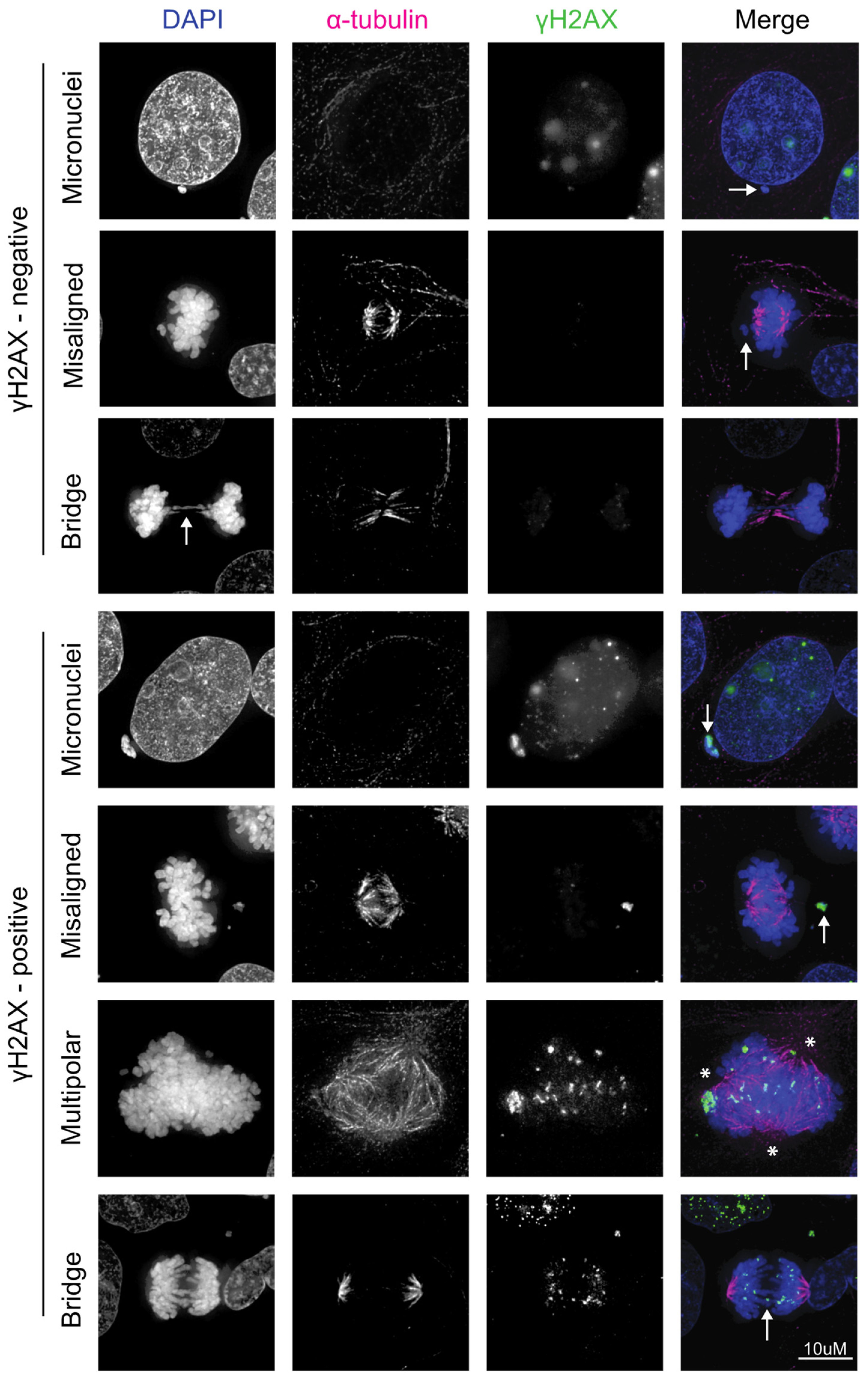{kind=link}
Abstract
Simple Summary
Abstract
1. Human Papillomavirus Genome and Lifecycle
2. Causes of Carcinogenesis
3. HPV Activates the DNA Damage Response
4. HPV Induces Chromosomal Instability, Which Can Lead to Further DNA Damage
5. HPV+ Cancers Use the Alternative End-Joining DNA Repair Pathway
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chesson, H.W.; Dunne, E.F.; Hariri, S.; Markowitz, L.E. The Estimated Lifetime Probability of Acquiring Human Papillomavirus in the United States. Sex. Transm. Dis. 2014, 41, 660–664. [Google Scholar] [CrossRef]
- Kreisel, K.M.; Spicknall, I.H.; Gargano, J.W.; Lewis, F.M.T.; Lewis, R.M.; Markowitz, L.E.; Roberts, H.; Johnson, A.S.; Song, R.; St Cyr, S.B.; et al. Sexually Transmitted Infections Among US Women and Men: Prevalence and Incidence Estimates, 2018. Sex. Transm. Dis. 2021, 48, 208–214. [Google Scholar] [CrossRef]
- Munger, K. The Role of Human Papillomaviruses in Human Cancers. Front. Biosci. 2002, 7, 641–649. [Google Scholar] [CrossRef]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human Papillomavirus Molecular Biology and Disease Association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef]
- Straub, E.; Fertey, J.; Dreer, M.; Iftner, T.; Stubenrauch, F. Characterization of the Human Papillomavirus 16 E8 Promoter. J. Virol. 2015, 89, 7304–7313. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human Papillomavirus Oncoproteins: Pathways to Transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- SShafti-Keramat, S.; Handisurya, A.; Kriehuber, E.; Meneguzzi, G.; Slupetzky, K.; Kirnbauer, R. Different Heparan Sulfate Proteoglycans Serve as Cellular Receptors for Human Papillomaviruses. J. Virol. 2003, 77, 13125–13135. [Google Scholar] [CrossRef]
- Popa, A.; Zhang, W.; Harrison, M.S.; Goodner, K.; Kazakov, T.; Goodwin, E.C.; Lipovsky, A.; Burd, C.G.; DiMaio, D. Direct Binding of Retromer to Human Papillomavirus Type 16 Minor Capsid Protein L2 Mediates Endosome Exit during Viral Infection. PLoS Pathog. 2015, 11, e1004699. [Google Scholar] [CrossRef]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Hilbig, L.; Sapp, M. The Nuclear Retention Signal of HPV16 L2 Protein Is Essential for Incoming Viral Genome to Transverse the Trans-Golgi Network. Virology 2014, 458–459, 93–105. [Google Scholar] [CrossRef]
- Aydin, I.; Villalonga-Planells, R.; Greune, L.; Bronnimann, M.P.; Calton, C.M.; Becker, M.; Lai, K.-Y.; Campos, S.K.; Schmidt, M.A.; Schelhaas, M. A Central Region in the Minor Capsid Protein of Papillomaviruses Facilitates Viral Genome Tethering and Membrane Penetration for Mitotic Nuclear Entry. PLoS Pathog. 2017, 13, e1006308. [Google Scholar] [CrossRef]
- Prabhakar, A.T.; James, C.D.; Das, D.; Otoa, R.; Day, M.; Burgner, J.; Fontan, C.T.; Wang, X.; Glass, S.H.; Wieland, A.; et al. CK2 Phosphorylation of Human Papillomavirus 16 E2 on Serine 23 Promotes Interaction with TopBP1 and Is Critical for E2 Interaction with Mitotic Chromatin and the Viral Life Cycle. mBio 2021, 12, e0116321. [Google Scholar] [CrossRef]
- Prabhakar, A.T.; James, C.D.; Das, D.; Fontan, C.T.; Otoa, R.; Wang, X.; Bristol, M.L.; Morgan, I.M. Interaction with TopBP1 Is Required for Human Papillomavirus 16 E2 Plasmid Segregation/Retention Function during Mitosis. J. Virol. 2022, 96, e0083022. [Google Scholar] [CrossRef]
- Hatterschide, J.; Bohidar, A.E.; Grace, M.; Nulton, T.J.; Kim, H.W.; Windle, B.; Morgan, I.M.; Munger, K.; White, E.A. PTPN14 Degradation by High-Risk Human Papillomavirus E7 Limits Keratinocyte Differentiation and Contributes to HPV-Mediated Oncogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7033–7042. [Google Scholar] [CrossRef]
- Bansal, A.; Singh, M.; Rai, B. Human Papillomavirus-Associated Cancers: A Growing Global Problem. Int. J. Appl. Basic. Med. Res. 2016, 6, 84–89. [Google Scholar] [CrossRef]
- Burnet, N.G.; Jefferies, S.J.; Benson, R.J.; Hunt, D.P.; Treasure, F.P. Years of Life Lost (YLL) from Cancer Is an Important Measure of Population Burden—And Should Be Considered When Allocating Research Funds. Br. J. Cancer 2005, 92, 241–245. [Google Scholar] [CrossRef]
- Lechner, M.; Jones, O.S.; Breeze, C.E.; Gilson, R. Gender-Neutral HPV Vaccination in the UK, Rising Male Oropharyngeal Cancer Rates, and Lack of HPV Awareness. Lancet Infect. Dis. 2019, 19, 131–132. [Google Scholar] [CrossRef]
- Cancer Trends Progress Report. National Cancer Institute, NIH, HHS, Bethesda, MD, March 2024. Available online: https://progressreport.cancer.gov (accessed on 18 April 2024).
- Lin, X.; Rodgers, L.; Zhu, L.; Stokley, S.; Meites, E.; Markowitz, L.E. Human Papillomavirus Vaccination Coverage Using Two-Dose or Three-Dose Schedule Criteria. Vaccine 2017, 35, 5759–5761. [Google Scholar] [CrossRef]
- Yoo, W.; Kim, S.; Huh, W.K.; Dilley, S.; Coughlin, S.S.; Partridge, E.E.; Chung, Y.; Dicks, V.; Lee, J.-K.; Bae, S. Recent Trends in Racial and Regional Disparities in Cervical Cancer Incidence and Mortality in United States. PLoS ONE 2017, 12, e0172548. [Google Scholar] [CrossRef]
- James, C.D.; Roberts, S. Viral Interactions with PDZ Domain-Containing Proteins-An Oncogenic Trait? Pathogens 2016, 5, 8. [Google Scholar] [CrossRef]
- Basukala, O.; Banks, L. The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis. Viruses 2021, 13, 1892. [Google Scholar] [CrossRef]
- Boyer, S.; Wazer, D.; Band, V. E7 Protein of Human Papilloma Virus-16 Induces Degradation of Retinoblastoma Protein through the Ubiquitin-Proteasome Pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar]
- Mehta, K.; Laimins, L. Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. mBio 2018, 9, e00064-18. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2019, 10, 3116. [Google Scholar] [CrossRef]
- Kaliff, M.; Karlsson, M.G.; Sorbe, B.; Bohr Mordhorst, L.; Helenius, G.; Lillsunde-Larsson, G. HPV-Negative Tumors in a Swedish Cohort of Cervical Cancer. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2019, 39, 279–288. [Google Scholar] [CrossRef]
- Meulendijks, D.; Tomasoa, N.B.; Dewit, L.; Smits, P.H.M.; Bakker, R.; van Velthuysen, M.-L.F.; Rosenberg, E.H.; Beijnen, J.H.; Schellens, J.H.M.; Cats, A. HPV-Negative Squamous Cell Carcinoma of the Anal Canal Is Unresponsive to Standard Treatment and Frequently Carries Disruptive Mutations in TP53. Br. J. Cancer 2015, 112, 1358–1366. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- van Gent, D.C.; Hoeijmakers, J.H.; Kanaar, R. Chromosomal Stability and the DNA Double-Stranded Break Connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef]
- Studstill, C.J.; Moody, C.A. For Better or Worse: Modulation of the Host DNA Damage Response by Human Papillomavirus. Annu. Rev. Virol. 2023, 10, 325–345. [Google Scholar] [CrossRef]
- Hustedt, N.; Durocher, D. The Control of DNA Repair by the Cell Cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human Papillomaviruses Recruit Cellular DNA Repair and Homologous Recombination Factors to Viral Replication Centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human Papillomaviruses Activate the ATM DNA Damage Pathway for Viral Genome Amplification upon Differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The Papillomavirus E1 Helicase Activates a Cellular DNA Damage Response in Viral Replication Foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef]
- Männik, A.; Rünkorg, K.; Jaanson, N.; Ustav, M.; Ustav, E. Induction of the Bovine Papillomavirus Origin “Onion Skin”-Type DNA Replication at High E1 Protein Concentrations In Vivo. J. Virol. 2002, 76, 5835–5845. [Google Scholar] [CrossRef]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of Genomic Instability in Cells Infected with the High-Risk Human Papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef]
- Prabhakar, A.T.; James, C.D.; Fontan, C.T.; Otoa, R.; Wang, X.; Bristol, M.L.; Hill, R.D.; Dubey, A.; Morgan, I.M. Human Papillomavirus 16 E2 Interaction with TopBP1 Is Required for E2 and Viral Genome Stability during the Viral Life Cycle. J. Virol. 2023, 97, e0006323. [Google Scholar] [CrossRef]
- Mehta, K.; Gunasekharan, V.; Satsuka, A.; Laimins, L.A. Human Papillomaviruses Activate and Recruit SMC1 Cohesin Proteins for the Differentiation-Dependent Life Cycle through Association with CTCF Insulators. PLoS Pathog. 2015, 11, e1004763. [Google Scholar] [CrossRef]
- Morgan, I.M.; DiNardo, L.J.; Windle, B. Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift? Viruses 2017, 9, 208. [Google Scholar] [CrossRef]
- Baedyananda, F.; Chaiwongkot, A.; Bhattarakosol, P. Elevated HPV16 E1 Expression Is Associated with Cervical Cancer Progression. Intervirology 2017, 60, 171–180. [Google Scholar] [CrossRef]
- Park, J.S.; Hwang, E.S.; Park, S.N.; Ahn, H.K.; Um, S.J.; Kim, C.J.; Kim, S.J.; Namkoong, S.E. Physical Status and Expression of HPV Genes in Cervical Cancers. Gynecol. Oncol. 1997, 65, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Seedorf, K.; Oltersdorf, T.; Krämmer, G.; Röwekamp, W. Identification of Early Proteins of the Human Papilloma Viruses Type 16 (HPV 16) and Type 18 (HPV 18) in Cervical Carcinoma Cells. EMBO J. 1987, 6, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, P.; Hong, S.; Kono, T.; Hoover, P.; Laimins, L. Topoisomerase 2β Induces DNA Breaks To Regulate Human Papillomavirus Replication. mBio 2021, 12, e00005-21. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Cheng, S.; Iovane, A.; Laimins, L.A. STAT-5 Regulates Transcription of the Topoisomerase IIβ-Binding Protein 1 (TopBP1) Gene To Activate the ATR Pathway and Promote Human Papillomavirus Replication. mBio 2015, 6, e02006-15. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jeon, S.H.; Han, M.G.; Kang, M.H.; Kim, I.A. BRD4 Inhibition Enhances the Antitumor Effects of Radiation Therapy in a Murine Breast Cancer Model. Int. J. Mol. Sci. 2023, 24, 13062. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Kim, H.J.; Dernburg, A.F. ATM Signaling Modulates Cohesin Behavior in Meiotic Prophase and Proliferating Cells. Nat. Struct. Mol. Biol. 2023, 30, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Mamberti, S.; Pabba, M.K.; Rapp, A.; Cardoso, M.C.; Scholz, M. The Chromatin Architectural Protein CTCF Is Critical for Cell Survival upon Irradiation-Induced DNA Damage. Int. J. Mol. Sci. 2022, 23, 3896. [Google Scholar] [CrossRef] [PubMed]
- Sowd, G.A.; Mody, D.; Eggold, J.; Cortez, D.; Friedman, K.L.; Fanning, E. SV40 Utilizes ATM Kinase Activity to Prevent Non-Homologous End Joining of Broken Viral DNA Replication Products. PLoS Pathog. 2014, 10, e1004536. [Google Scholar] [CrossRef] [PubMed]
- Templeton, C.W.; Laimins, L.A. P53-Dependent R-Loop Formation and HPV Pathogenesis. Proc. Natl. Acad. Sci. USA 2023, 120, e2305907120. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Münger, K. The Human Papillomavirus Type 16 E6 and E7 Oncoproteins Independently Induce Numerical and Structural Chromosome Instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar] [PubMed]
- Wallace, N.A.; Khanal, S.; Robinson, K.L.; Wendel, S.O.; Messer, J.J.; Galloway, D.A. High-Risk Alphapapillomavirus Oncogenes Impair the Homologous Recombination Pathway. J. Virol. 2017, 91, e01084-17. [Google Scholar] [CrossRef] [PubMed]
- Dust, K.; Carpenter, M.; Chen, J.C.; Grant, C.; McCorrister, S.; Westmacott, G.R.; Severini, A. Human Papillomavirus 16 E6 and E7 Oncoproteins Alter the Abundance of Proteins Associated with DNA Damage Response, Immune Signaling and Epidermal Differentiation. Viruses 2022, 14, 1764. [Google Scholar] [CrossRef] [PubMed]
- Wendel, S.O.; Snow, J.A.; Bastian, T.; Brown, L.; Hernandez, C.; Burghardt, E.; Kahn, A.; Murthy, V.; Neill, D.; Smith, Z.C.; et al. High Risk α-HPV E6 Impairs Translesion Synthesis by Blocking POLη Induction. Cancers 2020, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Pickering, M.T.; Kowalik, T.F. Rb Inactivation Leads to E2F1-Mediated DNA Double-Strand Break Accumulation. Oncogene 2006, 25, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Aloor, H.L.; Moody, C.A. The Rb Binding Domain of HPV31 E7 Is Required to Maintain High Levels of DNA Repair Factors in Infected Cells. Virology 2017, 500, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Sitz, J.; Blanchet, S.A.; Gameiro, S.F.; Biquand, E.; Morgan, T.M.; Galloy, M.; Dessapt, J.; Lavoie, E.G.; Blondeau, A.; Smith, B.C.; et al. Human Papillomavirus E7 Oncoprotein Targets RNF168 to Hijack the Host DNA Damage Response. Proc. Natl. Acad. Sci. USA 2019, 116, 19552–19562. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, T.; Altmeyer, M.; Savic, V.; Toledo, L.; Dinant, C.; Grøfte, M.; Bartkova, J.; Poulsen, M.; Oka, Y.; Bekker-Jensen, S.; et al. TRIP12 and UBR5 Suppress Spreading of Chromatin Ubiquitylation at Damaged Chromosomes. Cell 2012, 150, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Molkentine, D.P.; Molkentine, J.M.; Bridges, K.A.; Valdecanas, D.R.; Dhawan, A.; Bahri, R.; Hefner, A.J.; Kumar, M.; Yang, L.; Abdelhakiem, M.; et al. P16 Represses DNA Damage Repair via a Novel Ubiquitin-Dependent Signaling Cascade. Cancer Res. 2022, 82, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, C.C.; Blanco, L.Z.; Maniar, K.P.; Laimins, L.A. Expression of HPV-Induced DNA Damage Repair Factors Correlates With CIN Progression. Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2019, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kono, T.; Hoover, P.; Poropatich, K.; Paunesku, T.; Mittal, B.B.; Samant, S.; Laimins, L.A. Activation of DNA Damage Repair Factors in HPV Positive Oropharyngeal Cancers. Virology 2020, 547, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, A.J.; Brown, L.; Tawfik, O.; Madan, R.; Shnayder, Y.; Thomas, S.M.; Wallace, N.A. DNA Repair Gene Expression Is Increased in HPV Positive Head and Neck Squamous Cell Carcinomas. Virology 2020, 548, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.S.; Moore, D.; Parker, C.J.; Broker, T.R.; Chow, L.T. Targeting DNA Damage Response as a Strategy to Treat HPV Infections. Int. J. Mol. Sci. 2019, 20, 5455. [Google Scholar] [CrossRef] [PubMed]
- Dok, R.; Glorieux, M.; Bamps, M.; Nuyts, S. Effect of ATR Inhibition in RT Response of HPV-Negative and HPV-Positive Head and Neck Cancers. Int. J. Mol. Sci. 2021, 22, 1504. [Google Scholar] [CrossRef] [PubMed]
- Kimple, R.J.; Smith, M.A.; Blitzer, G.C.; Torres, A.D.; Martin, J.A.; Yang, R.Z.; Peet, C.R.; Lorenz, L.D.; Nickel, K.P.; Klingelhutz, A.J.; et al. Enhanced Radiation Sensitivity in HPV-Positive Head and Neck Cancer. Cancer Res. 2013, 73, 4791–4800. [Google Scholar] [CrossRef] [PubMed]
- Köcher, S.; Zech, H.B.; Krug, L.; Gatzemeier, F.; Christiansen, S.; Meyer, F.; Rietow, R.; Struve, N.; Mansour, W.Y.; Kriegs, M.; et al. A Lack of Effectiveness in the ATM-Orchestrated DNA Damage Response Contributes to the DNA Repair Defect of HPV-Positive Head and Neck Cancer Cells. Front. Oncol. 2022, 12, 765968. [Google Scholar] [CrossRef] [PubMed]
- Eldakhakhny, S.; Zhou, Q.; Crosbie, E.J.; Sayan, B.S. Human Papillomavirus E7 Induces P63 Expression to Modulate DNA Damage Response. Cell Death Dis. 2018, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Lopez, S.; Altwerger, G.; Bellone, S.; Bonazzoli, E.; Zammataro, L.; Manzano, A.; Manara, P.; Perrone, E.; Zeybek, B.; et al. PARP-1 Activity (PAR) Determines the Sensitivity of Cervical Cancer to Olaparib. Gynecol. Oncol. 2019, 155, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Pirotte, E.F.; Holzhauser, S.; Owens, D.; Quine, S.; Al-Hussaini, A.; Christian, A.D.; Giles, P.J.; Man, S.T.; Evans, M.; Powell, N.G. Sensitivity to Inhibition of DNA Repair by Olaparib in Novel Oropharyngeal Cancer Cell Lines Infected with Human Papillomavirus. PLoS ONE 2018, 13, e0207934. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of Chromosomal Instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef] [PubMed]
- Cosper, P.F.; Hrycyniak, L.C.F.; Paracha, M.; Lee, D.L.; Wan, J.; Jones, K.; Bice, S.A.; Nickel, K.; Mallick, S.; Taylor, A.M.; et al. HPV16 E6 Induces Chromosomal Instability Due to Polar Chromosomes Caused by E6AP-Dependent Degradation of the Mitotic Kinesin CENP-E. Proc. Natl. Acad. Sci. USA 2023, 120, e2216700120. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.; Gonzalez, S.; Crum, C.P.; Munger, K. The Human Papillomavirus Type 16 E6 and E7 Oncoproteins Cooperate to Induce Mitotic Defects and Genomic Instability by Uncoupling Centrosome Duplication from the Cell Division Cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Choi, Y.; Taylor, A.M.; Cosper, P.F. Human Papillomavirus-Induced Chromosomal Instability and Aneuploidy in Squamous Cell Cancers. Viruses 2024, 16, 501. [Google Scholar] [CrossRef]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A Mechanism Linking Extra Centrosomes to Chromosomal Instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef]
- Zhou, A.S.; Tucker, J.B.; Scribano, C.M.; Lynch, A.R.; Carlsen, C.L.; Pop-Vicas, S.T.; Pattaswamy, S.M.; Burkard, M.E.; Weaver, B.A. Diverse Microtubule-Targeted Anticancer Agents Kill Cells by Inducing Chromosome Missegregation on Multipolar Spindles. PLoS Biol. 2023, 21, e3002339. [Google Scholar] [CrossRef] [PubMed]
- Plug-DeMaggio, A.W.; Sundsvold, T.; Wurscher, M.A.; Koop, J.I.; Klingelhutz, A.J.; McDougall, J.K. Telomere Erosion and Chromosomal Instability in Cells Expressing the HPV Oncogene 16E6. Oncogene 2004, 23, 3561–3571. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dakic, A.; Zhang, Y.; Dai, Y.; Chen, R.; Schlegel, R. HPV E6 Protein Interacts Physically and Functionally with the Cellular Telomerase Complex. Proc. Natl. Acad. Sci. USA 2009, 106, 18780–18785. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.T.; Kyo, S.; Laimins, L.A. Telomerase Activation by Human Papillomavirus Type 16 E6 Protein: Induction of Human Telomerase Reverse Transcriptase Expression through Myc and GC-Rich Sp1 Binding Sites. J. Virol. 2001, 75, 5559–5566. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, R. Telomerase Induction in HPV Infection and Oncogenesis. Viruses 2017, 9, 180. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Chan, Y.W. Chromatin Bridges: Stochastic Breakage or Regulated Resolution? Trends Genet. 2023, 40, 69–82. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. THE STABILITY OF BROKEN ENDS OF CHROMOSOMES IN ZEA MAYS. Genetics 1941, 26, 234–282. [Google Scholar] [CrossRef] [PubMed]
- Dykhuizen, E.C.; Hargreaves, D.C.; Miller, E.L.; Cui, K.; Korshunov, A.; Kool, M.; Pfister, S.; Cho, Y.-J.; Zhao, K.; Crabtree, G.R. BAF Complexes Facilitate Decatenation of DNA by Topoisomerase IIα. Nature 2013, 497, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Chestukhin, A.; Pfeffer, C.; Milligan, S.; DeCaprio, J.A.; Pellman, D. Processing, Localization, and Requirement of Human Separase for Normal Anaphase Progression. Proc. Natl. Acad. Sci. USA 2003, 100, 4574–4579. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.P.L.; Medema, R.H. Chromosome Segregation Errors as a Cause of DNA Damage and Structural Chromosome Aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, N.T.; Zhang, C.-Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms Generating Cancer Genome Complexity from a Single Cell Division Error. Science 2020, 368, eaba0712. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.; Rossi, N.M.; Keskus, A.G.; Xie, Y.; Ahmad, T.; Bryant, A.; Lou, H.; Paredes, J.G.; Milano, R.; Rao, N.; et al. Insights into the Mechanisms and Structure of Breakage-Fusion-Bridge Cycles in Cervical Cancer Using Long-Read Sequencing. Am. J. Hum. Genet. 2024, 111, 544–561. [Google Scholar] [CrossRef] [PubMed]
- Pampalona, J.; Roscioli, E.; Silkworth, W.T.; Bowden, B.; Genescà, A.; Tusell, L.; Cimini, D. Chromosome Bridges Maintain Kinetochore-Microtubule Attachment throughout Mitosis and Rarely Break during Anaphase. PLoS ONE 2016, 11, e0147420. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Loewer, A.; Lahav, G.; Mitchison, T.J. Prolonged Mitotic Arrest Triggers Partial Activation of Apoptosis, Resulting in DNA Damage and P53 Induction. Mol. Biol. Cell 2012, 23, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human Papillomavirus 16 E7 Oncoprotein Attenuates DNA Damage Checkpoint Control by Increasing the Proteolytic Turnover of Claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.B.; Bonema, S.C.; García-Varela, R.; Denu, R.A.; Hu, Y.; McGregor, S.M.; Burkard, M.E.; Weaver, B.A. Misaligned Chromosomes Are a Major Source of Chromosomal Instability in Breast Cancer. Cancer Res. Commun. 2023, 3, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA Breaks and Chromosome Pulverization from Errors in Mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA Damage in Micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Kneissig, M.; Keuper, K.; De Pagter, M.S.; Van Roosmalen, M.J.; Martin, J.; Otto, H.; Passerini, V.; Campos Sparr, A.; Renkens, I.; Kropveld, F.; et al. Micronuclei-Based Model System Reveals Functional Consequences of Chromothripsis in Human Cells. eLife 2019, 8, e50292. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Helbling-Leclerc, A.; Kawasumi, R.; Jegadesan, N.K.; Xu, X.; Devulder, P.; Abe, T.; Takata, M.; Xu, D.; Rosselli, F.; et al. SMC5/6 Acts Jointly with Fanconi Anemia Factors to Support DNA Repair and Genome Stability. EMBO Rep. 2020, 21, e48222. [Google Scholar] [CrossRef] [PubMed]
- Leimbacher, P.-A.; Jones, S.E.; Shorrocks, A.-M.K.; de Marco Zompit, M.; Day, M.; Blaauwendraad, J.; Bundschuh, D.; Bonham, S.; Fischer, R.; Fink, D.; et al. MDC1 Interacts with TOPBP1 to Maintain Chromosomal Stability during Mitosis. Mol. Cell 2019, 74, 571–583.e8. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, C.C.; Laimins, L.A. FANCD2 Binds Human Papillomavirus Genomes and Associates with a Distinct Set of DNA Repair Proteins to Regulate Viral Replication. mBio 2017, 8, e02340-16. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.J.; Neary, G.J.; Williamson, F.S. The Relative Biological Efficiency of Single Doses of Fast Neutrons and Gamma-Rays on Vicia Faba Roots and the Effect of Oxygen: Part II. Chromosome Damage: The Production of Micronuclei. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1959, 1, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Morley, A.A. Measurement of Micronuclei in Lymphocytes. Mutat. Res. 1985, 147, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Cornforth, M.N.; Goodwin, E.H. Transmission of Radiation-Induced Acentric Chromosomal Fragments to Micronuclei in Normal Human Fibroblasts. Radiat. Res. 1991, 126, 210. [Google Scholar] [CrossRef]
- Ly, P.; Brunner, S.F.; Shoshani, O.; Kim, D.H.; Lan, W.; Pyntikova, T.; Flanagan, A.M.; Behjati, S.; Page, D.C.; Campbell, P.J.; et al. Chromosome Segregation Errors Generate a Diverse Spectrum of Simple and Complex Genomic Rearrangements. Nat. Genet. 2019, 51, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Terradas, M.; Martín, M.; Tusell, L.; Genescà, A. DNA Lesions Sequestered in Micronuclei Induce a Local Defective-Damage Response. DNA Repair. 2009, 8, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Zhang, H.; Chen, G.Z.; Shin, D.M.; Doetsch, P.W. HPV16 E6 and E7 Proteins Induce a Chronic Oxidative Stress Response via NOX2 That Causes Genomic Instability and Increased Susceptibility to DNA Damage in Head and Neck Cancer Cells. Carcinogenesis 2015, 36, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Cassel, A.P.R.; Barcellos, R.B.; da Silva, C.M.D.; de Matos Almeida, S.E.; Rossetti, M.L.R. Association between Human Papillomavirus (HPV) DNA and Micronuclei in Normal Cervical Cytology. Genet. Mol. Biol. 2014, 37, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Gutiérrez, E.I.; Dávila-Rodríguez, M.I.; Vargas-Villarreal, J.; Hernández-Garza, F.; Cerda-Flores, R.M. Association between Human Papilloma Virus-Type Infections with Micronuclei Frequencies. Prague Med. Rep. 2010, 111, 35–41. [Google Scholar] [PubMed]
- Adam, M.L.; Pini, C.; Túlio, S.; Cantalice, J.C.L.L.; Torres, R.A.; Dos Santos Correia, M.T. Assessment of the Association between Micronuclei and the Degree of Uterine Lesions and Viral Load in Women with Human Papillomavirus. Cancer Genom. Proteom. 2015, 12, 67–71. [Google Scholar]
- Gayathri, B.; Kalyani, R.; Hemalatha, A.; Vasavi, B. Significance of Micronucleus in Cervical Intraepithelial Lesions and Carcinoma. J. Cytol. 2012, 29, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.N.; Misra, J.S.; Ahmad, R. Assessment of Micronuclei Counts as Tumour Marker in Cervical Carcinogenesis: A Follow-up Study. Cytopathology 2020, 31, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Setayesh, T.; Kundi, M.; Nersesyan, A.; Stopper, H.; Fenech, M.; Krupitza, G.; Knasmüller, S. Use of Micronucleus Assays for the Prediction and Detection of Cervical Cancer: A Meta-Analysis. Carcinogenesis 2020, 41, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Dacus, D.; Stancic, S.; Pollina, S.R.; Rifrogiate, E.; Palinski, R.; Wallace, N.A. Beta Human Papillomavirus 8 E6 Induces Micronucleus Formation and Promotes Chromothripsis. J. Virol. 2022, 96, e0101522. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Kabeche, L.; Wood, M.D.; Laucius, C.D.; Qu, D.; Laughney, A.M.; Reynolds, G.E.; Louie, R.J.; Phillips, J.; Chan, D.A.; et al. Numerical Chromosomal Instability Mediates Susceptibility to Radiation Treatment. Nat. Commun. 2015, 6, 5990. [Google Scholar] [CrossRef] [PubMed]
- Cosper, P.F.; Copeland, S.E.; Tucker, J.B.; Weaver, B.A. Chromosome Missegregation as a Modulator of Radiation Sensitivity. Semin. Radiat. Oncol. 2022, 32, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Leeman, J.E.; Li, Y.; Bell, A.; Hussain, S.S.; Majumdar, R.; Rong-Mullins, X.; Blecua, P.; Damerla, R.; Narang, H.; Ravindran, P.T.; et al. Human Papillomavirus 16 Promotes Microhomology-Mediated End-Joining. Proc. Natl. Acad. Sci. USA 2019, 116, 21573–21579. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.A.; Carvajal-Garcia, J.; Gupta, G.P. Mechanism, Cellular Functions and Cancer Roles of Polymerase-Theta-Mediated DNA End Joining. Nat. Rev. Mol. Cell Biol. 2022, 23, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Koole, W.; van Schendel, R.; Karambelas, A.E.; van Heteren, J.T.; Okihara, K.L.; Tijsterman, M. A Polymerase Theta-Dependent Repair Pathway Suppresses Extensive Genomic Instability at Endogenous G4 DNA Sites. Nat. Commun. 2014, 5, 3216. [Google Scholar] [CrossRef]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of Microhomology-Mediated End-Joining Promoted by Human DNA Polymerase θ. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.-H.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-Mediated End Joining and Homologous Recombination Share the Initial End Resection Step to Repair DNA Double-Strand Breaks in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed]
- Kötter, A.; Cornils, K.; Borgmann, K.; Dahm-Daphi, J.; Petersen, C.; Dikomey, E.; Mansour, W.Y. Inhibition of PARP1-Dependent End-Joining Contributes to Olaparib-Mediated Radiosensitization in Tumor Cells. Mol. Oncol. 2014, 8, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Goff, J.P.; Shields, D.S.; Seki, M.; Choi, S.; Epperly, M.W.; Dixon, T.; Wang, H.; Bakkenist, C.J.; Dertinger, S.D.; Torous, D.K.; et al. Lack of DNA Polymerase Theta (POLQ) Radiosensitizes Bone Marrow Stromal Cells in Vitro and Increases Reticulocyte Micronuclei after Total-Body Irradiation. Radiat. Res. 2009, 172, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Brambati, A.; Sacco, O.; Porcella, S.; Heyza, J.; Kareh, M.; Schmidt, J.C.; Sfeir, A. RHINO Directs MMEJ to Repair DNA Breaks in Mitosis. Science 2023, 381, eadh3694. [Google Scholar] [CrossRef] [PubMed]
- Arana, M.E.; Seki, M.; Wood, R.D.; Rogozin, I.B.; Kunkel, T.A. Low-Fidelity DNA Synthesis by Human DNA Polymerase Theta. Nucleic Acids Res. 2008, 36, 3847–3856. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Marini, F.; Wood, R.D. POLQ (Pol Theta), a DNA Polymerase and DNA-Dependent ATPase in Human Cells. Nucleic Acids Res. 2003, 31, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Hogg, M.; Seki, M.; Wood, R.D.; Doublié, S.; Wallace, S.S. Lesion Bypass Activity of DNA Polymerase θ (POLQ) Is an Intrinsic Property of the Pol Domain and Depends on Unique Sequence Inserts. J. Mol. Biol. 2011, 405, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Garcia, J.; Cho, J.-E.; Carvajal-Garcia, P.; Feng, W.; Wood, R.D.; Sekelsky, J.; Gupta, G.P.; Roberts, S.A.; Ramsden, D.A. Mechanistic Basis for Microhomology Identification and Genome Scarring by Polymerase Theta. Proc. Natl. Acad. Sci. USA 2020, 117, 8476–8485. [Google Scholar] [CrossRef]
- Saito, S.; Maeda, R.; Adachi, N. Dual Loss of Human POLQ and LIG4 Abolishes Random Integration. Nat. Commun. 2017, 8, 16112. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Lescale, C.; Babin, L.; Bedora-Faure, M.; Lenden-Hasse, H.; Baron, L.; Demangel, C.; Yelamos, J.; Brunet, E.; Deriano, L. Repair of G1 Induced DNA Double-Strand Breaks in S-G2/M by Alternative NHEJ. Nat. Commun. 2020, 11, 5239. [Google Scholar] [CrossRef]
- Zelensky, A.N.; Schimmel, J.; Kool, H.; Kanaar, R.; Tijsterman, M. Inactivation of Pol θ and C-NHEJ Eliminates off-Target Integration of Exogenous DNA. Nat. Commun. 2017, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase θ-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Ma, L.; Jones, T.; Palomero, L.; Pujana, M.A.; Martinez-Ruiz, H.; Ha, P.K.; Murnane, J.; Cuartas, I.; Seoane, J.; et al. Subjugation of TGFβ Signaling by Human Papilloma Virus in Head and Neck Squamous Cell Carcinoma Shifts DNA Repair from Homologous Recombination to Alternative End Joining. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 6001–6014. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Palomero, L.; Moore, J.; Guix, I.; Espín, R.; Aytés, A.; Mao, J.-H.; Paulovich, A.G.; Whiteaker, J.R.; Ivey, R.G.; et al. Loss of TGFβ Signaling Increases Alternative End-Joining DNA Repair That Sensitizes to Genotoxic Therapies across Cancer Types. Sci. Transl. Med. 2021, 13, eabc4465. [Google Scholar] [CrossRef] [PubMed]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and Host Genome Interactions in Primary Head and Neck Cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-Wide Profiling of HPV Integration in Cervical Cancer Identifies Clustered Genomic Hot Spots and a Potential Microhomology-Mediated Integration Mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Bugbee, T.; Palinski, R.; Akinyemi, I.A.; McIntosh, M.T.; MacCarthy, T.; Bhaduri-McIntosh, S.; Wallace, N. Beta Human Papillomavirus 8E6 Promotes Alternative End Joining. eLife 2023, 12, e81923. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A First-in-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, K.M.; Bryan, A.; McCunn, E.; Lantz, P.E.; Blalock, H.; Ojeda, I.C.; Mehta, K.; Cosper, P.F. The Causes and Consequences of DNA Damage and Chromosomal Instability Induced by Human Papillomavirus. Cancers 2024, 16, 1662. https://doi.org/10.3390/cancers16091662
Jones KM, Bryan A, McCunn E, Lantz PE, Blalock H, Ojeda IC, Mehta K, Cosper PF. The Causes and Consequences of DNA Damage and Chromosomal Instability Induced by Human Papillomavirus. Cancers. 2024; 16(9):1662. https://doi.org/10.3390/cancers16091662
Chicago/Turabian StyleJones, Kathryn M., Ava Bryan, Emily McCunn, Pate E. Lantz, Hunter Blalock, Isabel C. Ojeda, Kavi Mehta, and Pippa F. Cosper. 2024. "The Causes and Consequences of DNA Damage and Chromosomal Instability Induced by Human Papillomavirus" Cancers 16, no. 9: 1662. https://doi.org/10.3390/cancers16091662
APA StyleJones, K. M., Bryan, A., McCunn, E., Lantz, P. E., Blalock, H., Ojeda, I. C., Mehta, K., & Cosper, P. F. (2024). The Causes and Consequences of DNA Damage and Chromosomal Instability Induced by Human Papillomavirus. Cancers, 16(9), 1662. https://doi.org/10.3390/cancers16091662