

EMT Dynamics in Lymph Node Metastasis of Oral Squamous Cell Carcinoma

 ,
,  ,
,  ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
Study Validation
3. Results
3.1. Cross-Tab Analysis: Advanced Stage vs. Early Stage (Table 1)
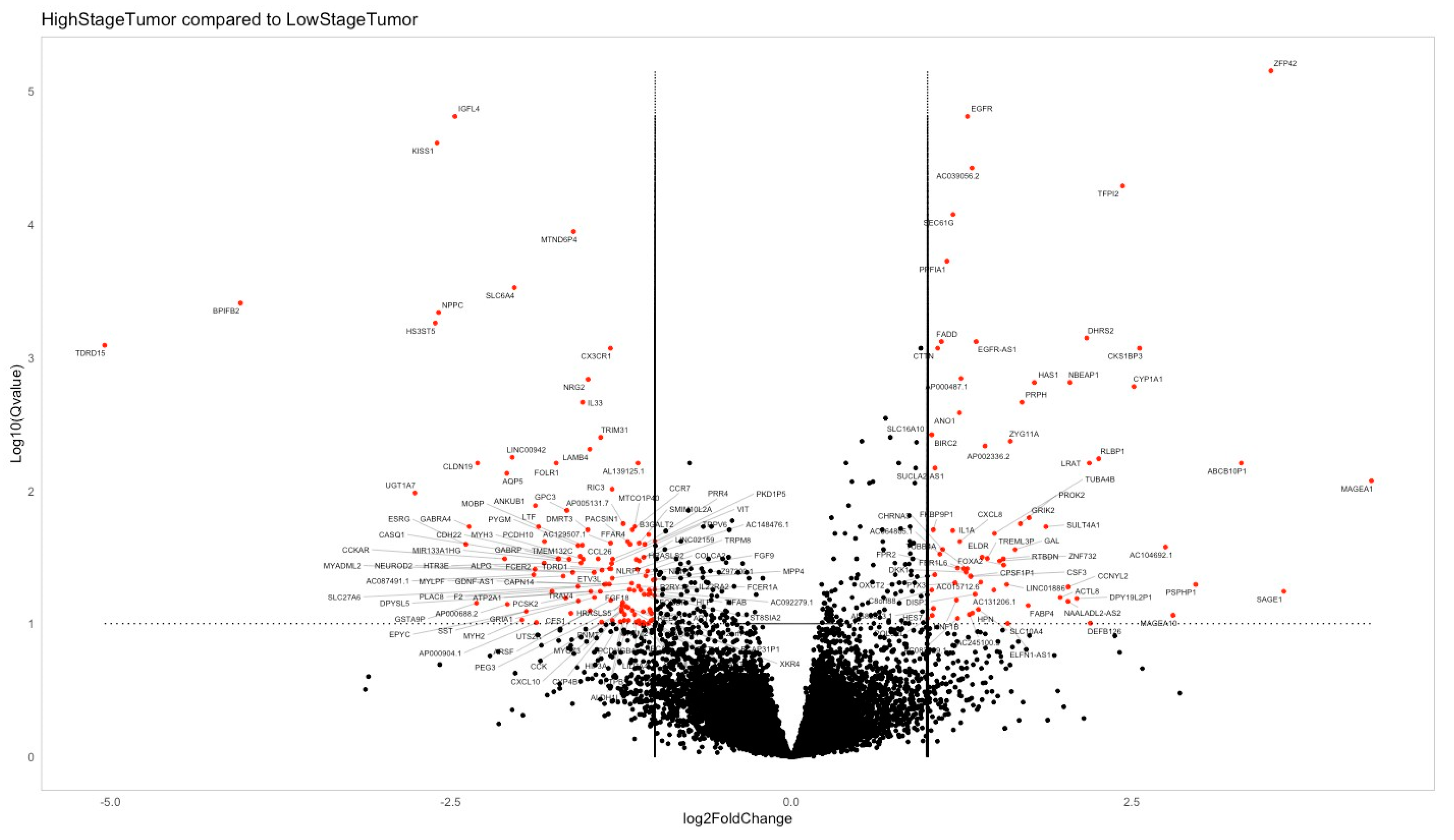
3.2. Genetic Profiling Analysis
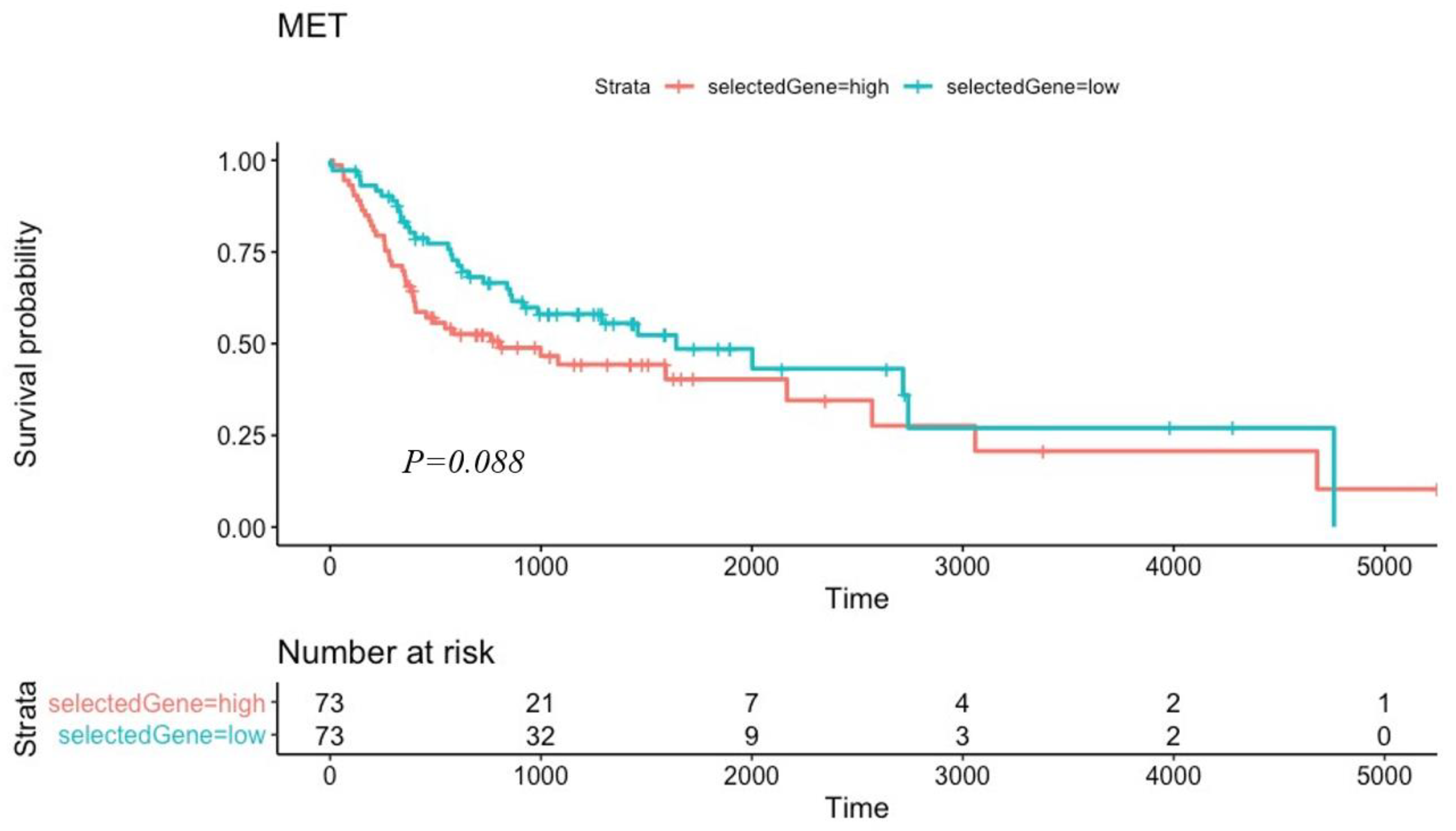
3.3. Kaplan–Meier Survival Testing
3.4. Independent Validation Using IHC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Montero, P.H.; Patel, S.G. Cancer of the oral cavity. Surg. Oncol. Clin. 2015, 24, 491–508. [Google Scholar] [CrossRef]
- Karlsson, M.C.; Gonzalez, S.F.; Welin, J.; Fuxe, J. Epithelial-mesenchymal transition in cancer metastasis through the lymphatic system. Mol. Oncol. 2017, 11, 781–791. [Google Scholar] [CrossRef]
- Park, M.; Kim, D.; Ko, S.; Kim, A.; Mo, K.; Yoon, H. Breast cancer metastasis: Mechanisms and therapeutic implications. Int. J. Mol. Sci. 2022, 23, 6806. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Blick, T.; Widodo, E.; Hugo, H.; Waltham, M.; Lenburg, M.E.; Neve, R.M.; Thompson, E.W. Epithelial-mesenchymal transition traits in human breast cancer cell lines. Clin. Exp. Metastasis 2008, 25, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P. Leaving the neighborhood: Molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays 2001, 23, 912–923. [Google Scholar] [CrossRef]
- Zhai, X.; Zhu, H.; Wang, W.; Zhang, S.; Zhang, Y.; Mao, G. Abnormal expression of EMT-related proteins, S100A4, vimentin and E-cadherin, is correlated with clinicopathological features and prognosis in HCC. Med. Oncol. 2014, 31, 970. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFβ signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Waite, K.A.; Eng, C. From developmental disorder to heritable cancer: It’s all in the BMP/TGF-β family. Nat. Rev. Genet. 2003, 4, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Derynck, R. TGF-β signaling in cancer–a double-edged sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar]
- De Kruijf, E.M.; Dekker, T.J.A.; Hawinkels, L.J.A.C.; Putter, H.; Smit, V.T.H.B.M.; Kroep, J.R.; Mesker, W.E. The prognostic role of TGF-β signaling pathway in breast cancer patients. Ann. Oncol. 2013, 24, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Deckers, M.; van Dinther, M.; Buijs, J.; Que, I.; Lowik, C.; van der Pluijm, G.; ten Dijke, P. The tumor suppressor Smad4 is required for transforming growth factor β–induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 2006, 66, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Robinson, M.; Smith, E.; Huntley, S.; Prime, S.; Paterson, I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-β1 involves MAPK, Smad and AP-1 signalling path-ways. J. Cell. Biochem. 2005, 95, 918–931. [Google Scholar] [CrossRef]
- Sticht, C.; Freier, K.; Knöpfle, K.; Flechtenmacher, C.; Pungs, S.; Hofele, C.; Lichter, P. Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC). Neoplasia 2008, 10, 462-IN4. [Google Scholar] [CrossRef]
- Shi, Y.; Hata, A.; Lo, R.S.; Massagué, J.; Pavletich, N.P. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature 1997, 388, 87–93. [Google Scholar] [CrossRef]
- Angadi, P.V.; Patil, P.V.; Angadi, V.; Mane, D.; Shekar, S.; Hallikerimath, S.; Kardesai, S.G. Immunoexpression of epithelial-mesenchymal transition proteins E-cadherin, β-catenin, and N-cadherin in oral squamous cell carcinoma. Int. J. Surg. Pathol. 2016, 24, 696–703. [Google Scholar] [CrossRef]
- Kaur, J.; Sawhney, M.; DattaGupta, S.; Shukla, N.K.; Srivastava, A.; Walfish, P.G.; Ralhan, R. Clinical significance of altered expression of β-catenin and E-cadherin in oral dysplasia and cancer: Potential link with ALCAM expression. PLoS ONE 2013, 8, e67361. [Google Scholar] [CrossRef]
- González-Moles, M.A.; Ruiz-Ávila, I.; Gil-Montoya, J.A.; Plaza-Campillo, J.; Scully, C. β-catenin in oral cancer: An update on current knowledge. Oral Oncol. 2014, 50, 818–824. [Google Scholar] [CrossRef]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adhes. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef]
- Collins, C.; Nelson, W.J. Running with neighbors: Coordinating cell migration and cell–cell adhesion. Curr. Opin. Cell Biol. 2015, 36, 62–70. [Google Scholar] [CrossRef]
- Wu, J.S.; Jiang, J.; Chen, B.J.; Wang, K.; Tang, Y.L.; Liang, X.H. Plasticity of cancer cell invasion: Patterns and mechanisms. Transl. Oncol. 2021, 14, 100899. [Google Scholar] [CrossRef]
- Jeschke, U.; Mylonas, I.; Kuhn, C.; Shabani, N.; Kunert-Keil, C.; Schindlbeck, C.; Gerber, B.; Friese, K. Expression of E-cadherin in human ductal breast cancer carcinoma in situ, invasive carcinomas, their lymph node metastases, their distant metastases, carcinomas with recurrence and in recurrence. Anticancer Res. 2007, 27, 1969–1974. [Google Scholar] [PubMed]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for meta-static colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef]
- Yokoyama, K.; Kamata, N.; Hayashi, E.; Hoteiya, T.; Ueda, N.; Fujimoto, R.; Nagayama, M. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 2001, 37, 65–71. [Google Scholar] [CrossRef]
- Nakamura, R.; Ishii, H.; Endo, K.; Hotta, A.; Fujii, E.; Miyazawa, K.; Saitoh, M. Reciprocal expression of Slug and Snail in human oral cancer cells. PLoS ONE 2018, 13, e0199442. [Google Scholar] [CrossRef]
- Bu, W.; Wang, Z.; Meng, L.; Li, X.; Liu, X.; Chen, Y.; Sun, H. Disulfiram inhibits epithelial–mesenchymal transition through TGFβ–ERK–Snail pathway independently of Smad4 to decrease oral squamous cell carcinoma metastasis. Cancer Manag. Res. 2019, 11, 3887–3898. [Google Scholar] [CrossRef]
- Franz, M.; Spiegel, K.; Umbreit, C.; Richter, P.; Codina-Canet, C.; Berndt, A.; Berndt, A. Expression of Snail is associated with myofibroblast phenotype development in oral squamous cell carcinoma. Histochem. Cell Biol. 2009, 131, 651–660. [Google Scholar] [CrossRef]
- Villarejo, A.; Cortés-Cabrera, Á.; Molina-Ortíz, P.; Portillo, F.; Cano, A. Differential role of Snail1 and Snail2 zinc fingers in E-cadherin repression and epithelial to mesenchymal transition. J. Biol. Chem. 2014, 289, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Yu, Y.; Zhang, T.; Zhou, X.; Zhou, J.; Jia, L.; Feng, Y. Snail is critical for tumor growth and metastasis of ovarian carcinoma. Int. J. Cancer 2010, 126, 2102–2111. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, P.B. The role of snail in EMT and tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef]
- Gao, L.; Xu, T.; Huang, G.; Jiang, S.; Gu, Y.; Chen, F. Oral microbiomes: More and more importance in oral cavity and whole body. Protein Cell 2018, 9, 488–500. [Google Scholar] [CrossRef]
- Lee, J.; Roberts, J.S.; Atanasova, K.R.; Chowdhury, N.; Han, K.; Yilmaz, Ö. Human primary epithelial cells acquire an epithelialmesenchymal-transition phenotype during long-term infection by the oral opportunistic pathogen, Porphyromonas gingivalis. Front. Cell. Infect. Microbiol. 2017, 7, 493. [Google Scholar] [CrossRef]
- Inaba, H.; Sugita, H.; Kuboniwa, M.; Iwai, S.; Hamada, M.; Noda, T.; Morisaki, I.; Lamont, R.J.; Amano, A. Porphyromonas gingivalis promotes invasion of oral squamous cell carcinoma through induction of proMMP9 and its activation. Cell. Microbiol. 2014, 16, 131–145. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef]
- Davis, R.; Rizwani, W.; Banerjee, S.; Kovacs, M.; Haura, E.; Coppola, D.; Chellappan, S. Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS ONE 2009, 4, e7524. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.A.; Kiang, A.; Wang-Rodriguez, J.; Rahimy, E.; Haas, M.; Yu, V.; Ellies, L.G.; Chen, J.; Fan, J.B.; Brumund, K.T.; et al. Nicotine promotes acquisition of stem cell and epithelial-to-mesenchymal properties in head and neck squamous cell carcinoma. PLoS ONE 2012, 7, e51967. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Q.; Lv, Y.E.; Lin, B.H.; Luo, L.M.; Lv, S.L.; Bi, A.H.; Jia, Y.S. Silencing of periostin inhibits nicotine-mediated tumor cell growth and epithelial-mesenchymal transition in lung cancer cells. Mol. Med. Rep. 2013, 7, 875–880. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Study Cohort | “Early Stage” Group | “Advanced Stage” Group | p Value |
|---|---|---|---|---|
| Num. | 159 | 47 | 112 | |
| Mean age (±STD) | 62 ± 13 years | 61 ± 17 years | 62 ± 12 years | 0.3 |
| Male/female | 105/54 | 27/21 | 78/34 | 0.06 |
| Tobacco exposure | 0.001 | |||
| Num. (%) | 82 (51%) | 5 (10%) | 77 (69%) | |
| Av. pack/year | 47 | 16.13 | 27.91 | |
| Alcohol consumption (%) | 101 (63%) | 24 (51%) | 77 (68%) | 0.2 |
| Primary tumor site (%) | 0.04 | |||
| Oral tongue | 70 (44%) | 24 (51%) | 46 (41%) | |
| Floor of mouth | 25 (15%) | 4 (8%) | 21 (19%) | |
| Buccal mucosa | 8 (4%) | 2 (4%) | 6 (5%) | |
| Alveolar ridge | 5 (2%) | 0 | 5 (4%) | |
| Hard palate | 4 (2%) | 0 | 4 (3%) | |
| Lip | 1 (0.6%) | 0 | 1 (0.8%) | |
| Oral cavity * | 46 (28%) | 17 (36%) | 29 (26%) | |
| p N staging (by H&E) | - | |||
| N0 (%) | 89 (56%) | 47 (100%) | 42 (37%) | |
| N1 (%) | 35 (22%) | 0 | 35 (31%) | |
| N2A (%) | 2 (1%) | 0 | 2 (2%) | |
| N2B (%) | 20 (12%) | 0 | 20 (18%) | |
| N2C (%) | 12 (7%) | 0 | 12 (10%) | |
| N3 (%) | 1 (0.6%) | 0 | 1 (0.8%) | |
| pT staging | - | |||
| T1 (%) | 9 (5%) | 7 (15%) | 2 (1.7%) | |
| T2 (%) | 54 (33%) | 40 (85%) | 14 (13%) | |
| T3 (%) | 45 (28%) | 0 | 45 (40%) | |
| T4a (%) | 50 (31%) | 0 | 50 (44%) | |
| T4b (%) | 1 (0.6%) | 0 | 1 (0.8%) | |
| TNM staging ** | ||||
| Stage 1 (%) | 7 (4%) | 7 (15%) | 0 | |
| Stage 2 (%) | 40 (25%) | 40 (85%) | 0 | |
| Stage 3 (%) | 41 (26%) | 0 | 41 (36%) | |
| Stage 4a (%) | 69 (43%) | 0 | 69 (61%) | |
| Stage 4b (%) | 2 (1%) | 0 | 2 (2%) | |
| Surgical margins status | 0.06 | |||
| Negative margins (%) | 103 (64%) | 35 (74%) | 69 (61%) | |
| Positive margins ¥ (%) | 19 (12%) | 5 (10%) | 14 (12%) | |
| Close margins ¥ (%) | 37 (23%) | 7 (16%) | 30 (27%) | |
| Overall survival (months) | 32 months | 36 months | 19 months | 0.01 |
| Characteristic | Study Cohort |
|---|---|
| Num. | 28 |
| Mean age (±STD) | 67 ± 13.9 years |
| Male/Female | 16/12 |
| Primary tumor site (%) | |
| Oral tongue | 15 |
| Floor of mouth | 2 |
| Buccal mucosa | 3 |
| Alveolar ridge | 5 |
| Hard palate | 0 |
| Lip | 3 |
| Oral cavity * | - |
| p N staging (by H&E) | |
| N0 | 13 |
| N1 | 6 |
| N2A | 4 |
| N2B | 4 |
| N2C | - |
| N3 | 1 |
| pT staging | |
| T1 | 14 |
| T2 | 9 |
| T3 | 2 |
| T4 | 3 |
| TNM staging ** | |
| Stage 1 | 10 |
| Stage 2 | 6 |
| Stage 3 | 5 |
| Stage 4 | 7 |
| IHC staining (ave. %T area) | |
| E-cadherin (cohort) | 23.091 (SD 4.474) |
| E-cadherin (N negative) | 25.919 (SD 4.790) |
| E-cadherin (N positive) | 20.263 (SD 4.582) |
| N-cadherin (cohort) | 0.323 (SD 0.420) |
| N-cadherin (N negative) | 0.171 (SD 0.201) |
| N-cadherin (N positive) | 0.446 (SD 0.442) |
| Overall survival (months) | 33 months |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghantous, Y.; Mozalbat, S.; Nashef, A.; Abdol-Elraziq, M.; Sudri, S.; Araidy, S.; Tadmor, H.; Abu El-naaj, I. EMT Dynamics in Lymph Node Metastasis of Oral Squamous Cell Carcinoma. Cancers 2024, 16, 1185. https://doi.org/10.3390/cancers16061185
Ghantous Y, Mozalbat S, Nashef A, Abdol-Elraziq M, Sudri S, Araidy S, Tadmor H, Abu El-naaj I. EMT Dynamics in Lymph Node Metastasis of Oral Squamous Cell Carcinoma. Cancers. 2024; 16(6):1185. https://doi.org/10.3390/cancers16061185
Chicago/Turabian StyleGhantous, Yasmine, Shiraz Mozalbat, Aysar Nashef, Murad Abdol-Elraziq, Shiran Sudri, Shareef Araidy, Hagar Tadmor, and Imad Abu El-naaj. 2024. "EMT Dynamics in Lymph Node Metastasis of Oral Squamous Cell Carcinoma" Cancers 16, no. 6: 1185. https://doi.org/10.3390/cancers16061185
APA StyleGhantous, Y., Mozalbat, S., Nashef, A., Abdol-Elraziq, M., Sudri, S., Araidy, S., Tadmor, H., & Abu El-naaj, I. (2024). EMT Dynamics in Lymph Node Metastasis of Oral Squamous Cell Carcinoma. Cancers, 16(6), 1185. https://doi.org/10.3390/cancers16061185