

Harnessing ctDNA in Advanced Melanoma: A Promising Tool for Informed Clinical Decisions


Abstract
Simple Summary
Abstract
1. Introduction
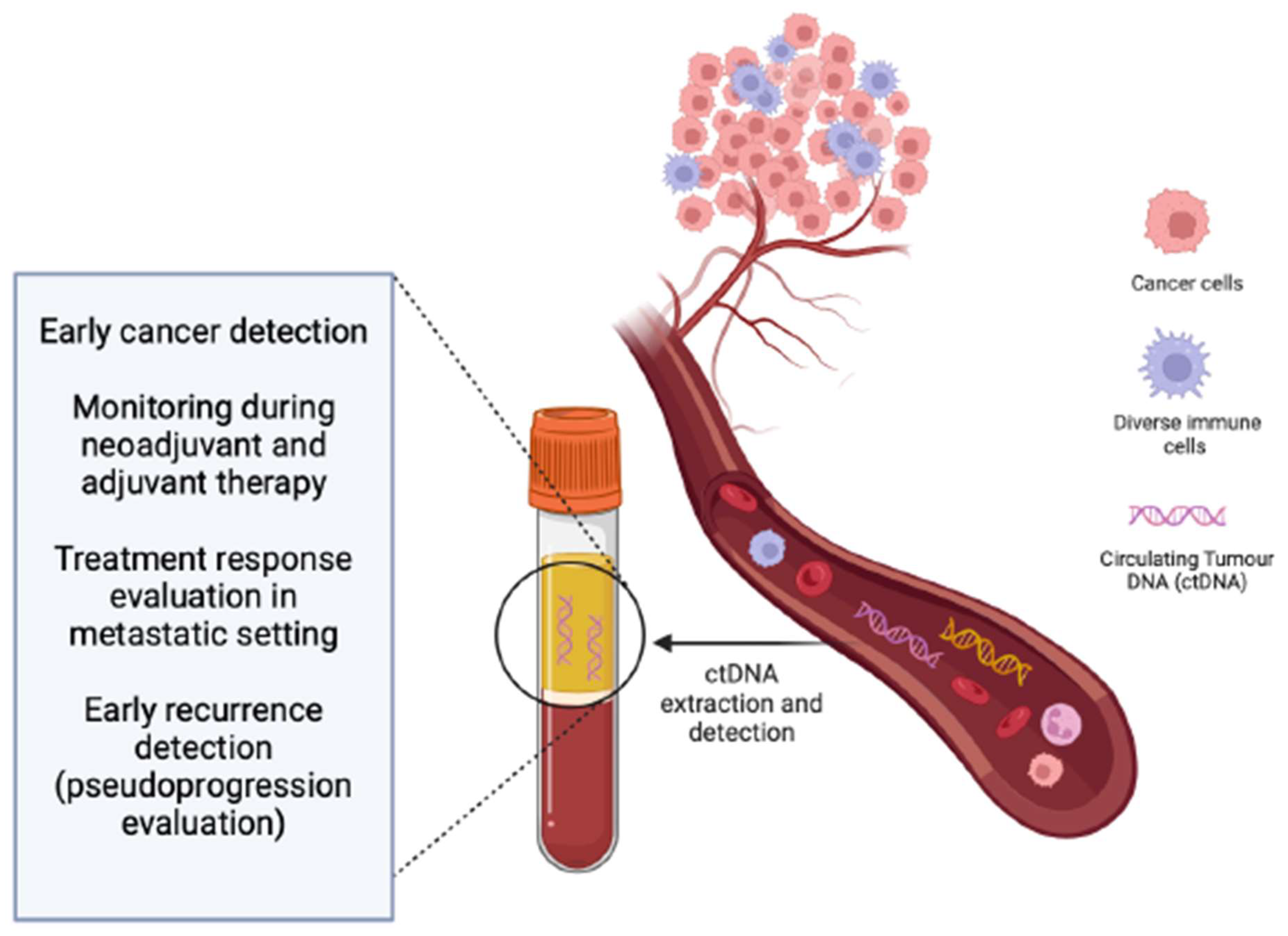
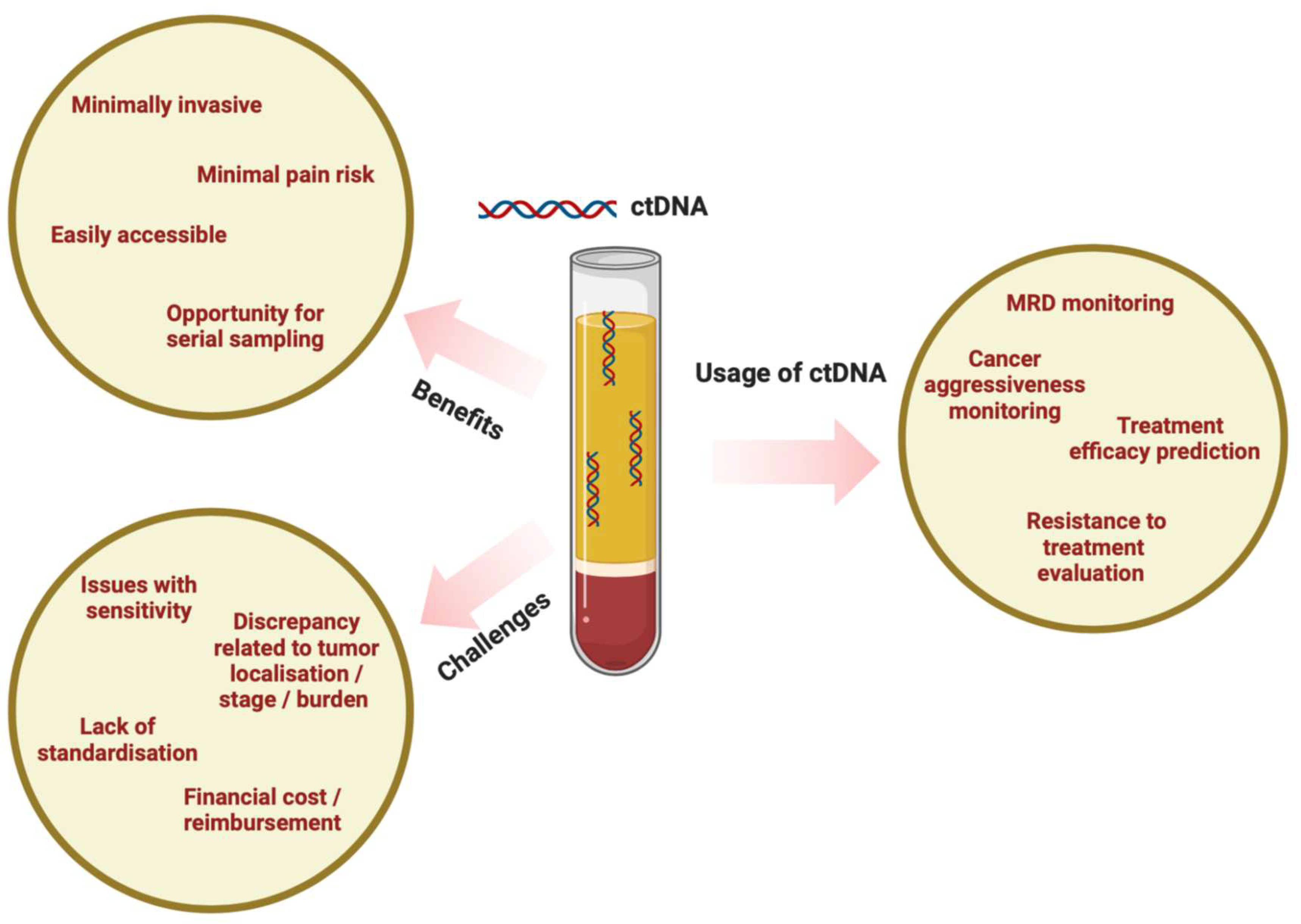
2. ctDNA as a Blood-Based Biomarker
3. Detection of ctDNA in Locally Advanced Melanoma: Early Recurrence Monitoring and Determination of Minimal Residual Disease
4. Detection of ctDNA in Metastatic Melanoma: Treatment Guide for Patients Receiving Immunotherapy and Targeted Therapy
5. Mutations in ctDNA and Their Significance for Melanoma Prognosis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; Vries, E.D.; Whiteman, D.C.; Bray, F. Global burden of cutaneous melanoma in 2020 and projections to 2040. JAMA Dermatol. 2022, 158, 495–503. [Google Scholar] [CrossRef]
- Singh, S.R.K.; Malapati, S.J.; Kumar, R.; Willner, C.; Wang, D. NCDB Analysis of Melanoma 2004–2015: Epidemiology and Outcomes by Subtype, Sociodemographic Factors Impacting Clinical Presentation, and Real-World Survival Benefit of Immunotherapy Approval. Cancers 2021, 13, 1455. [Google Scholar] [CrossRef]
- Adams, R.; Coumbe, J.E.M.; Coumbe, B.G.T.; Thomas, J.; Willsmore, Z.; Dimitrievska, M.; Yasuzawa-Parker, M.; Hoyle, M.; Ingar, S.; Geh, J.L.C.; et al. BRAF inhibitors and their immunological effects in malignant melanoma. Expert Rev. Clin. Immunol. 2022, 18, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Aide, N.; Iravani, A.; Prigent, K.; Kottler, D.; Alipour, R.; Hicks, R.J. PET/CT variants and pitfalls in malignant melanoma. Cancer Imaging 2022, 22, 3. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Eckburg, A.; Gantiwala, S.; Hart, Z.; Dein, J.; Lam, K.; Puri, N. Resistance to molecularly targeted therapies in melanoma. Cancers 2021, 13, 1115. [Google Scholar] [CrossRef]
- Swetter, S.M.; Thompson, J.A.; Albertini, M.R.; Barker, C.A.; Baumgartner, J.; Boland, G.; Chmielowski, B.; DiMaio, D.; Durham, A.; Fields, R.C.; et al. NCCN Guidelines® Insights: Melanoma: Cutaneous, Version 2.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.D. Melanoma radiological surveillance: A review of current evidence and clinical challenges. Yale J. Biol. Med. 2020, 93, 207–213. [Google Scholar] [PubMed]
- Van Wilpe, S.; Koornstra, R.; Den Brok, M.; De Groot, J.W.; Blank, C.; De Vries, J.; Gerritsen, W.; Mehra, N. Lactate dehydrogenase: A marker of diminished antitumor immunity. OncoImmunology 2020, 9, 1731942. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.B.; Forschner, A.; Leiter, U.; Garbe, C.; Eigentler, T.K. S100B and LDH as early prognostic markers for response and overall survival in melanoma patients treated with anti-PD-1 or combined anti-PD-1 plus anti-CTLA-4 antibodies. Br. J. Cancer 2018, 119, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Olhagen, B.; Thorell, B.; Wising, P. The Endocellular Nucleic Acid Distribution and Plasma Protein Formation in Myelomatosis. Scand. J. Clin. Lab. Investig. 1949, 1, 49–59. [Google Scholar] [CrossRef]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Saucier, J.B.; Feng, Y.; Jiang, Y.; Sinsin, J.; McCombs, A.K.; Schmitt, E.S.; Peacock, S.; Chen, S.; et al. Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA. Nat. Med. 2019, 25, 439–447. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Reich, C.F.; Pisetsky, D.S. The role of macrophages in the in vitro generation of extracellular DNA from apoptotic and necrotic cells. Immunology 2005, 115, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.A.; Yeung, S.-W.; Lui, W.-B.; Rainer, T.H.; Lo, Y.D. Effects of Preanalytical Factors on the Molecular Size of Cell-Free DNA in Blood. Clin. Chem. 2005, 51, 781–784. [Google Scholar] [CrossRef]
- Jung, M.; Klotzek, S.; Lewandowski, M.; Fleischhacker, M.; Jung, K. Changes in Concentration of DNA in Serum and Plasma during Storage of Blood Samples. Clin. Chem. 2003, 49, 1028–1029. [Google Scholar] [CrossRef]
- Lui, Y.Y.; Chik, K.W.; Chiu, R.W. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709. [Google Scholar] [CrossRef]
- Husain, H.; Melnikova, V.O.; Kosco, K.; Woodward, B.; More, S.; Pingle, S.C.; Weihe, E.; Park, B.H.; Tewari, M.; Erlander, M.G.; et al. Monitoring Daily Dynamics of Early Tumor Response to Targeted Therapy by Detecting Circulating Tumor DNA in Urine. Clin. Cancer Res. 2017, 23, 4716–4723. [Google Scholar] [CrossRef]
- Higgins, M.J.; Jelovac, D.; Barnathan, E.; Blair, B.; Slater, S.; Powers, P.; Zorzi, J.; Jeter, S.C.; Oliver, G.R.; Fetting, J.; et al. Detection of Tumor PIK3CA Status in Metastatic Breast Cancer Using Peripheral Blood. Clin. Cancer Res. 2012, 18, 3462–3469. [Google Scholar] [CrossRef]
- Dong, L.; Yoo, H.-B.; Wang, J.; Park, S.-R. Accurate quantification of supercoiled DNA by digital PCR. Sci. Rep. 2016, 6, 24230. [Google Scholar] [CrossRef]
- Johansson, G.; Andersson, D.; Filges, S.; Li, J.; Muth, A.; Godfrey, T.E.; Stahlberg, A. Considerations and quality controls when analyzing cell-free tumor DNA. Biomol. Detect. Quantif. 2019, 17, 100078. [Google Scholar] [CrossRef] [PubMed]
- Warton, K.; Yuwono, N.L.; Cowley, M.J.; McCabe, M.J.; So, A.; Ford, C.E. Evaluation of Streck BCT and PAXgene Stabilised Blood Collection Tubes for Cell-Free Circulating DNA Studies in Plasma. Mol. Diagn. Ther. 2017, 21, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Pallisgaard, N.; Spindler, K.-L.G.; Andersen, R.F.; Brandslund, I.; Jakobsen, A. Controls to validate plasma samples for cell free DNA quantification. Clin. Chim. Acta 2015, 446, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Marczynski, G.T.; Laus, A.C.; dos Reis, M.B.; Reis, R.M.; Vazquez, V.d.L. Circulating tumor DNA (ctDNA) detection is associated with shorter progression-free survival in advanced melanoma patients. Sci. Rep. 2020, 10, 18682. [Google Scholar] [CrossRef]
- Aoude, L.G.; Brosda, S.; Ng, J.; Lonie, J.M.; Belle, C.J.; Patel, K.; Koufariotis, L.T.; Wood, S.; Atkinsin, V.; Smithers, B.M.; et al. Circulating Tumor DNA. J. Mol. Diagn. 2023, 25, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Seremet, T.; Jansen, Y.; Planken, S.; Njimi, H.; DElaunoy, M.; Housni, H.; Awada, G.; Schwarze, J.K.; Keyaerts, M.; Everaert, H.; et al. Undetectable circulating tumor DNA (ctDNA) levels correlate with favorable outcome in metastatic melanoma patients treated with anti-PD1 therapy. J. Transl. Med. 2019, 17, 303. [Google Scholar] [CrossRef] [PubMed]
- Passiglia, F.; Rizzo, S.; Rolfo, C.; Galvano, A.; Bronte, E.; Incorvaia, L.; Listi, A.; Barraco, N.; Castiglia, M.; Calo, V.; et al. Metastatic site location influences the diagnostic accuracy of ctDNA EGFR—Mutation testing in NSCLC patients: A pooled analysis. Curr. Cancer Drug Targets 2018, 18, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical relevance of blood-based ctDNA analysis: Mutation detection and beyond. Br. J. Cancer 2020, 124, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Semenkovich, N.P.; Szymanski, J.J.; Earland, N.; Chauhan, P.S.; Pellini, B.; Chaudhuri, A.A. Genomic approaches to cancer and minimal residual disease detection using circulating tumor DNA. J. ImmunoTher. Cancer 2023, 11, e006284. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Mei, W.; Ma, K.; Zeng, C. Circulating Tumor DNA and Minimal Residual Disease (MRD) in Solid Tumors: Current Horizons and Future Perspectives. Front. Oncol. 2021, 11, 763790. [Google Scholar] [CrossRef] [PubMed]
- Tivey, A.; Britton, F.; Scott, J.-A.; Rothwell, D.; Lorigan, P.; Lee, R. Circulating Tumour DNA in Melanoma—Clinic Ready? Curr. Oncol. Rep. 2022, 24, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kao, Y.; Zhou, Q.; Wuethrich, A.; Stark, M.S.; Schaider, H.; Soyer, H.P.; Lin, L.L.; Trau, M. An Integrated Microfluidic-SERS Platform Enables Sensitive Phenotyping of Serum Extracellular Vesicles in Early Stage Melanomas. Adv. Funct. Mater. 2021, 32, 2010296. [Google Scholar] [CrossRef]
- Cheng, Y.; Lu, J.; Chen, G.; Ardekani, G.S.; Rotte, A.; Martinka, M.; Xu, X.; McElwee, K.J.; Zhang, G.; Zhou, Y. Stage-specific prognostic biomarkers in melanoma. Oncotarget 2015, 6, 4180–4189. [Google Scholar] [CrossRef]
- Gandini, S.; Zanna, I.; De Angelis, S.P.; Cocorocchio, E.; Queirolo, P.; Lee, H.J.; Carlino, M.S.; Mazzarella, L.; Duso, B.A.; Palli, D.; et al. Circulating tumour DNA and melanoma survival: A systematic literature review and meta-analysis. Crit. Rev. Oncol. /Hematol. 2020, 157, 103187. [Google Scholar] [CrossRef]
- Lee, J.H.; Saw, R.P.; Thompson, J.F.; Lo, S.; Spillane, A.J.; Stretch, J.R.; Howle, J.; Menzies, A.M.; Carlino, M.S.; Kefford, R.F.; et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann. Oncol. 2019, 30, 815–822. [Google Scholar] [CrossRef]
- Tan, L.; Sandhu, S.; Lee, R.J.; Li, J.; Callahan, J.; Ftouni, S.; Dhomen, N.; Middlehurst, P.; Wallace, A.; Raleigh, J.; et al. Prediction and monitoring of relapse in stage III melanoma using circulating tumor DNA. Ann. Oncol. 2019, 30, 804–814. [Google Scholar] [CrossRef]
- Forschner, A.; Niessner, H.; Sinnberg, T.; Eigentler, T.; Amaral, T.; Seith, F.; Garve, C.; Biskup, S.; Battke, F. Circulating tumor DNA (ctDNA) in the detection of relapse in melanoma patients with adjuvant anti-PD-1 therapy. JDDG J. Der Dtsch. Dermatol. Ges. 2022, 20, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Raleigh, J.M.; Callahan, J.; Vergana, I.A.; Ftouni, S.; Hatzimihalis, A.; Colebatch, A.J.; Li, J.; Semple, T.; Doig, F.; et al. Circulating Tumor DNA Analysis and Functional Imaging Provide Complementary Approaches for Comprehensive Disease Monitoring in Metastatic Melanoma. JCO Precis. Oncol. 2017, 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Lee, K.J.; Stark, M.S. Current Trends in Circulating Biomarkers for Melanoma Detection. Front. Med. 2022, 9, 873728. [Google Scholar] [CrossRef] [PubMed]
- Chang-Hao, T.S.; Weiss, J.; Hudson, C.; Christophi, C.; Cebon, J.; Behren, A.; Dobrovic, A. Monitoring response to therapy in melanoma by quantifying circulating tumour DNA with droplet digital PCR for BRAF and NRAS mutations. Sci. Rep. 2015, 5, 11198. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.; Kefford, R.; Hauschild, A.; Hwu, P. Correlation of BRAF Mutation Status in Circulating-Free DNA and Tumor and Association with Clinical Outcome across Four BRAFi and MEKi Clinical Trials. Clin. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Velculescu, V.E.; Pritchard, T.S.; Sausen, M.; Pardoll, D.M.; Topalian, S.L.; Diaz, L.A., Jr. Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J. ImmunoTherapy Cancer 2014, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Marsavela, G.; Johansson, P.A.; Pereira, M.R.; McEvoy, A.C.; Reid, A.; Robinson, C.; Warburton, L.; Khattak, M.A.; Meniawy, T.M.; Amanuel, B.; et al. The prognostic impact of circulating tumor DNA in melanoma patients treated with systematic therapies-beyond BRAF mutant detection. Cancers 2020, 12, 3793. [Google Scholar] [CrossRef] [PubMed]
- Marsavela, G.; McEvoy, A.C.; Pereira, M.R.; Reid, A.L.; Al-Ogaili, Z.; Warburton, L.; Khattak, M.A.; Abed, A.; Meniawy, T.M.; Millward, M. Detection of clinical progression through plasma ctDNA in metastatic melanoma patients: A comparison to radiological progression. Br. J. Cancer 2022, 126, 401–408. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Gutierrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Marsavela, G.; Lee, J.; Calapre, L.; Wong, S.Q.; Pereira, M.R.; McEvoy, A.C.; Reid, A.L.; Robinson, C.; Warburton, L.; Abed, A.; et al. Circulating Tumor DNA Predicts Outcome from First-, but not Second-line Treatment and Identifies Melanoma Patients Who May Benefit from Combination Immunotherapy. Clin. Cancer Res. 2020, 26, 5926–5933. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.; Rizos, H.; et al. Association Between Circulating Tumor DNA and Pseudoprogression in Patients with Metastatic Melanoma Treated with Anti–Programmed Cell Death 1 Antibodies. JAMA Oncol. 2018, 4, 717. [Google Scholar] [CrossRef]
- Eroglu, Z.; Krinshpun, S.; Kalashnikova, E.; Sudhaman, S.; Topcu, T.O.; Nichols, M.; Martin, J.; Bui, K.M.; Palsuledesai, C.C.; Malhotra, M.; et al. Circulating tumor DNA-based molecular residual disease detection for treatment monitoring in advanced melanoma patients. Cancer 2023, 129, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.A.; Tadepalli, J.S.; Shao, Y.; Zhang, Y.; Weiss, S.; Robinson, E.; Spittle, C.; Furtado, M.; Shelton, D.N.; Karlin-Neumann, G.; et al. Sensitivity of plasma BRAFmutant and NRASmutant cell-free DNA assays to detect metastatic melanoma in patients with low RECIST scores and non-RECIST disease progression. Mol. Oncol. 2015, 10, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, S.; Blenkiron, C.; Stephens, R.; Mathy, J.A.; Somers-Edgar, T.; Rolfe, G.; Martin, R.; Jackson, C.; Eccles, M.; Robb, T.; et al. Dynamic ctDNA Mutational Complexity in Patients with Melanoma Receiving Immunotherapy. Mol. Diagn. Ther. 2023, 27, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sun, H.; Cong, L.; Liu, C.; Sun, Q.; Wu, N.; Cong, X. Prognostic Value of ctDNA Mutation in Melanoma: A Meta-Analysis. J. Oncol. 2021, 2021, 6660571. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Drugs Approved for Melanoma. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/melanoma (accessed on 29 November 2023).
- Syeda, M.M.; Wiggins, J.M.; Corless, B.C.; Long, G.V.; Flaherty, K.T.; Schadendorf, D.; Nathan, P.D.; Robert, C.; Ribas, A.; Davies, M.A.; et al. Circulating tumour DNA in patients with advanced melanoma treated with dabrafenib or dabrafenib plus trametinib: A clinical validation study. Lancet Oncol. 2021, 22, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.; Liu, Y.; Wang, M.; Li, Y.; Xu, T.; Wei, Y. The Predictive Value of Tumor Mutation Burden on Clinical Efficacy of Immune Checkpoint Inhibitors in Melanoma: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2022, 13, 748674. [Google Scholar] [CrossRef]
- Kisistók, J.; Christensen, D.S.; Rasmussen, M.H.; Duval, L.; Aggerholm-Pedersen, N.; Luczak, A.A.; Sorensen, B.S.; Jakobsen, M.R.; Oellegaard, T.H.; Birkbak, N.J. Analysis of circulating tumor DNA during checkpoint inhibition in metastatic melanoma using a tumor-agnostic panel. Melanoma Res. 2023, 33, 364–374. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| ctDNA in Locally Advanced Melanoma | ctDNA in Metastatic Melanoma |
|---|---|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pikturniene, R.; Cesas, A.; Jarmalaite, S.; Razbadauskas, A.; Urbonas, V. Harnessing ctDNA in Advanced Melanoma: A Promising Tool for Informed Clinical Decisions. Cancers 2024, 16, 1197. https://doi.org/10.3390/cancers16061197
Pikturniene R, Cesas A, Jarmalaite S, Razbadauskas A, Urbonas V. Harnessing ctDNA in Advanced Melanoma: A Promising Tool for Informed Clinical Decisions. Cancers. 2024; 16(6):1197. https://doi.org/10.3390/cancers16061197
Chicago/Turabian StylePikturniene, Rugile, Alvydas Cesas, Sonata Jarmalaite, Arturas Razbadauskas, and Vincas Urbonas. 2024. "Harnessing ctDNA in Advanced Melanoma: A Promising Tool for Informed Clinical Decisions" Cancers 16, no. 6: 1197. https://doi.org/10.3390/cancers16061197
APA StylePikturniene, R., Cesas, A., Jarmalaite, S., Razbadauskas, A., & Urbonas, V. (2024). Harnessing ctDNA in Advanced Melanoma: A Promising Tool for Informed Clinical Decisions. Cancers, 16(6), 1197. https://doi.org/10.3390/cancers16061197