
Revitalizing Cancer Treatment: Exploring the Role of Drug Repurposing
 ,
,  , and
, and 
Abstract
Simple Summary
Abstract
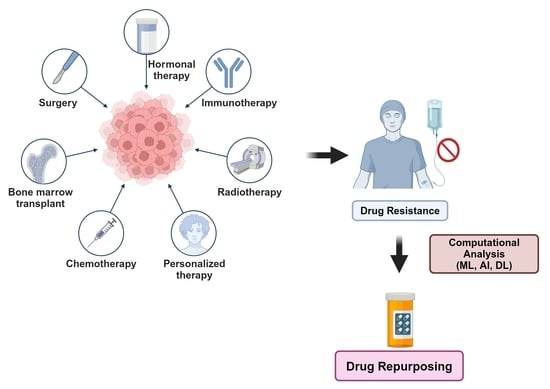
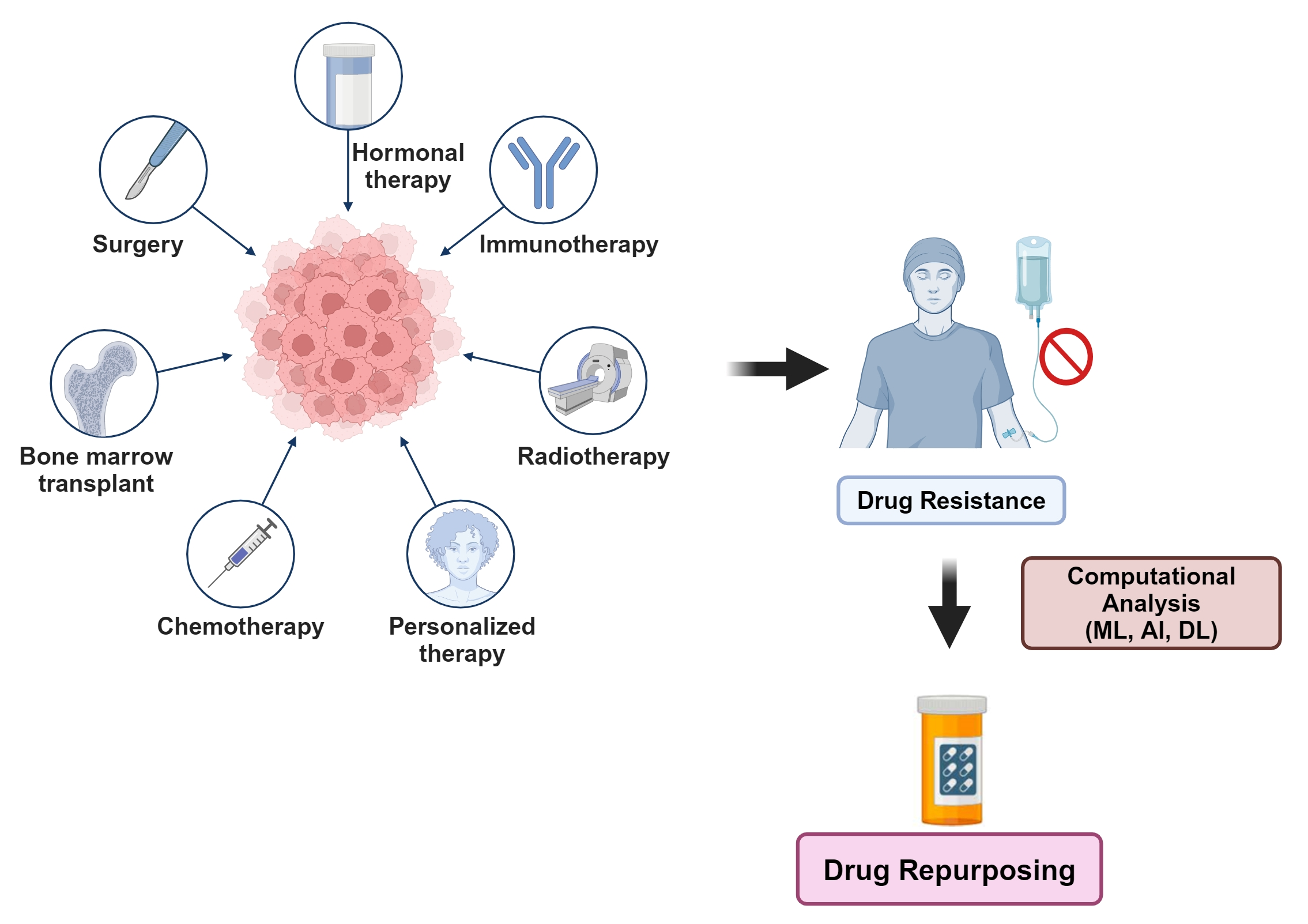{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
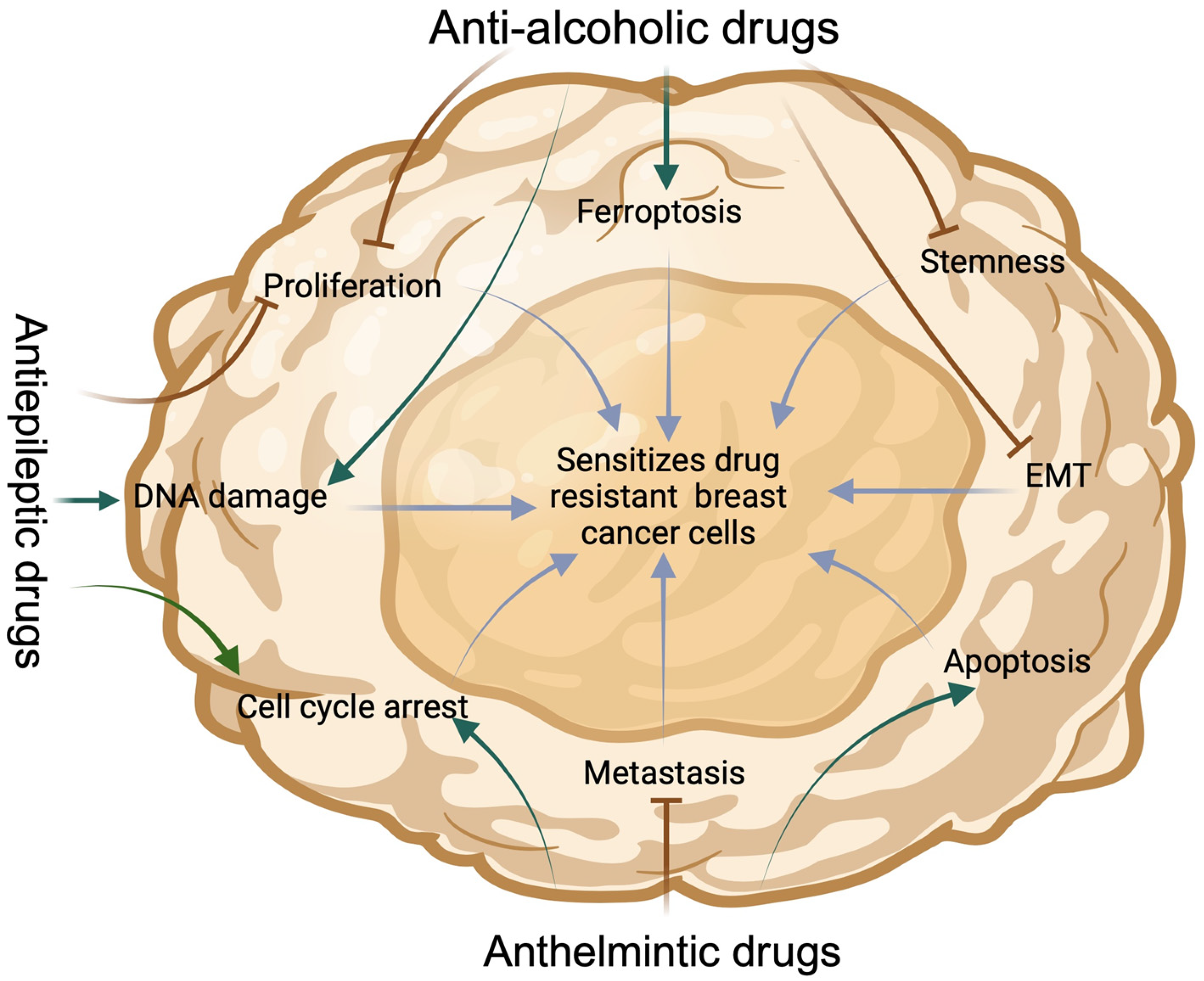
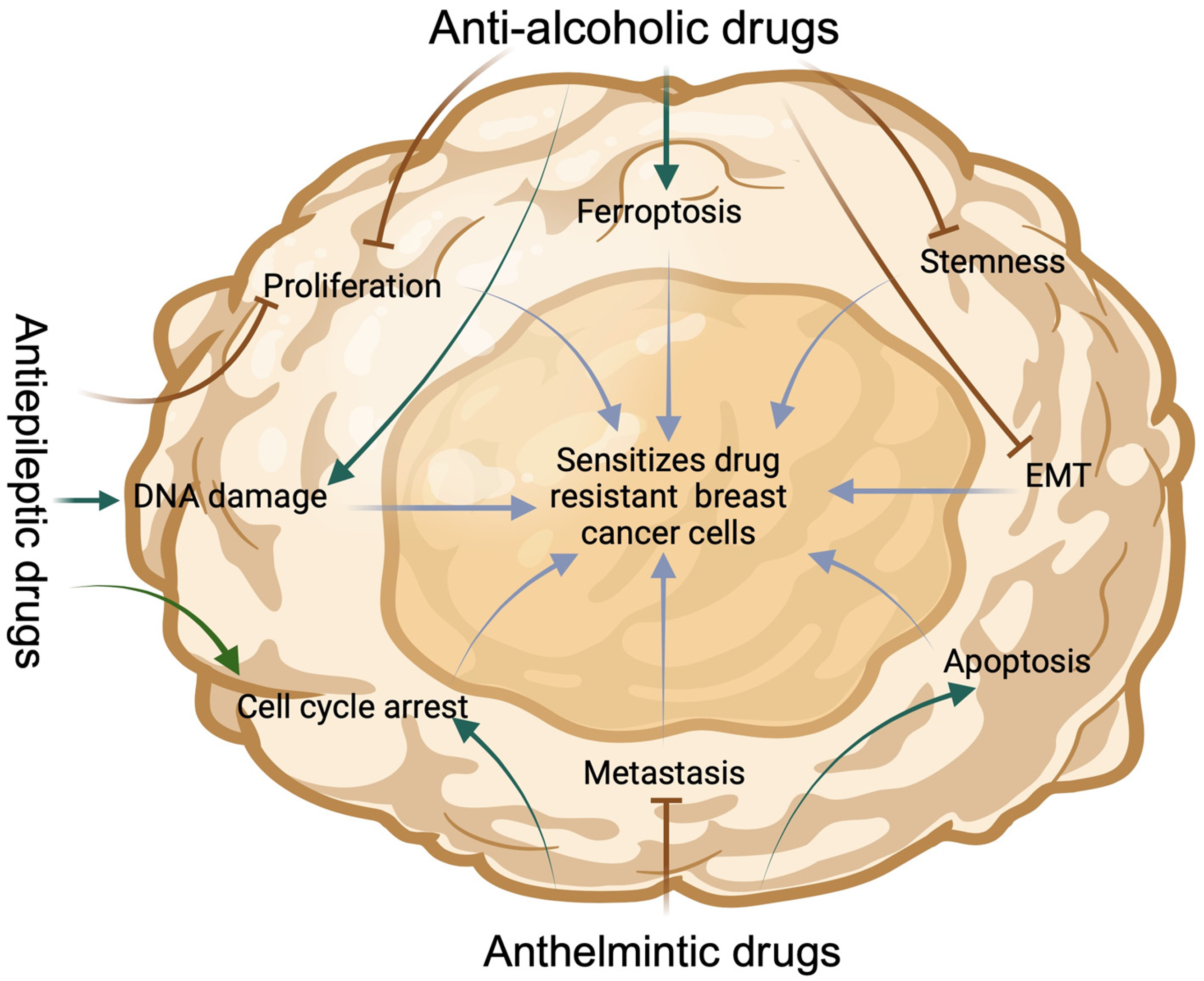
2. Experimental Approach: Repurposing Some Anti-Alcoholic, Anthelmintic and Antiepileptic Drugs for Cancer, Using Breast Cancer as a Case Study
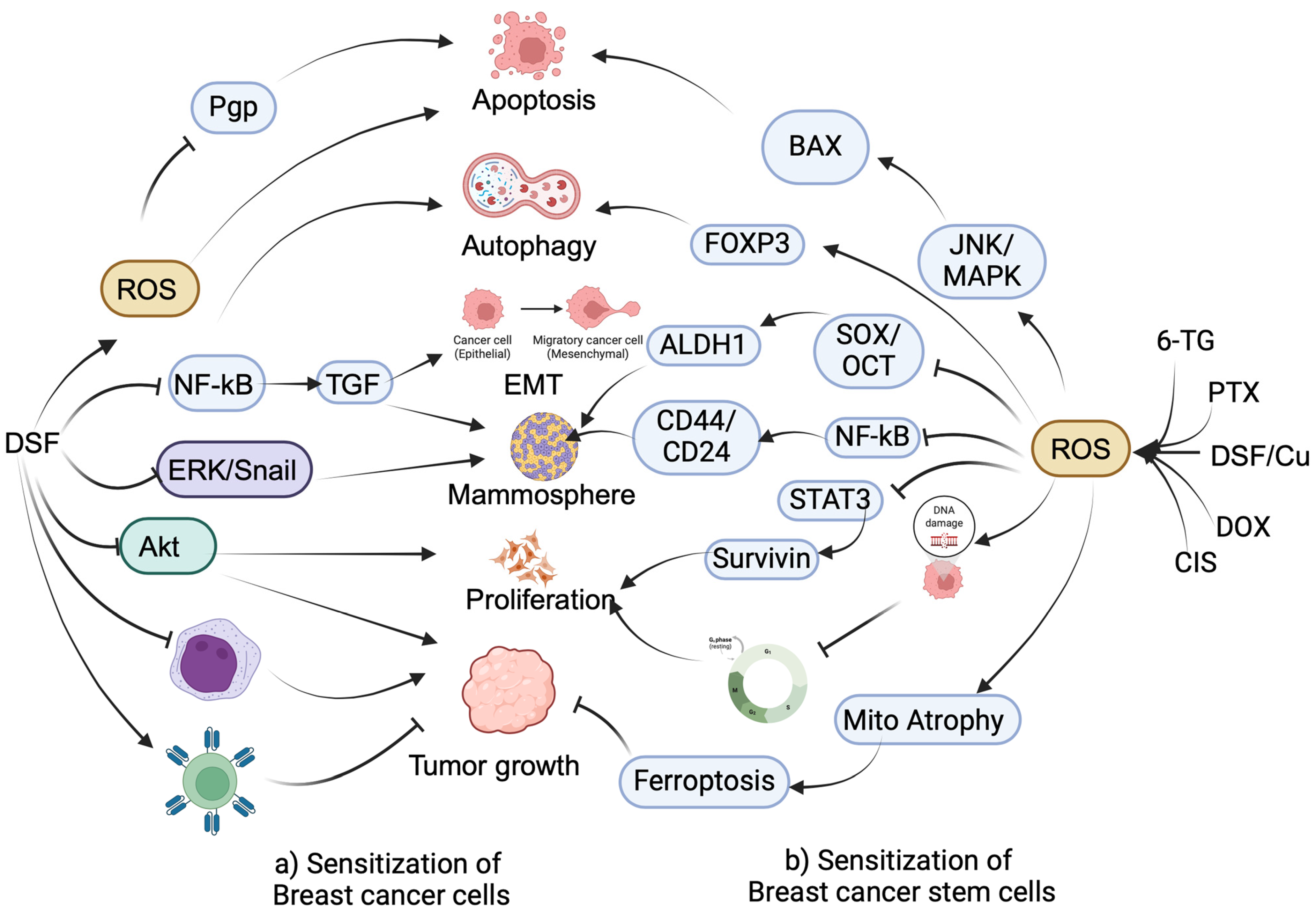
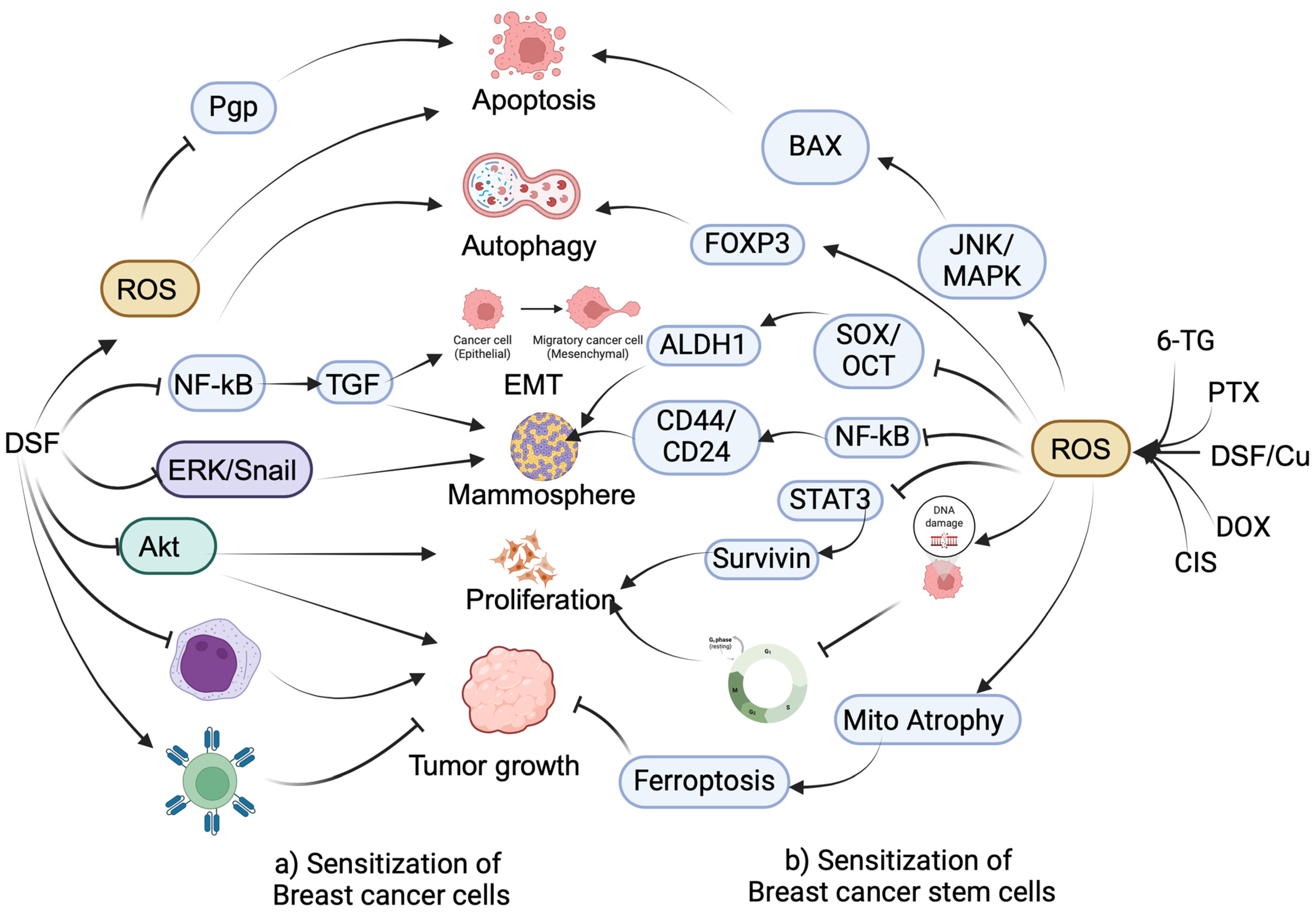
2.1. Sensitization of Drug-Resistant BC Cells by Anti-Alcoholic Drugs
2.2. Sensitization of BC Cells with Anthelmintic Drugs
2.3. Sensitization of BC Cells with Antiepileptic Drugs
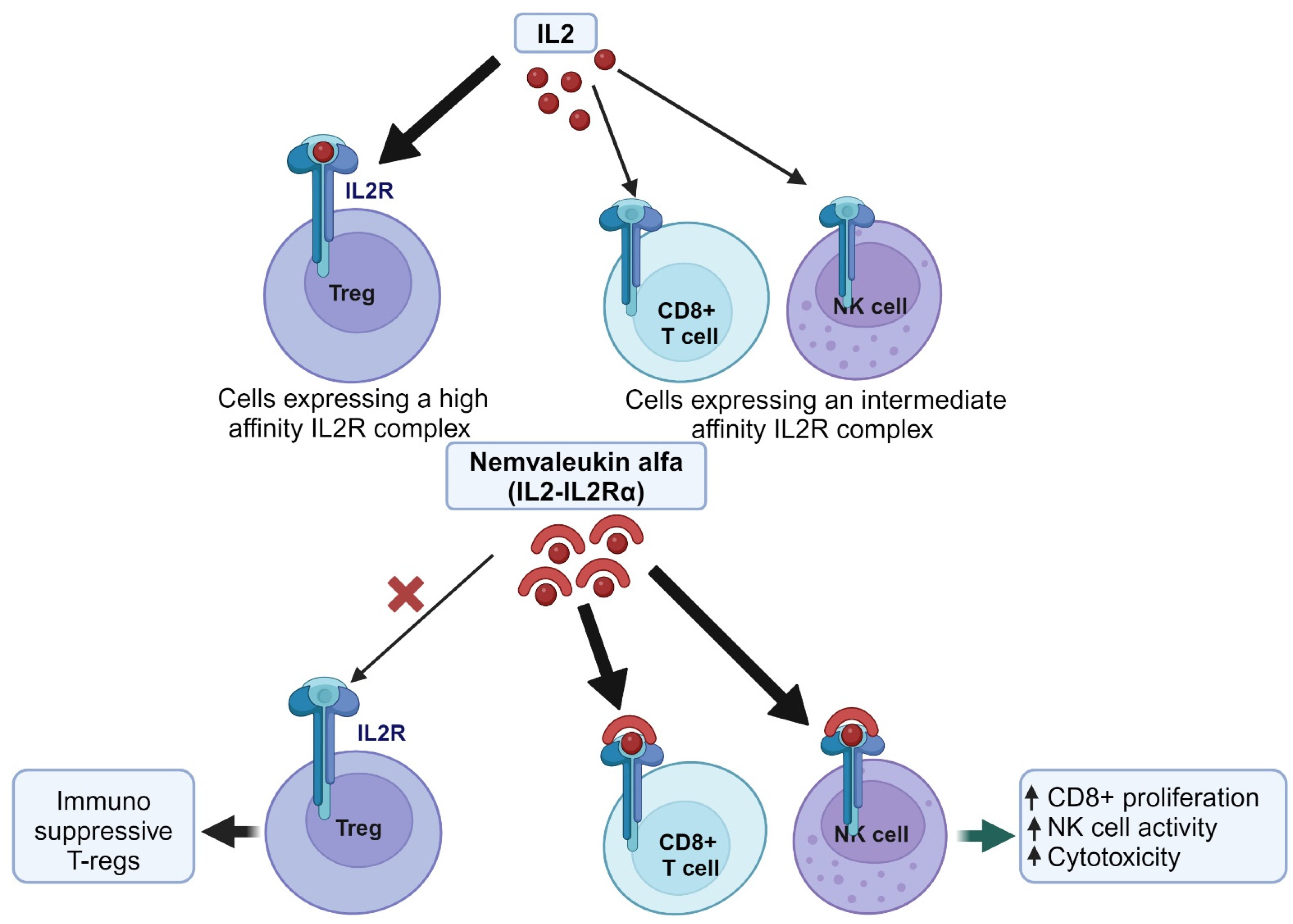
3. Re-Engineering Proteins for Drug Repurposing Using Nemvaleukin Alfa as a Case Study
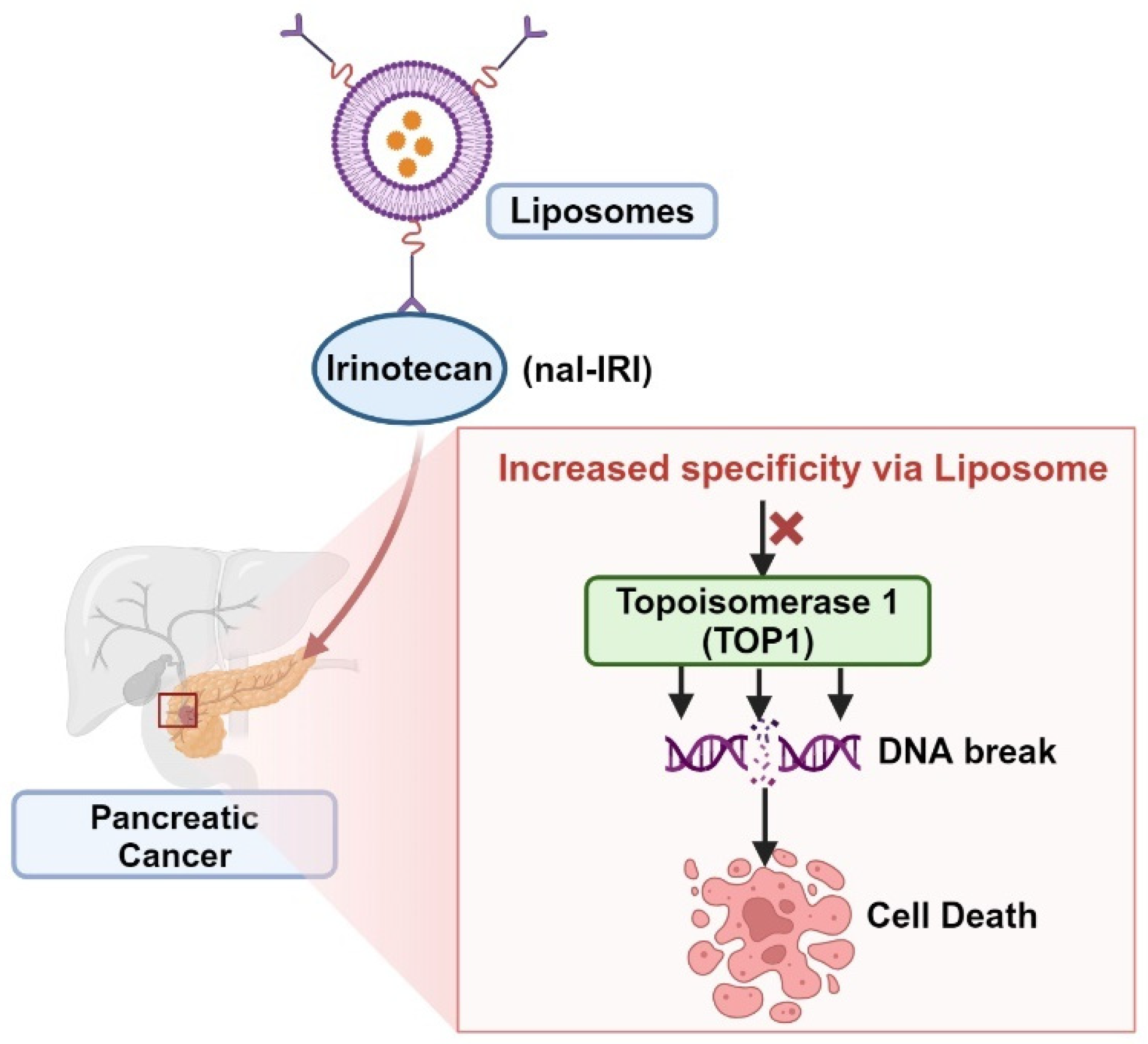
4. Repurposing Drugs with Nanoliposomes Using Pancreatic Cancer as a Case Study
5. Computational Approaches for Cancer Drug Repurposing
5.1. Genome-Wide Association Studies (GWAS)
5.2. Structure-Based Repurposing
5.3. Transcriptome-Based Drug Repurposing, Using Gastrointestinal Stromal Tumors (GISTs) as an Example
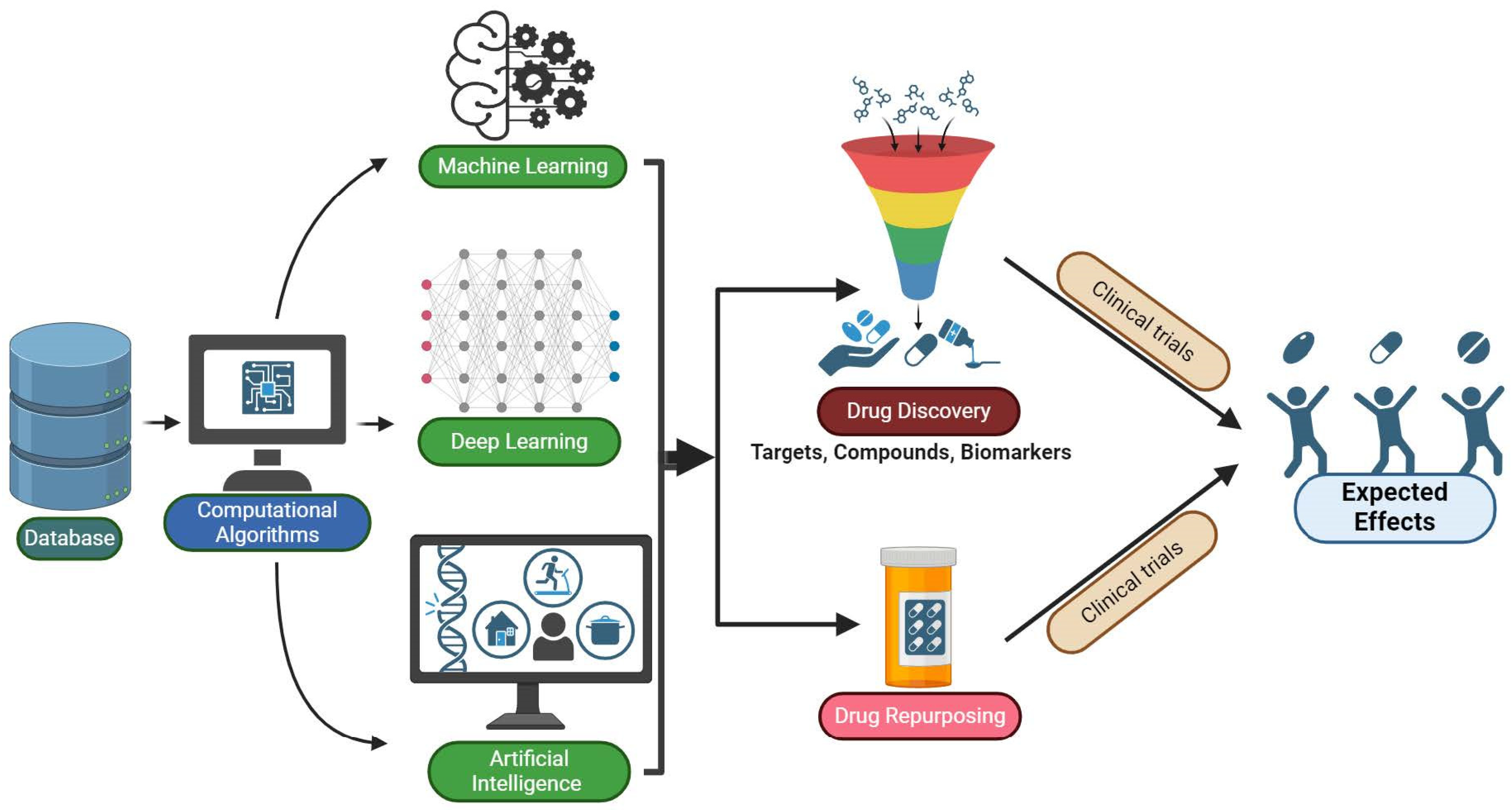
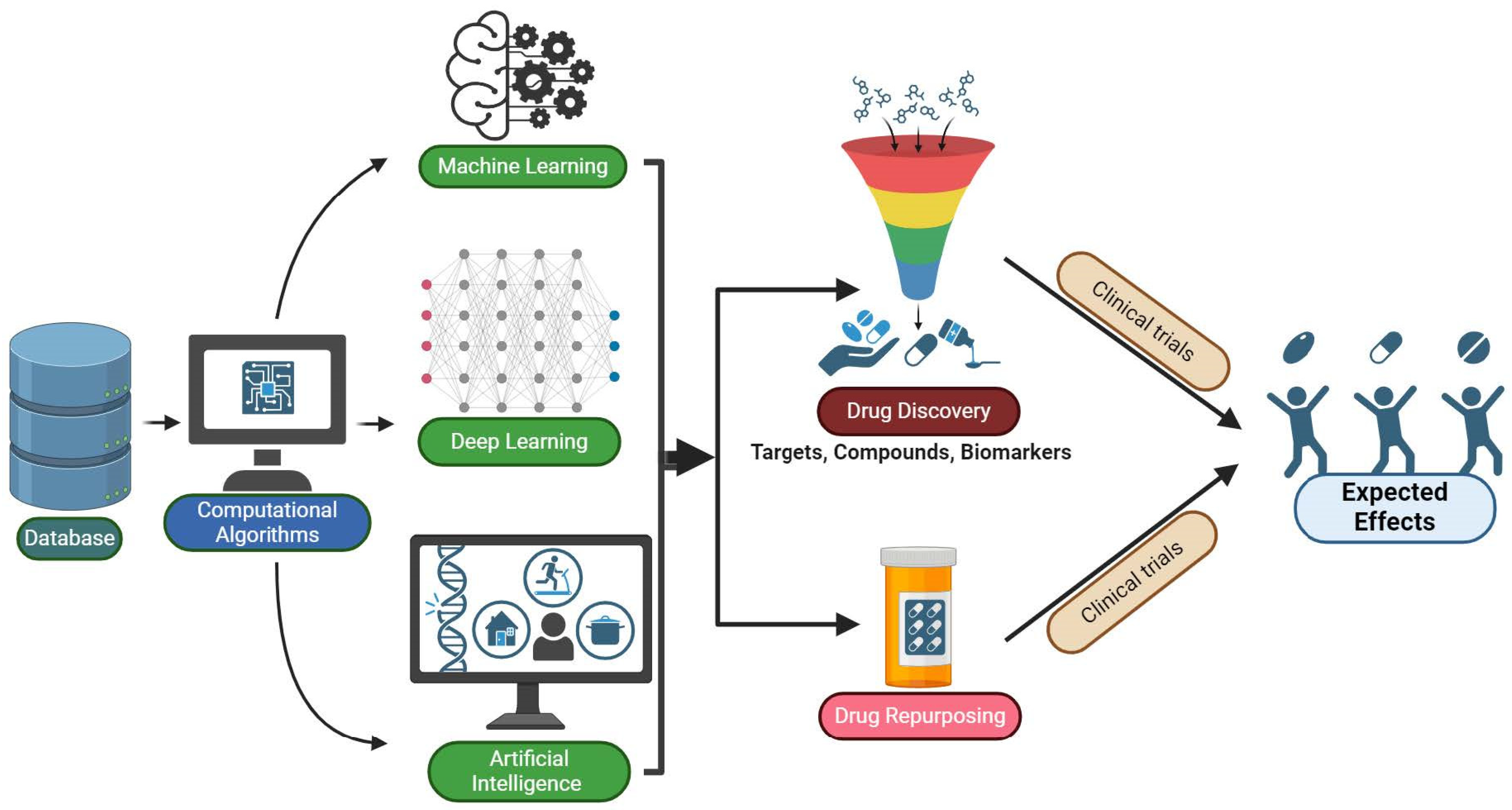
5.4. Machine Learning (ML), Artificial Intelligence (AI), and Deep Learning (DL)
6. Future Direction
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef]
- Elmore, L.W.; Greer, S.F.; Daniels, E.C.; Saxe, C.C.; Melner, M.H.; Krawiec, G.M.; Cance, W.G.; Phelps, W.C. Blueprint for cancer research: Critical gaps and opportunities. CA Cancer J. Clin. 2021, 71, 107–139. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Puhl, A.C.; Zorn, K.M.; Lane, T.R.; Russo, D.P.; Klein, J.J.; Hickey, A.J.; Clark, A.M. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 2019, 18, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S. Mining for therapeutic gold. Nat. Rev. Drug Discov. 2011, 10, 397. [Google Scholar] [CrossRef] [PubMed]
- Al-Dimassi, S.; Abou-Antoun, T.; El-Sibai, M. Cancer cell resistance mechanisms: A mini review. Clin. Transl. Oncol. 2014, 16, 511–516. [Google Scholar] [CrossRef]
- Doan, T.L.; Pollastri, M.; Walters, M.A.; Georg, G.I. The future of drug repositioning: Old drugs, new opportunities. In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2011; Volume 46, pp. 385–401. [Google Scholar]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar]
- Nosengo, N. New tricks for old drugs. Nature 2016, 534, 314–316. [Google Scholar] [CrossRef]
- Weth, F.R.; Hoggarth, G.B.; Weth, A.F.; Paterson, E.; White, M.P.; Tan, S.T.; Peng, L.; Gray, C. Unlocking hidden potential: Advancements, approaches, and obstacles in repurposing drugs for cancer therapy. Br. J. Cancer 2023, 130, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Sertkaya, A.; Wong, H.-H.; Jessup, A.; Beleche, T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin. Trials 2016, 13, 117–126. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.; Alagarsamy, V.; Solomon, V.; Jose, P.; Murugesan, S. Drug repurposing: An effective tool in modern drug discovery. Russ. J. Bioorganic Chem. 2023, 49, 157–166. [Google Scholar] [CrossRef]
- Quirke, V.M. Tamoxifen from failed contraceptive pill to best-selling breast cancer medicine: A case-study in pharmaceutical innovation. Front. Pharmacol. 2017, 8, 620. [Google Scholar] [CrossRef] [PubMed]
- Mottini, C.; Napolitano, F.; Li, Z.; Gao, X.; Cardone, L. Computer-aided drug repurposing for cancer therapy: Approaches and opportunities to challenge anticancer targets. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 68, pp. 59–74. [Google Scholar]
- Vatansever, S.; Schlessinger, A.; Wacker, D.; Kaniskan, H.Ü.; Jin, J.; Zhou, M.M.; Zhang, B. Artificial intelligence and machine learning-aided drug discovery in central nervous system diseases: State-of-the-arts and future directions. Med. Res. Rev. 2021, 41, 1427–1473. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, J.; Liddell, K.; Darrow, J.; Aboy, M.; Jordan, M.; Crespo, C.; Minssen, T. Repositioning Generic Drugs: Empirical Findings and Policy Implications. IIC Int. Rev. Intellect. Prop. Compet. Law 2022, 53, 1287–1322. [Google Scholar] [CrossRef]
- Murteira, S.; Millier, A.; Ghezaiel, Z.; Lamure, M. Drug reformulations and repositioning in the pharmaceutical industry and their impact on market access: Regulatory implications. J. Mark. Access Health Policy 2014, 2, 22813. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Dai, X.; Xu, Y.; Xing, G.; Liu, H.; Lu, T.; Chen, Y.; Zhang, Y. Drug repositioning: Progress and challenges in drug discovery for various diseases. Eur. J. Med. Chem. 2022, 234, 114239. [Google Scholar] [CrossRef]
- Zannetti, A. Breast Cancer: From Pathophysiology to Novel Therapeutic Approaches 2.0. Int. J. Mol. Sci. 2023, 24, 2542. [Google Scholar] [CrossRef] [PubMed]
- Nolan, E.; Lindeman, G.J.; Visvader, J.E. Deciphering breast cancer: From biology to the clinic. Cell 2023, 186, 1708–1728. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Rao, R. Subtype specific targeting of calcium signaling in breast cancer. Cell Calcium 2020, 85, 102109. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, M.A.; Gessner, A.; El-Najjar, N. Repurposing of Chronically Used Drugs in Cancer Therapy: A Chance to Grasp. Cancers 2023, 15, 3199. [Google Scholar] [CrossRef]
- Gales, L.; Forsea, L.; Mitrea, D.; Stefanica, I.; Stanculescu, I.; Mitrica, R.; Georgescu, M.; Trifanescu, O.; Anghel, R.; Serbanescu, L. Antidiabetics, Anthelmintics, Statins, and Beta-Blockers as Co-Adjuvant Drugs in Cancer Therapy. Medicina 2022, 58, 1239. [Google Scholar] [CrossRef]
- Duan, L.; Shen, H.; Zhao, G.; Yang, R.; Cai, X.; Zhang, L.; Jin, C.; Huang, Y. Inhibitory effect of Disulfiram/copper complex on non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 1010–1016. [Google Scholar] [CrossRef]
- Allensworth, J.L.; Evans, M.K.; Bertucci, F.; Aldrich, A.J.; Festa, R.A.; Finetti, P.; Ueno, N.T.; Safi, R.; McDonnell, D.P.; Thiele, D.J.; et al. Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer. Mol. Oncol. 2015, 9, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Shen, J.; Yan, H.; Gao, X.; Dong, T.; Wang, P.; Zhou, J. The Evolving Role of Disulfiram in Radiobiology and the Treatment of Breast Cancer. Onco Targets Ther. 2020, 13, 10441–10446. [Google Scholar] [CrossRef] [PubMed]
- Askgaard, G.; Friis, S.; Hallas, J.; Thygesen, L.C.; Pottegård, A. Use of disulfiram and risk of cancer: A population-based case-control study. Eur. J. Cancer Prev. 2014, 23, 225–232. [Google Scholar] [CrossRef]
- Han, D.; Wu, G.; Chang, C.; Zhu, F.; Xiao, Y.; Li, Q.; Zhang, T.; Zhang, L. Disulfiram inhibits TGF-β-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-κB/Snail pathway. Oncotarget 2015, 6, 40907–40919. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, D.; Ringler, J.; Chen, W.; Cui, Q.C.; Ethier, S.P.; Dou, Q.P.; Wu, G. Disulfiram treatment facilitates phosphoinositide 3-kinase inhibition in human breast cancer cells in vitro and in vivo. Cancer Res. 2010, 70, 3996–4004. [Google Scholar] [CrossRef] [PubMed]
- Swetha, K.L.; Sharma, S.; Chowdhury, R.; Roy, A. Disulfiram potentiates docetaxel cytotoxicity in breast cancer cells through enhanced ROS and autophagy. Pharmacol. Rep. PR 2020, 72, 1749–1765. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, H.L.; Wymant, J.M.; Solfa, F.; Hiscox, S.E.; Taylor, K.M.; Westwell, A.D.; Jones, A.T. Disulfiram-induced cytotoxicity and endo-lysosomal sequestration of zinc in breast cancer cells. Biochem. Pharmacol. 2015, 93, 332–342. [Google Scholar] [CrossRef]
- Lafi, Z.; Alshaer, W.; Gharaibeh, L.; Alqudah, D.A.; AlQuaissi, B.; Bashaireh, B.; Ibrahim, A.A. Synergistic combination of doxorubicin with hydralazine, and disulfiram against MCF-7 breast cancer cell line. PLoS ONE 2023, 18, e0291981. [Google Scholar] [CrossRef]
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A.P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165339. [Google Scholar] [CrossRef]
- Ko, M.; Makena, M.R.; Schiapparelli, P.; Suarez-Meade, P.; Mekile, A.X.; Lal, B.; Lopez-Bertoni, H.; Kozielski, K.L.; Green, J.J.; Laterra, J. The endosomal pH regulator NHE9 is a driver of stemness in glioblastoma. Proc. Natl. Acad. Sci. USA Nexus 2022, 1, pgac013. [Google Scholar] [CrossRef]
- Yip, N.C.; Fombon, I.S.; Liu, P.; Brown, S.; Kannappan, V.; Armesilla, A.L.; Xu, B.; Cassidy, J.; Darling, J.L.; Wang, W. Disulfiram modulated ROS-MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br. J. Cancer 2011, 104, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; An, X.; Zhang, D.; Li, Q.; Dai, X.; Yu, H.; Li, Z. Combination of the 6-thioguanine and disulfiram/Cu synergistically inhibits proliferation of triple-negative breast cancer cells by enhancing DNA damage and disrupting DNA damage checkpoint. Biochim. Biophys. Acta. Mol. Cell Res. 2022, 1869, 119169. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Guo, F.; Albers, A.E.; Sehouli, J.; Kaufmann, A.M. Disulfiram modulates ROS accumulation and overcomes synergistically cisplatin resistance in breast cancer cell lines. Biomed. Pharmacother. 2019, 113, 108727. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, J.Y.; Lee, N.; Oh, E.; Sung, D.; Cho, T.M.; Seo, J.H. Disulfiram suppresses cancer stem-like properties and STAT3 signaling in triple-negative breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 486, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Cho, Y.; Oh, E.; Lee, N.; An, H.; Sung, D.; Cho, T.M.; Seo, J.H. Disulfiram targets cancer stem-like properties and the HER2/Akt signaling pathway in HER2-positive breast cancer. Cancer Lett. 2016, 379, 39–48. [Google Scholar] [CrossRef]
- Fasehee, H.; Dinarvand, R.; Ghavamzadeh, A.; Esfandyari-Manesh, M.; Moradian, H.; Faghihi, S.; Ghaffari, S.H. Delivery of disulfiram into breast cancer cells using folate-receptor-targeted PLGA-PEG nanoparticles: In vitro and in vivo investigations. J. Nanobiotechnol. 2016, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Z.; Brown, S.; Kannappan, V.; Tawari, P.E.; Jiang, W.; Irache, J.M.; Tang, J.Z.; Armesilla, A.L.; Darling, J.L.; et al. Liposome encapsulated Disulfiram inhibits NFκB pathway and targets breast cancer stem cells in vitro and in vivo. Oncotarget 2014, 5, 7471–7485. [Google Scholar] [CrossRef]
- Liu, C.; Qiang, J.; Deng, Q.; Xia, J.; Deng, L.; Zhou, L.; Wang, D.; He, X.; Liu, Y.; Zhao, B.; et al. ALDH1A1 Activity in Tumor-Initiating Cells Remodels Myeloid-Derived Suppressor Cells to Promote Breast Cancer Progression. Cancer Res. 2021, 81, 5919–5934. [Google Scholar] [CrossRef]
- Xiao, P.; Tao, X.; Wang, H.; Liu, H.; Feng, Y.; Zhu, Y.; Jiang, Z.; Yin, T.; Zhang, Y.; He, H.; et al. Enzyme/pH dual stimuli-responsive nanoplatform co-deliver disulfiram and doxorubicin for effective treatment of breast cancer lung metastasis. Expert Opin. Drug Deliv. 2023, 20, 1015–1031. [Google Scholar] [CrossRef] [PubMed]
- Solak, K.; Mavi, A.; Yılmaz, B. Disulfiram-loaded functionalized magnetic nanoparticles combined with copper and sodium nitroprusside in breast cancer cells. Mater. Sci. Eng. C Mater Biol. Appl. 2021, 119, 111452. [Google Scholar] [CrossRef] [PubMed]
- Rolle, F.; Bincoletto, V.; Gazzano, E.; Rolando, B.; Lollo, G.; Stella, B.; Riganti, C.; Arpicco, S. Coencapsulation of disulfiram and doxorubicin in liposomes strongly reverses multidrug resistance in breast cancer cells. Int. J. Pharm. 2020, 580, 119191. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; An, X.; Fu, C.; Yu, H.; Zhang, D.; Li, Q.; Man, X.; Dai, X.; Li, Z. Disulfiram/Copper Induce Ferroptosis in Triple-Negative Breast Cancer Cell Line MDA-MB-231. Front. Biosci. 2023, 28, 186. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, N.; Kim, Y.J.; Cho, Y.; An, H.; Oh, E.; Cho, T.M.; Sung, D.; Seo, J.H. Disulfiram induces anoikis and suppresses lung colonization in triple-negative breast cancer via calpain activation. Cancer Lett. 2017, 386, 151–160. [Google Scholar] [CrossRef] [PubMed]
- El Fawal, G.; Abu-Serie, M.M.; El-Gendi, H.; El-Fakharany, E.M. Fabrication, characterization and in vitro evaluation of disulfiram-loaded cellulose acetate/poly(ethylene oxide) nanofiber scaffold for breast and colon cancer cell lines treatment. Int. J. Biol. Macromol. 2022, 204, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Drum, D.L.; Sun, R.; Zhang, Y.; Chen, F.; Sun, F.; Dal, E.; Yu, L.; Jia, J.; Arya, S.; et al. Stressed target cancer cells drive nongenetic reprogramming of CAR T cells and solid tumor microenvironment. Nat. Commun. 2023, 14, 5727. [Google Scholar] [CrossRef]
- Couto, R.D.; Fernandes, B.J.D. Low Doses Naltrexone: The Potential Benefit Effects for its Use in Patients with Cancer. Curr Drug Res. Rev. 2021, 13, 86–89. [Google Scholar] [CrossRef]
- Vijayakumar, J.; Haddad, T.; Gupta, K.; Sauers, J.; Yee, D. An open label phase II study of safety and clinical activity of naltrexone for treatment of hormone refractory metastatic breast cancer. Investig. New Drugs 2023, 41, 70–75. [Google Scholar] [CrossRef]
- Murugan, S.; Rousseau, B.; Sarkar, D.K. Beta 2 Adrenergic Receptor Antagonist Propranolol and Opioidergic Receptor Antagonist Naltrexone Produce Synergistic Effects on Breast Cancer Growth Prevention by Acting on Cancer Cells and Immune Environment in a Preclinical Model of Breast Cancer. Cancers 2021, 13, 4858. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.Y.; Jung, B.K.; Hong, S.J. Albendazole and Mebendazole as Anti-Parasitic and Anti-Cancer Agents: An Update. Korean J. Parasitol. 2021, 59, 189–225. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Baird, S.K. Treatment of breast and colon cancer cell lines with anti-helmintic benzimidazoles mebendazole or albendazole results in selective apoptotic cell death. J. Cancer Res. Clin. Oncol. 2021, 147, 2945–2953. [Google Scholar] [CrossRef] [PubMed]
- Jubie, S.; Durai, U.; Latha, S.; Ayyamperumal, S.; Wadhwani, A.; Prabha, T. Repurposing of Benzimidazole Scaffolds for HER2 Positive Breast Cancer Therapy: An In-Silico Approach. Curr Drug Res Rev 2021, 13, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Racoviceanu, R.; Trandafirescu, C.; Voicu, M.; Ghiulai, R.; Borcan, F.; Dehelean, C.; Watz, C.; Aigner, Z.; Ambrus, R.; Coricovac, D.E.; et al. Solid Polymeric Nanoparticles of Albendazole: Synthesis, Physico-Chemical Characterization and Biological Activity. Molecules 2020, 25, 5130. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sun, H.; Zhang, B.; Liu, S.; Deng, S.; Weng, Z.; Zuo, B.; Yang, J.; He, Y. (18)F-FDG PET imaging for monitoring the early anti-tumor effect of albendazole on triple-negative breast cancer. Breast Cancer 2020, 27, 372–380. [Google Scholar] [CrossRef]
- Priotti, J.; Baglioni, M.V.; García, A.; Rico, M.J.; Leonardi, D.; Lamas, M.C.; Menacho Márquez, M. Repositioning of Anti-parasitic Drugs in Cyclodextrin Inclusion Complexes for Treatment of Triple-Negative Breast Cancer. AAPS PharmSciTech 2018, 19, 3734–3741. [Google Scholar] [CrossRef]
- Joe, N.S.; Godet, I.; Milki, N.; Ain, N.U.I.; Oza, H.H.; Riggins, G.J.; Gilkes, D.M. Mebendazole prevents distant organ metastases in part by decreasing ITGβ4 expression and cancer stemness. Breast Cancer Res. BCR 2022, 24, 98. [Google Scholar] [CrossRef] [PubMed]
- Joe, N.S.; Wang, Y.; Oza, H.H.; Godet, I.; Milki, N.; Riggins, G.J.; Gilkes, D.M. Mebendazole Treatment Disrupts the Transcriptional Activity of Hypoxia-Inducible Factors 1 and 2 in Breast Cancer Cells. Cancers 2023, 15, 1330. [Google Scholar] [CrossRef] [PubMed]
- Kefayat, A.; Hosseini, M.; Ghahremani, F.; Jolfaie, N.A.; Rafienia, M. Biodegradable and biocompatible subcutaneous implants consisted of pH-sensitive mebendazole-loaded/folic acid-targeted chitosan nanoparticles for murine triple-negative breast cancer treatment. J. Nanobiotechnol. 2022, 20, 169. [Google Scholar] [CrossRef] [PubMed]
- Elmaaty, A.A.; Darwish, K.M.; Chrouda, A.; Boseila, A.A.; Tantawy, M.A.; Elhady, S.S.; Shaik, A.B.; Mustafa, M.; Al-Karmalawy, A.A. In Silico and In Vitro Studies for Benzimidazole Anthelmintics Repurposing as VEGFR-2 Antagonists: Novel Mebendazole-Loaded Mixed Micelles with Enhanced Dissolution and Anticancer Activity. ACS Omega 2022, 7, 875–899. [Google Scholar] [CrossRef]
- Zhang, L.; Bochkur Dratver, M.; Yazal, T.; Dong, K.; Nguyen, A.; Yu, G.; Dao, A.; Bochkur Dratver, M.; Duhachek-Muggy, S.; Bhat, K.; et al. Mebendazole Potentiates Radiation Therapy in Triple-Negative Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 195–207. [Google Scholar] [CrossRef]
- Choi, H.S.; Ko, Y.S.; Jin, H.; Kang, K.M.; Ha, I.B.; Jeong, H.; Song, H.N.; Kim, H.J.; Jeong, B.K. Anticancer Effect of Benzimidazole Derivatives, Especially Mebendazole, on Triple-Negative Breast Cancer (TNBC) and Radiotherapy-Resistant TNBC In Vivo and In Vitro. Molecules 2021, 26, 5118. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Ko, Y.S.; Jin, H.; Kang, K.M.; Ha, I.B.; Jeong, H.; Lee, J.H.; Jeong, B.K.; Kim, H.J. Mebendazole Increases Anticancer Activity of Radiotherapy in Radiotherapy-Resistant Triple-Negative Breast Cancer Cells by Enhancing Natural Killer Cell-Mediated Cytotoxicity. Int. J. Mol. Sci. 2022, 23, 15493. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Yuan, Z.; Zhang, J.; Chen, Y.; Fu, Y.; Liu, Y.; Fu, L.; Zhang, L.; Zhou, X.L. Flubendazole induces mitochondrial dysfunction and DRP1-mediated mitophagy by targeting EVA1A in breast cancer. Cell Death Dis. 2022, 13, 375. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Zhao, R.; Wang, M.; Jiang, X.; Gao, F.; Fu, L.; Zhang, L.; Zhou, X.L. Flubendazole elicits anti-cancer effects via targeting EVA1A-modulated autophagy and apoptosis in Triple-negative Breast Cancer. Theranostics 2020, 10, 8080–8097. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Guo, M.; Li, J.; Zheng, Y.; Zhang, S.; Xie, T.; Liu, B. Systems biology-based discovery of a potential Atg4B agonist (Flubendazole) that induces autophagy in breast cancer. Mol. Biosyst. 2015, 11, 2860–2866. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.J.; Luo, X.; Zhang, W.; Peng, F.; Cui, B.; Wu, S.J.; Zheng, F.M.; Xu, J.; Xu, L.Z.; Long, Z.J.; et al. Flubendazole, FDA-approved anthelmintic, targets breast cancer stem-like cells. Oncotarget 2015, 6, 6326–6340. [Google Scholar] [CrossRef]
- Kim, Y.J.; Sung, D.; Oh, E.; Cho, Y.; Cho, T.M.; Farrand, L.; Seo, J.H.; Kim, J.Y. Flubendazole overcomes trastuzumab resistance by targeting cancer stem-like properties and HER2 signaling in HER2-positive breast cancer. Cancer Lett. 2018, 412, 118–130. [Google Scholar] [CrossRef]
- Oh, E.; Kim, Y.J.; An, H.; Sung, D.; Cho, T.M.; Farrand, L.; Jang, S.; Seo, J.H.; Kim, J.Y. Flubendazole elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Int J Cancer 2018, 143, 1978–1993. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liao, M.; Li, Z.; Ye, J.; Wu, L.; Mou, Y.; Fu, L.; Zhen, Y. Flubendazole Enhances the Inhibitory Effect of Paclitaxel via HIF1α/PI3K/AKT Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 15121. [Google Scholar] [CrossRef]
- Gatla, H.R.; Muniraj, N.; Thevkar, P.; Yavvari, S.; Sukhavasi, S.; Makena, M.R. Regulation of chemokines and cytokines by histone deacetylases and an update on histone decetylase inhibitors in human diseases. Int. J. Mol. Sci. 2019, 20, 1110. [Google Scholar] [CrossRef] [PubMed]
- Aroosa, M.; Malik, J.A.; Ahmed, S.; Bender, O.; Ahemad, N.; Anwar, S. The evidence for repurposing anti-epileptic drugs to target cancer. Mol. Biol. Rep. 2023, 50, 7667–7680. [Google Scholar] [CrossRef] [PubMed]
- Caponigro, F.; Di Gennaro, E.; Ionna, F.; Longo, F.; Aversa, C.; Pavone, E.; Maglione, M.G.; Di Marzo, M.; Muto, P.; Cavalcanti, E.; et al. Phase II clinical study of valproic acid plus cisplatin and cetuximab in recurrent and/or metastatic squamous cell carcinoma of Head and Neck-V-CHANCE trial. BMC Cancer 2016, 16, 918. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Halasa, M.; Okon, E.; Kukula-Koch, W.; Stepulak, A. Valproic Acid and Breast Cancer: State of the Art in 2021. Cancers 2021, 13, 3409. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, L.; Zhang, S.; Song, J.; Sun, H.; Shan, C.; Wang, D.; Liu, S. Valproic acid Suppresses Breast Cancer Cell Growth Through Triggering Pyruvate Kinase M2 Isoform Mediated Warburg Effect. Cell Transpl. 2021, 30, 9636897211027524. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.; Paolì, A.; Forastiero, M.; Marsico, S.; De Amicis, F.; Marrelli, M.; Naimo, G.D.; Mauro, L.; Panno, M.L. Valproic acid inhibits cell growth in both MCF-7 and MDA-MB231 cells by triggering different responses in a cell type-specific manner. J. Transl. Med. 2023, 21, 165. [Google Scholar] [CrossRef] [PubMed]
- Injinari, N.; Amini-Farsani, Z.; Yadollahi-Farsani, M.; Teimori, H. Apoptotic effects of valproic acid on miR-34a, miR-520h and HDAC1 gene in breast cancer. Life Sci. 2021, 269, 119027. [Google Scholar] [CrossRef] [PubMed]
- Meschi, M.; Khorsandi, K.; Kianmehr, Z. The Effect of Berberine Follow by Blue Light Irradiation and Valproic Acid on the Growth Inhibition of MDA-MB-231 Breast Cancer Cells. Appl. Biochem. Biotechnol. 2023, 195, 6752–6767. [Google Scholar] [CrossRef]
- Granit, A.; Mishra, K.; Barasch, D.; Peretz-Yablonsky, T.; Eyal, S.; Kakhlon, O. Metabolomic profiling of triple negative breast cancer cells suggests that valproic acid can enhance the anticancer effect of cisplatin. Front. Cell Dev. Biol. 2022, 10, 1014798. [Google Scholar] [CrossRef] [PubMed]
- Granit Mizrahi, A.; Gugenheim, A.; Hamad, H.; Hamed, R.; Tetro, N.; Maimon, O.; Khutsurauli, S.; Nechushtan, H.; Nisman, B.; Duran, D.; et al. Valproic acid reprograms the metabolic aberration of cisplatin treatment via ALDH modulation in triple-negative breast cancer cells. Front. Cell Dev. Biol. 2023, 11, 1217149. [Google Scholar] [CrossRef]
- El Said, H.H.; Badary, O.A.; Shouman, S.A.; Elmazar, M.M.; El-Khatib, A.S. Enhanced antitumor activity of combined methotrexate and histone deacetylase inhibitor valproic acid on mammary cancer in vitro and in vivo. Can. J. Physiol. Pharmacol. 2022, 100, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Laengle, J.; Kabiljo, J.; Hunter, L.; Homola, J.; Prodinger, S.; Egger, G.; Bergmann, M. Histone deacetylase inhibitors valproic acid and vorinostat enhance trastuzumab-mediated antibody-dependent cell-mediated phagocytosis. J. Immunother. Cancer 2020, 8, e000195. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Lim, D.; Tian, Z.; Liu, G.; Ding, C.; Cai, Z.; Chen, C.; Zhang, F.; Feng, Z. Valproic Acid Regulates HR and Cell Cycle Through MUS81-pRPA2 Pathway in Response to Hydroxyurea. Front. Oncol. 2021, 11, 681278. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Duan, W.; Cai, Z.; Lim, D.; Feng, Z. Valproic acid triggers radiation-induced abscopal effect by modulating the unirradiated tumor immune microenvironment in a rat model of breast cancer. J. Radiat. Res. 2021, 62, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Rege, J. Re-engineering proteins to develop novel immunotherapies. Eur. Pharm. Rev. 2023, 28, 41–42. [Google Scholar]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Ko, B.; Takebe, N.; Andrews, O.; Makena, M.R.; Chen, A.P. Rethinking Oncologic Treatment Strategies with Interleukin-2. Cells 2023, 12, 1316. [Google Scholar] [CrossRef]
- Vaishampayan, U.N.; Tomczak, P.; Muzaffar, J.; Winer, I.S.; Rosen, S.D.; Hoimes, C.J.; Chauhan, A.; Spreafico, A.; Lewis, K.D.; Bruno, D.S. Nemvaleukin alfa monotherapy and in combination with pembrolizumab in patients (pts) with advanced solid tumors: ARTISTRY-1. Am. Soc. Clin. Oncol. 2022, 40, 2500. [Google Scholar] [CrossRef]
- Pramanik, K.C.; Makena, M.R.; Bhowmick, K.; Pandey, M.K. Advancement of NF-κB signaling pathway: A novel target in pancreatic cancer. Int. J. Mol. Sci. 2018, 19, 3890. [Google Scholar] [CrossRef]
- Ram Makena, M.; Gatla, H.; Verlekar, D.; Sukhavasi, S.; Pandey, M.K.; Pramanik, K.C. Wnt/β-catenin signaling: The culprit in pancreatic carcinogenesis and therapeutic resistance. Int. J. Mol. Sci. 2019, 20, 4242. [Google Scholar] [CrossRef] [PubMed]
- Milano, G.; Innocenti, F.; Minami, H. Liposomal irinotecan (Onivyde): Exemplifying the benefits of nanotherapeutic drugs. Cancer Sci. 2022, 113, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Wang, J.; Makena, M.R.; Lee, J.-S.; Paz, N.; Hall, C.P.; Song, M.M.; Calderon, R.I.; Cruz, R.E.; Hindle, A. Activity of MM-398, nanoliposomal irinotecan (nal-IRI), in Ewing’s family tumor xenografts is associated with high exposure of tumor to drug and high SLFN11 expression. Clin. Cancer Res. 2015, 21, 1139–1150. [Google Scholar] [CrossRef] [PubMed]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.-S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; De Jesus-Acosta, A.; Zheng, L.; Lee, V.; Kamel, I.; Le, D.; Pishvaian, M.; Laheru, D. Clinical outcomes of liposomal irinotecan in advanced pancreatic adenocarcinoma patients previously treated with conventional irinotecan-based chemotherapy: A real-world study. Front. Oncol. 2023, 13, 1250136. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Melisi, D.; Macarulla, T.; Pazo-Cid, R.; Chandana, S.R.; De La Fouchardiere, C.; Dean, A.P.; Kiss, I.; Lee, W.; Goetze, T.O. NAPOLI-3: A randomized, open-label phase 3 study of liposomal irinotecan+ 5-fluorouracil/leucovorin+ oxaliplatin (NALIRIFOX) versus nab-paclitaxel+ gemcitabine in treatment-naïve patients with metastatic pancreatic ductal adenocarcinoma (mPDAC). Am. Soc. Clin. Oncol. 2023, 41, LBA661. [Google Scholar] [CrossRef]
- Liu, A. With FDA Nod for Onivyde Combo, Ipsen Ends 10-Year Drought in Newly Diagnosed Pancreatic Cancer; FIERCE PHARMA: New York, NY, USA, 2024; Available online: https://www.fiercepharma.com/pharma/ipsen-ends-10-year-drought-fda-nod-onivyde-combo-newly-diagnosed-pancreatic-cancer (accessed on 13 February 2024).
- Reay, W.R.; Cairns, M.J. Advancing the use of genome-wide association studies for drug repurposing. Nat. Rev. Genet. 2021, 22, 658–671. [Google Scholar] [CrossRef]
- Cheng, F.; Lu, W.; Liu, C.; Fang, J.; Hou, Y.; Handy, D.E.; Wang, R.; Zhao, Y.; Yang, Y.; Huang, J. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat. Commun. 2019, 10, 3476. [Google Scholar] [CrossRef]
- Shaikh, N.; Linthoi, R.; Swamy, K.; Karthikeyan, M.; Vyas, R. Comprehensive molecular docking and dynamic simulations for drug repurposing of clinical drugs against multiple cancer kinase targets. J. Biomol. Struct. Dyn. 2023, 41, 7735–7743. [Google Scholar] [CrossRef]
- Oyedele, A.-Q.K.; Ogunlana, A.T.; Boyenle, I.D.; Adeyemi, A.O.; Rita, T.O.; Adelusi, T.I.; Abdul-Hammed, M.; Elegbeleye, O.E.; Odunitan, T.T. Docking covalent targets for drug discovery: Stimulating the computer-aided drug design community of possible pitfalls and erroneous practices. Mol. Divers. 2023, 27, 1879–1903. [Google Scholar] [CrossRef] [PubMed]
- John Harris, C.; D Hill, R.; W Sheppard, D.; J Slater, M.; FW Stouten, P. The design and application of target-focused compound libraries. Comb. Chem. High Throughput Screen. 2011, 14, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.-h.; Liu, J.-x.; Liu, Y.; Chen, S.-w.; Dai, W.-t.; Xiao, Z.-X.; Cao, Y. DrugRep: An automatic virtual screening server for drug repurposing. Acta Pharmacol. Sin. 2023, 44, 888–896. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Duo, H.; Hao, Y.; Zhang, X.; Zhou, X.; Zeng, Y.; Li, Y.; Li, B. Computational drug repurposing by exploiting large-scale gene expression data: Strategy, methods and applications. Comput. Biol. Med. 2023, 155, 106671. [Google Scholar] [CrossRef]
- Iorio, F.; Rittman, T.; Ge, H.; Menden, M.; Saez-Rodriguez, J. Transcriptional data: A new gateway to drug repositioning? Drug Discov. Today 2013, 18, 350–357. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Quek, R.; George, S. Update on the treatment of gastrointestinal stromal tumors (GISTs): Role of imatinib. Biol. Targets Ther. 2010, 4, 19–31. [Google Scholar]
- Lopes, L.F.; Bacchi, C.E. Imatinib treatment for gastrointestinal stromal tumour (GIST). J. Cell. Mol. Med. 2010, 14, 42–50. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, R.; Payra, S.; Singh, S.K. Artificial Intelligence and Machine Learning in Pharmacological Research: Bridging the Gap Between Data and Drug Discovery. Cureus 2023, 15, e44359. [Google Scholar] [CrossRef] [PubMed]
- Taye, M.M. Understanding of Machine Learning with Deep Learning: Architectures, Workflow, Applications and Future Directions. Computers 2023, 12, 91. [Google Scholar] [CrossRef]
- Kolluri, S.; Lin, J.; Liu, R.; Zhang, Y.; Zhang, W. Machine learning and artificial intelligence in pharmaceutical research and development: A review. AAPS J. 2022, 24, 19. [Google Scholar] [CrossRef] [PubMed]
- Tanoli, Z.; Vähä-Koskela, M.; Aittokallio, T. Artificial intelligence, machine learning, and drug repurposing in cancer. Expert Opin. Drug Discov. 2021, 16, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Soomro, A.M.; Salih, A.R.C.; Samantasinghar, A.; Asif, A.; Kang, I.S.; Choi, K.H. A comprehensive review of artificial intelligence and network based approaches to drug repurposing in COVID-19. Biomed. Pharmacother. 2022, 153, 113350. [Google Scholar] [CrossRef]
- Wieder, R.; Adam, N. Drug repositioning for cancer in the era of AI, big omics, and real-world data. Crit. Rev. Oncol. Hematol. 2022, 175, 103730. [Google Scholar] [CrossRef]
- Bhinder, B.; Gilvary, C.; Madhukar, N.S.; Elemento, O. Artificial intelligence in cancer research and precision medicine. Cancer Discov. 2021, 11, 900–915. [Google Scholar] [CrossRef]
- Zeng, J.; Cruz Pico, C.X.; Saridogan, T.; Shufean, M.A.; Kahle, M.; Yang, D.; Shaw, K.; Meric-Bernstam, F. Natural language processing–assisted literature retrieval and analysis for combination therapy in cancer. JCO Clin. Cancer Inform. 2022, 6, e2100109. [Google Scholar] [CrossRef]
- Ryu, A.J.; Kumar, S.; Dispenzieri, A.; Kyle, R.A.; Rajkumar, S.V.; Kingsley, T.C. Artificial intelligence-enabled screening strategy for drug repurposing in monoclonal gammopathy of undetermined significance. Blood Cancer J. 2023, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Mayers, M.; Li, T.S.; Queralt-Rosinach, N.; Su, A.I. Time-resolved evaluation of compound repositioning predictions on a text-mined knowledge network. BMC Bioinform. 2019, 20, 653. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malla, R.; Viswanathan, S.; Makena, S.; Kapoor, S.; Verma, D.; Raju, A.A.; Dunna, M.; Muniraj, N. Revitalizing Cancer Treatment: Exploring the Role of Drug Repurposing. Cancers 2024, 16, 1463. https://doi.org/10.3390/cancers16081463
Malla R, Viswanathan S, Makena S, Kapoor S, Verma D, Raju AA, Dunna M, Muniraj N. Revitalizing Cancer Treatment: Exploring the Role of Drug Repurposing. Cancers. 2024; 16(8):1463. https://doi.org/10.3390/cancers16081463
Chicago/Turabian StyleMalla, RamaRao, Sathiyapriya Viswanathan, Sree Makena, Shruti Kapoor, Deepak Verma, Alluri Ashok Raju, Manikantha Dunna, and Nethaji Muniraj. 2024. "Revitalizing Cancer Treatment: Exploring the Role of Drug Repurposing" Cancers 16, no. 8: 1463. https://doi.org/10.3390/cancers16081463
APA StyleMalla, R., Viswanathan, S., Makena, S., Kapoor, S., Verma, D., Raju, A. A., Dunna, M., & Muniraj, N. (2024). Revitalizing Cancer Treatment: Exploring the Role of Drug Repurposing. Cancers, 16(8), 1463. https://doi.org/10.3390/cancers16081463