




Effects of Peptide Receptor Radiotherapy in Patients with Advanced Paraganglioma and Pheochromocytoma: A Nation-Wide Cohort Study
 , ,
, ,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Patients
2.2. PRRT
2.3. Outcome
2.4. Ethics
2.5. Statistics
3. Results
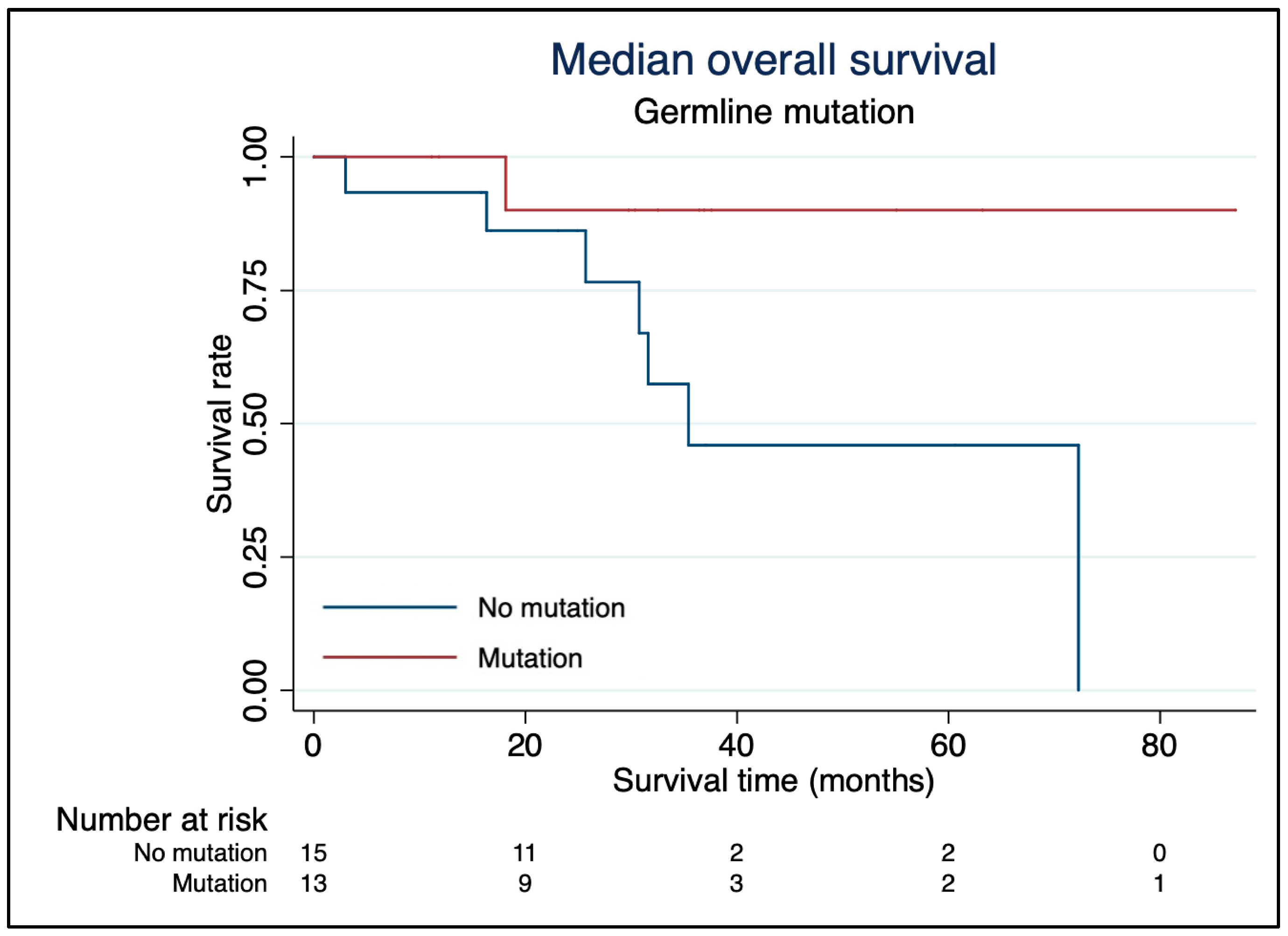
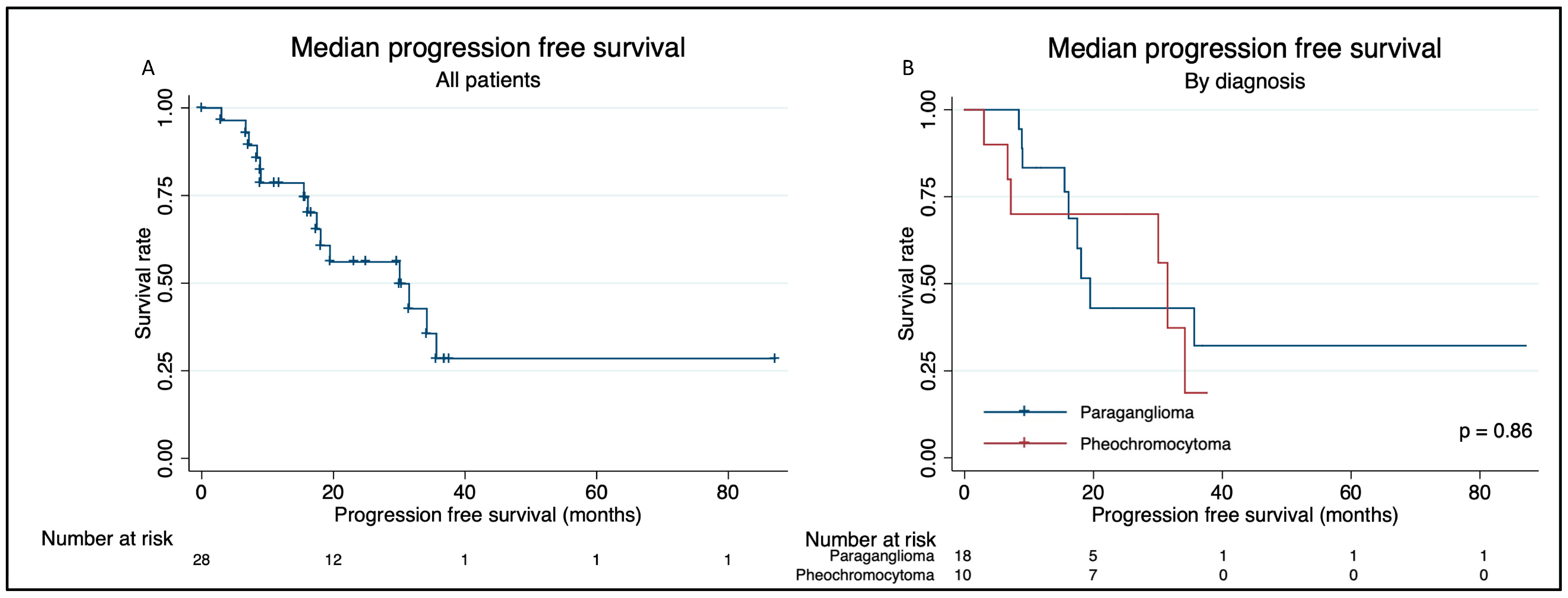
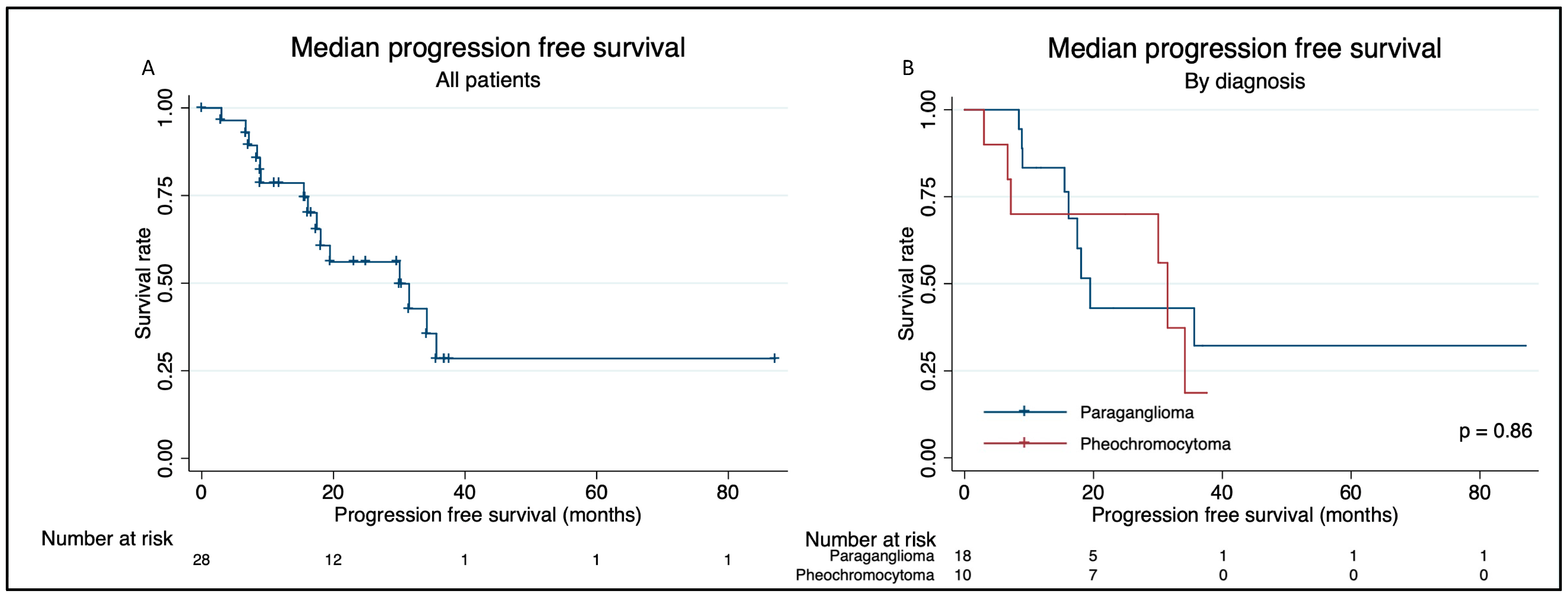
Overall Survival and Progression-Free Survival
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ebbehoj, A.; Stochholm, K.; Jacobsen, S.F.; Trolle, C.; Jepsen, P.; Robaczyk, M.G.; Rasmussen, Å.K.; Feldt-Rasmussen, U.; Thomsen, R.W.; Søndergaard, E.; et al. Incidence and Clinical Presentation of Pheochromocytoma and Sympathetic Paraganglioma: A Population-based Study. J. Clin. Endocrinol. Metab. 2021, 106, e2251–e2261. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.M.; Duh, Q.-Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.-P.; Grebe, S.K.G.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Goldner, W.S.; Benson, A.B.; Bergsland, E.; Blaszkowsky, L.S.; Brock, P.; Chan, J.; Das, S.; Dickson, P.V.; Fanta, P.; et al. Neuroendocrine and Adrenal Tumors, Version 2.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 839–868. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Phay, J.E.; Yen, T.W.F.; Dickson, P.V.; Wang, T.S.; Garcia, R.; Yang, A.D.; Solórzano, C.C.; Kim, L.T. Update on Pheochromocytoma and Paraganglioma from the SSO Endocrine/Head and Neck Disease-Site Work Group. Part 1 of 2: Advances in Pathogenesis and Diagnosis of Pheochromocytoma and Paraganglioma. Ann. Surg. Oncol. 2020, 27, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar] [CrossRef]
- Patel, D.; Phay, J.E.; Yen, T.W.F.; Dickson, P.V.; Wang, T.S.; Garcia, R.; Yang, A.D.; Kim, L.T.; Solórzano, C.C. Update on Pheochromocytoma and Paraganglioma from the SSO Endocrine and Head and Neck Disease Site Working Group, Part 2 of 2: Perioperative Management and Outcomes of Pheochromocytoma and Paraganglioma. Ann. Surg. Oncol. 2020, 27, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cui, Y.; Zhang, D.; Tong, A. Efficacy and Safety of Tyrosine Kinase Inhibitors in Patients with Metastatic Pheochromocytomas/Paragangliomas. J. Clin. Endocrinol. Metab. 2023, 108, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Baudin, E.; Goichot, B.; Berruti, A.; Hadoux, J.; Moalla, S.; Laboureau, S.; Noelting, S.; de la Fouchardière, C.; Kienitz, T.; Deutschbein, T.; et al. First International Randomized Study in Malignant Progressive Pheochromocytoma and Paragangliomas (FIRSTMAPPP): An academic double-blind trial investigating sunitinib. Ann. Oncol. 2021, 32 (Suppl. S5), S621. [Google Scholar] [CrossRef]
- Rufini, V.; Shulkin, B. The evolution in the use of MIBG in more than 25 years of experimental and clinical applications. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 341–350. Available online: http://www.ncbi.nlm.nih.gov/pubmed/19088689 (accessed on 11 December 2023). [PubMed]
- Kjaer, A.; Knigge, U. Use of radioactive substances in diagnosis and treatment of neuroendocrine tumors. Scand. J. Gastroenterol. 2015, 50, 740–747. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Prado-Wohlwend, S.; del Olmo-García, M.I.; Bello-Arques, P.; Merino-Torres, J.F. Response to targeted radionuclide therapy with [131I]MIBG AND [177Lu]Lu-DOTA-TATE according to adrenal vs. extra-adrenal primary location in metastatic paragangliomas and pheochromocytomas: A systematic review. Front. Endocrinol. 2022, 13, 957172. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Taïeb, D.; Carrasquillo, J.A.; Pryma, D.A.; Patel, M.; Millo, C.; de Herder, W.W.; Del Rivero, J.; Crona, J.; Shulkin, B.L.; et al. High-Specific-Activity-131I-MIBG versus 177Lu-DOTATATE Targeted Radionuclide Therapy for Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2021, 27, 2989–2995. [Google Scholar] [CrossRef]
- Zandee, W.T.; Feelders, R.A.; Smit Duijzentkunst, D.A.; Hofland, J.; Metselaar, R.M.; A Oldenburg, R.; van Linge, A.; Kam, B.L.R.; Teunissen, J.J.M.; Korpershoek, E.; et al. Treatment of inoperable or metastatic paragangliomas and pheochromocytomas with peptide receptor radionuclide therapy using 177Lu-DOTATATE. Eur. J. Endocrinol. 2019, 181, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Severi, S.; Bongiovanni, A.; Ferrara, M.; Nicolini, S.; Di Mauro, F.; Sansovini, M.; Lolli, I.; Tardelli, E.; Cittanti, C.; Di Iorio, V.; et al. Peptide receptor radionuclide therapy in patients with metastatic progressive pheochromocytoma and paraganglioma: Long-term toxicity, efficacy and prognostic biomarker data of phase II clinical trials. ESMO Open 2021, 6, 100171. [Google Scholar] [CrossRef] [PubMed]
- Vyakaranam, A.R.; Crona, J.; Norlén, O.; Granberg, D.; Garske-Román, U.; Sandström, M.; Fröss-Baron, K.; Thiis-Evensen, E.; Hellman, P.; Sundin, A. Favorable Outcome in Patients with Pheochromocytoma and Paraganglioma Treated with 177Lu-DOTATATE. Cancers 2019, 11, 909. [Google Scholar] [CrossRef] [PubMed]
- Kolasinska-ćwikła, A.; Pęczkowska, M.; Ćwikła, J.B.; Michałowska, I.; Pałucki, J.M.; Bodei, L.; Lewczuk-Myślicka, A.; Januszewicz, A. A clinical efficacy of prrt in patients with advanced, nonresectable, paraganglioma-pheochromocytoma, related to sdhx gene mutation. J. Clin. Med. 2019, 8, 952. [Google Scholar] [CrossRef] [PubMed]
- Imhof, A.; Brunner, P.; Marincek, N.; Briel, M.; Schindler, C.; Rasch, H.; Mäcke, H.R.; Rochlitz, C.; Müller-Brand, J.; Walter, M.A. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J. Clin. Oncol. 2011, 29, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, S.; Mittal, B.R.; Bhansali, A. ‘Peptide receptor radionuclide therapy in the management of advanced pheochromocytoma and paraganglioma: A systematic review and meta-analysis’. Clin. Endocrinol. 2019, 91, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Kumar Jaiswal, S.; Sarathi, V.; Samad Memon, S.; Garg, R.; Malhotra, G.; Verma, P.; Shah, R.; Sehemby, M.K.; Patil, V.A.; Jadhav, S.; et al. 177Lu-DOTATATE therapy in metastatic/inoperable pheochromocytoma-paraganglioma. Endocr. Connect. 2020, 9, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Puranik, A.D.; Kulkarni, H.R.; Singh, A.; Baum, R.P. Peptide receptor radionuclide therapy with 90Y/177Lu-labelled peptides for inoperable head and neck paragangliomas (glomus tumours). Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Zovato, S.; Kumanova, A.; Demattè, S.; Sansovini, M.; Bodei, L.; Di Sarra, D.; Casagranda, E.; Severi, S.; Ambrosetti, A.; Schiavi, F.; et al. Peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE in individuals with neck or mediastinal paraganglioma (PGL). Horm. Metab. Res. 2012, 44, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Forrer, F.; Riedweg, I.; Maecke, H.R.; Mueller-Brand, J. Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 334–340. Available online: http://www.ncbi.nlm.nih.gov/pubmed/18480742 (accessed on 13 March 2024). [PubMed]
- Pinato, D.J.; Black, J.R.M.; Ramaswami, R.; Tan, T.M.; Adjogatse, D.; Sharma, R. Peptide receptor radionuclide therapy for metastatic paragangliomas. Med. Oncol. 2016, 33, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.P.; Ballal, S.; Bal, C. Concomitant 177Lu-DOTATATE and capecitabine therapy in malignant paragangliomas. EJNMMI Res. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Grozinsky-Glasberg, S.; Hofman, M.S.; Callahan, J.; Meirovitz, A.; Maimon, O.; Pattison, D.A.; Gross, D.J.; Hicks, R.J. Efficacy of peptide receptor radionuclide therapy for functional metastatic paraganglioma and pheochromocytoma. J. Clin. Endocrinol. Metab. 2017, 102, 3278–3287. [Google Scholar] [CrossRef] [PubMed]
- Jochmanova, I.; Pacak, K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer 2018, 4, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Assadipour, Y.; Sadowski, S.M.; Alimchandani, M.; Quezado, M.; Steinberg, S.M.; Nilubol, N.; Patel, D.; Prodanov, T.; Pacak, K.; Kebebew, E. SDHB mutation status and tumor size but not tumor grade are important predictors of clinical outcome in pheochromocytoma and abdominal paraganglioma. Surgery 2017, 161, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.H.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct Clinical Features of Paraganglioma Syndromes Associated With SDHB and SDHD Gene Mutations. JAMA 2004, 292, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Crona, J.; Taïeb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef] [PubMed]
- Van Hulsteijn, L.T.; Niemeijer, N.D.; Dekkers, O.M.; Corssmit, E.P.M. 131I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. 2014, 80, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Pryma, D.A.; Chin, B.B.; Noto, R.B.; Dillon, J.S.; Perkins, S.; Solnes, L.; Kostakoglu, L.; Serafini, A.N.; Pampaloni, M.H.; Jensen, J.; et al. Efficacy and Safety of High-Specific-Activity 131I-MIBG Therapy in Patients with Advanced Pheochromocytoma or Paraganglioma. J. Nucl. Med. 2019, 60, 623–630. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Characteristics | All (N = 28) | Pheochromocytoma (N = 10) | Paraganglioma (N = 18) | p |
|---|---|---|---|---|
| Gender, n (%) | 0.80 | |||
| Men | 17 (61) | 6 (60) | 11 (61) | |
| Women | 11 (39) | 4 (36) | 7 (41) | |
| Median age at diagnosis, years (range) | 47 (15–75) | 46 (19–75) | 49 (15–72) | 0.76 |
| Men | 47 (19–75) | 31 (19–53) | 50 (25–75) | 0.056 |
| Women | 46 (15–65) | 54 (34–63) | 39 (15–65) | 0.45 |
| T-classification | 0.97 | |||
| Tx | 2 | 1 | 1 | |
| T1 | 6 | 2 | 4 | |
| T2 | 8 | 3 | 5 | |
| T3 | 12 | 4 | 8 | |
| N-classification | 0.18 | |||
| Nx | 5 | 0 | 5 | |
| N0 | 9 | 4 | 5 | |
| N1 | 14 | 6 | 8 | |
| M-classification | 0.46 | |||
| M0 | 8 | 3 | 5 | |
| M1a | 1 | 1 | 0 | |
| M1b | 7 | 3 | 4 | |
| M1c | 12 | 3 | 9 | |
| Median pCgA (range), pmol/L a | 254 (74–31,400) | 254 (89–1780) | 303 (74–31,400) | 0.71 |
| Median p-metanephrine (range), nmol/L a | 0.19 (0.01–41) | 0.41 (0.01–15.1) | 0.17 (0.01–41) | 0.66 |
| Median p-normetanephrine (range), nmol/L a | 2.5 (0.27–131) | 2.82 (0.27–71.1) | 1.25 (0.34–131) | 0.42 |
| Mutations | 13 | 5 | 8 | 0.93 |
| No. | Gender | Age at First PRRT | Primary Tumour | Primary Tumour Localisation | Metastases | Germline Mutation | Ki-67 (%) | Surgery | External Radio Therapy | MIBG Therapy | Chemo Therapy | TKI | PRRT Cycles |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 33 | PGL | Chest | LN neck | None | 5–15 | x * | x | 2 | |||
| 2 | M | 49 | PGL | Paravertebral | Adrenal gland, LN neck | None | NA | x * | x | x * | 4 | ||
| 3 | M | 76 | PGL | Head, neck, chest wall | Liver, bones | None | 10 | x * | x * | 6 | |||
| 4 | M | 25 | PGL | Neck, glomus caroticus, abdomen | SDHB | 4 | x * | 4 | |||||
| 5 | M | 52 | PGL | Pelvic | SDHB | 20 | x * | 14 | |||||
| 6 | M | 54 | PCC | Bones | RET | 1 | x | 4 | |||||
| 7 | F | 33 | PGL | Glomus jugularis | None | 3 | x | x | 4 | ||||
| 8 | M | 55 | PCC | RET | 0 | x | 4 | ||||||
| 9 | M | 58 | PCC | RET | NA | x | x | 4 | |||||
| 10 | F | 65 | PGL | Chest, left atrium | None | 50 | x | x | 4 | ||||
| 11 | F | 23 | PGL | Retroperitoneum | Mediastinum, abdomen | None | 1 | 4 | |||||
| 12 | M | 26 | PCC | Liver, bones | None | 25 | 4 | ||||||
| 13 | M | 68 | PGL | Bladder | None | x | x | x | 6 | ||||
| 14 | M | 55 | PGL | Abdomen | Liver, lungs, bones | None | 22 | x | x | 4 | |||
| 15 | M | 66 | PCC | LN mediastinum and abdomen | None | NA | 6 | ||||||
| 16 | F | 47 | PCC | SDHB | 20 | x | x | 4 | |||||
| 17 | F | 47 | PCC | Liver, bones, LN | NF1 | NA | x | x | x | 4 | |||
| 18 | F | 64 | PCC | LN | None | NA | x | 1 | |||||
| 19 | F | 65 | PCC | None | 50 | 4 | |||||||
| 20 | F | 67 | PGL | Glomus jugularis | SDHD | <1 | 4 | ||||||
| 21 | F | 49 | PGL | Abdomen | SDHB | 10 | x | 4 | |||||
| 22 | M | 56 | PGL | SDHB | 3 | 4 | |||||||
| 23 | M | 57 | PCC | None | 2 | x | 4 | ||||||
| 24 | M | 75 | PGL | None | 10 | x | 4 | ||||||
| 25 | F | 67 | PGL | Neck | None | 17 | 4 | ||||||
| 26 | M | 41 | PGL | Neck | Liver, bones, LN | SDHD | 1 | 4 | |||||
| 27 | F | 68 | PGL | SDHB | 20 | 4 | |||||||
| 28 | M | 52 | PGL | Abdomen | Liver, bones, LN | SDHB | 60 | x | x | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kornerup, L.S.; Andreassen, M.; Knigge, U.; Arveschoug, A.K.; Poulsen, P.L.; Kjær, A.; Oturai, P.S.; Grønbæk, H.; Dam, G. Effects of Peptide Receptor Radiotherapy in Patients with Advanced Paraganglioma and Pheochromocytoma: A Nation-Wide Cohort Study. Cancers 2024, 16, 1349. https://doi.org/10.3390/cancers16071349
Kornerup LS, Andreassen M, Knigge U, Arveschoug AK, Poulsen PL, Kjær A, Oturai PS, Grønbæk H, Dam G. Effects of Peptide Receptor Radiotherapy in Patients with Advanced Paraganglioma and Pheochromocytoma: A Nation-Wide Cohort Study. Cancers. 2024; 16(7):1349. https://doi.org/10.3390/cancers16071349
Chicago/Turabian StyleKornerup, Linda Skibsted, Mikkel Andreassen, Ulrich Knigge, Anne Kirstine Arveschoug, Per Løgstup Poulsen, Andreas Kjær, Peter Sandor Oturai, Henning Grønbæk, and Gitte Dam. 2024. "Effects of Peptide Receptor Radiotherapy in Patients with Advanced Paraganglioma and Pheochromocytoma: A Nation-Wide Cohort Study" Cancers 16, no. 7: 1349. https://doi.org/10.3390/cancers16071349
APA StyleKornerup, L. S., Andreassen, M., Knigge, U., Arveschoug, A. K., Poulsen, P. L., Kjær, A., Oturai, P. S., Grønbæk, H., & Dam, G. (2024). Effects of Peptide Receptor Radiotherapy in Patients with Advanced Paraganglioma and Pheochromocytoma: A Nation-Wide Cohort Study. Cancers, 16(7), 1349. https://doi.org/10.3390/cancers16071349