
Enhancing Leukemia Treatment: The Role of Combined Therapies Based on Amino Acid Starvation

Abstract
Simple Summary
Abstract
1. Introduction
2. Glutamine, a Versatile Precursor for Biosynthesis and Bioenergetics
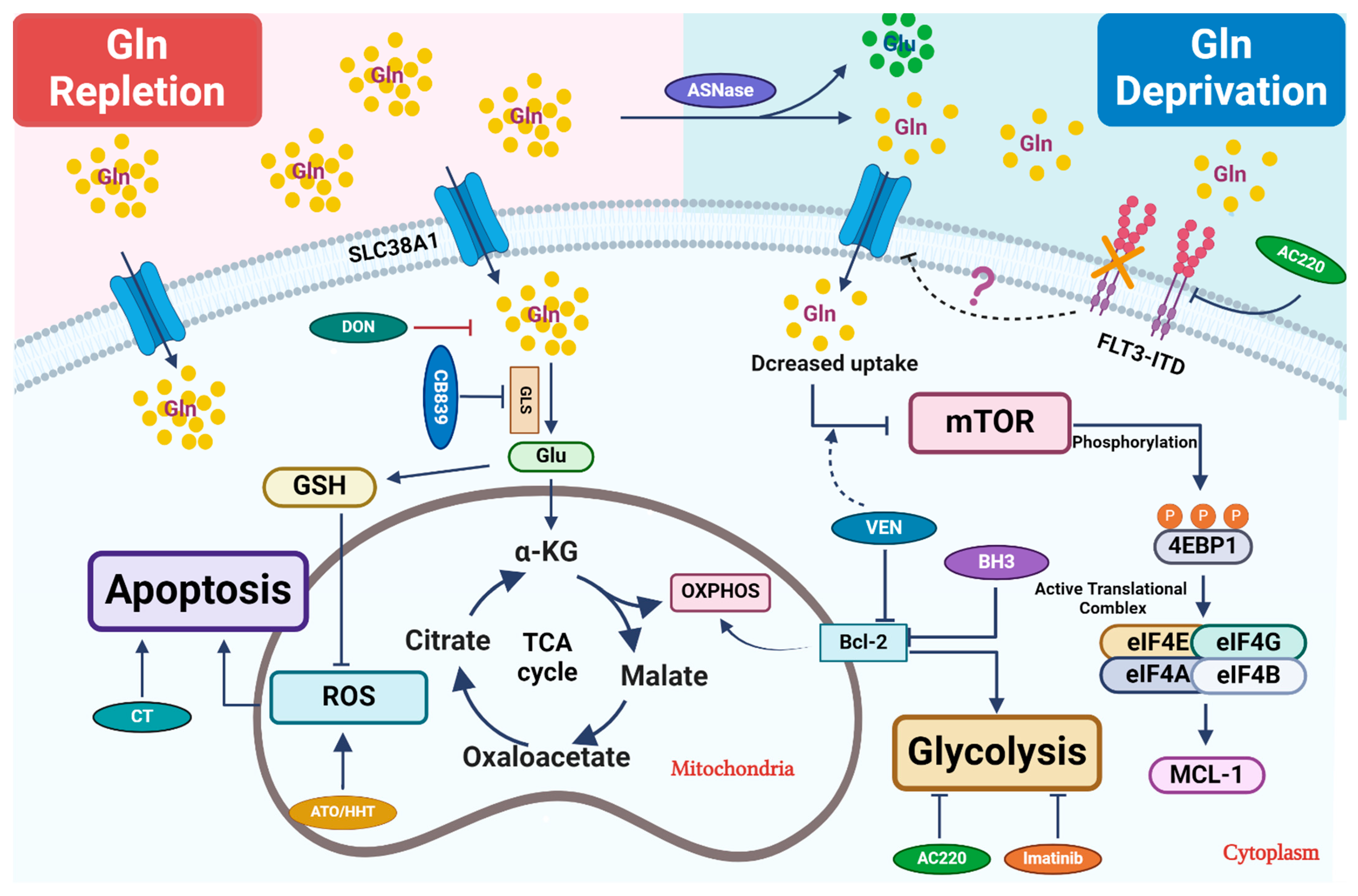
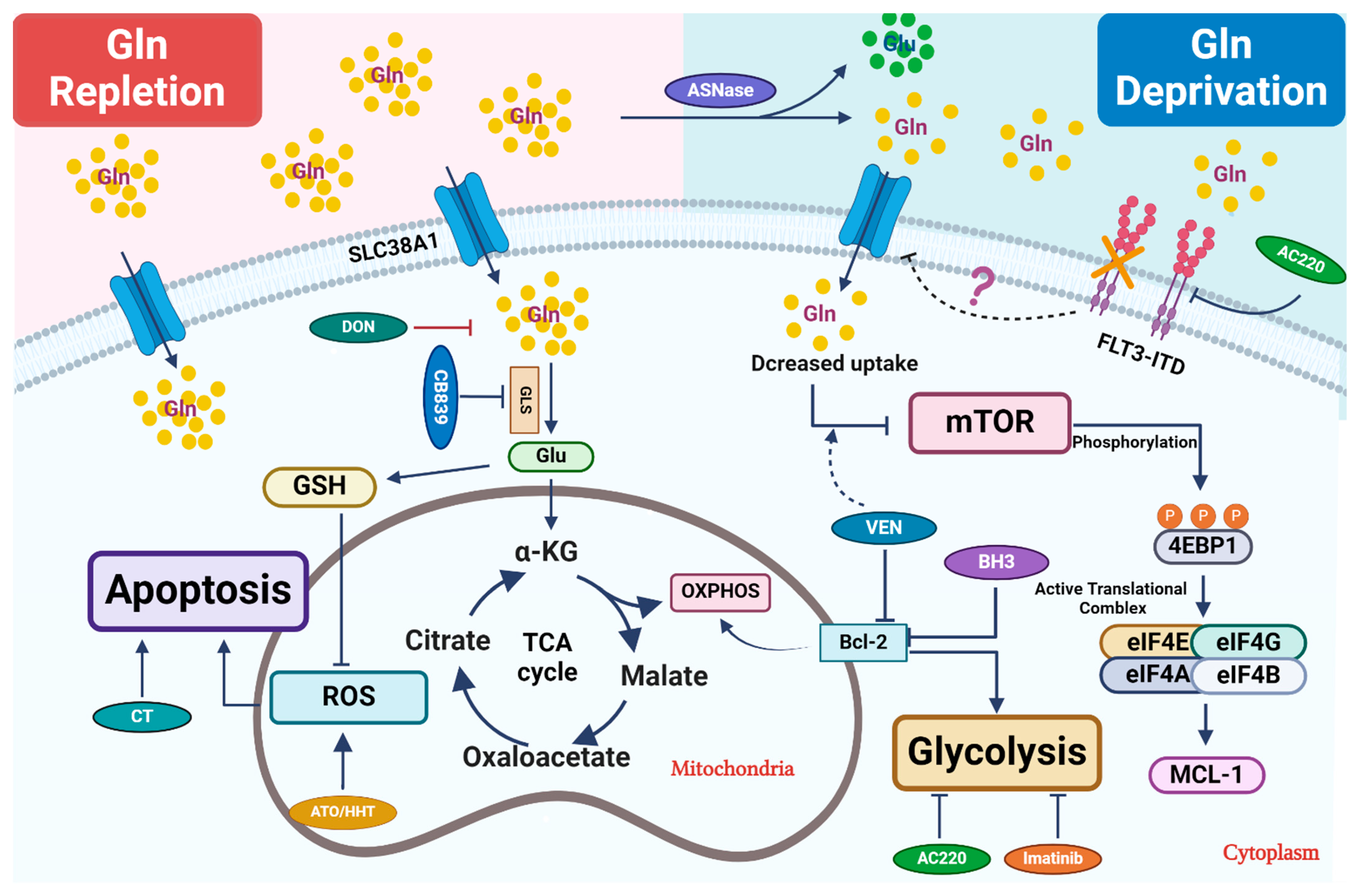
2.1. Glutamine Catabolism and Its Regulation
2.2. Targeting Glutamine Metabolism in AML
2.3. Combination Therapeutics Involving Glutamine Perturbation and Traditional Chemo-Agents
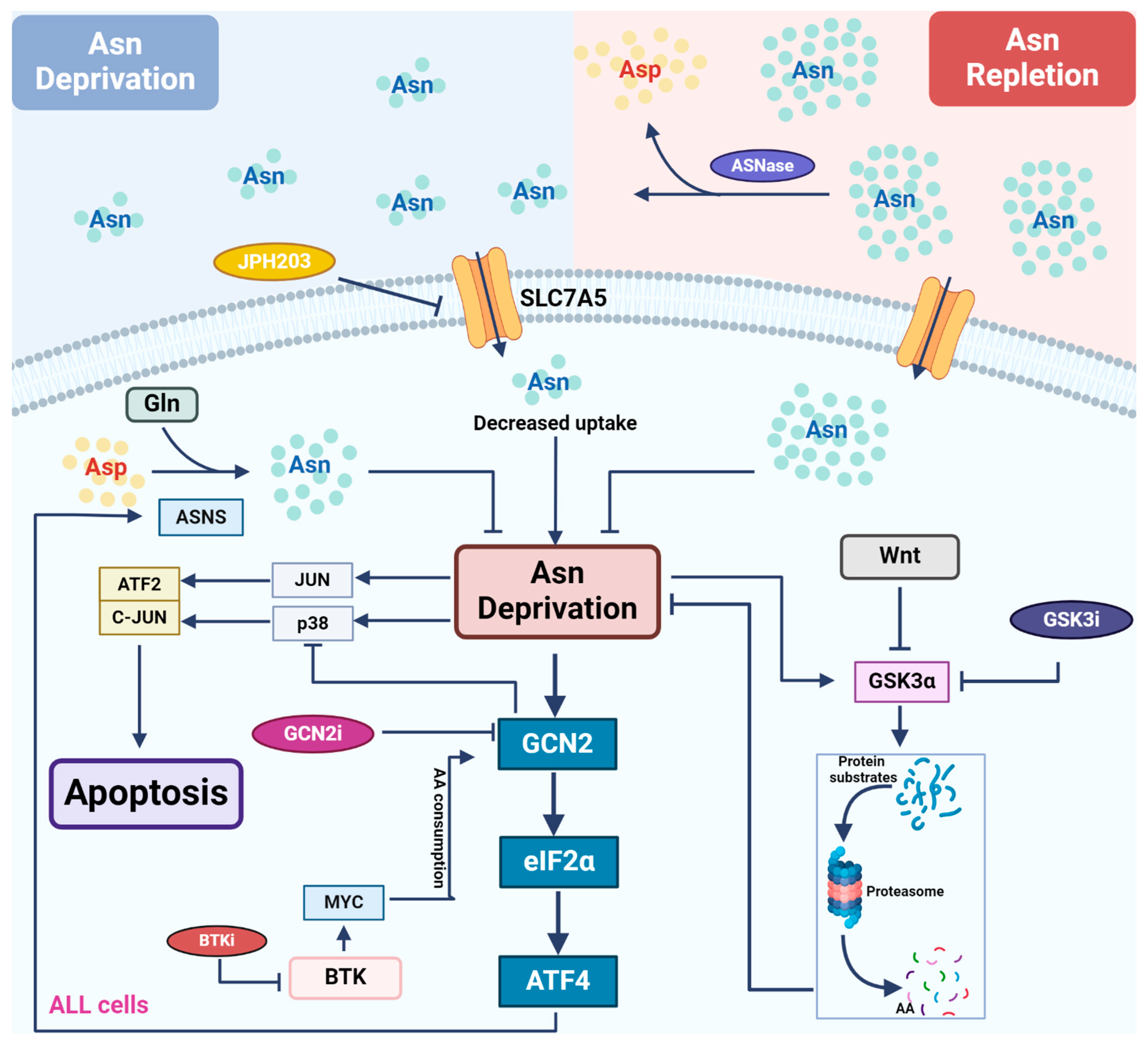
3. Asparagine and Its Depletion-Based Therapy in ALL Patients
3.1. Asparagine Metabolism and the Regulation of Its Biosynthesis
3.2. Application of L-Asparaginase and Its Potential Synergy with Other Therapeutics in Leukemias
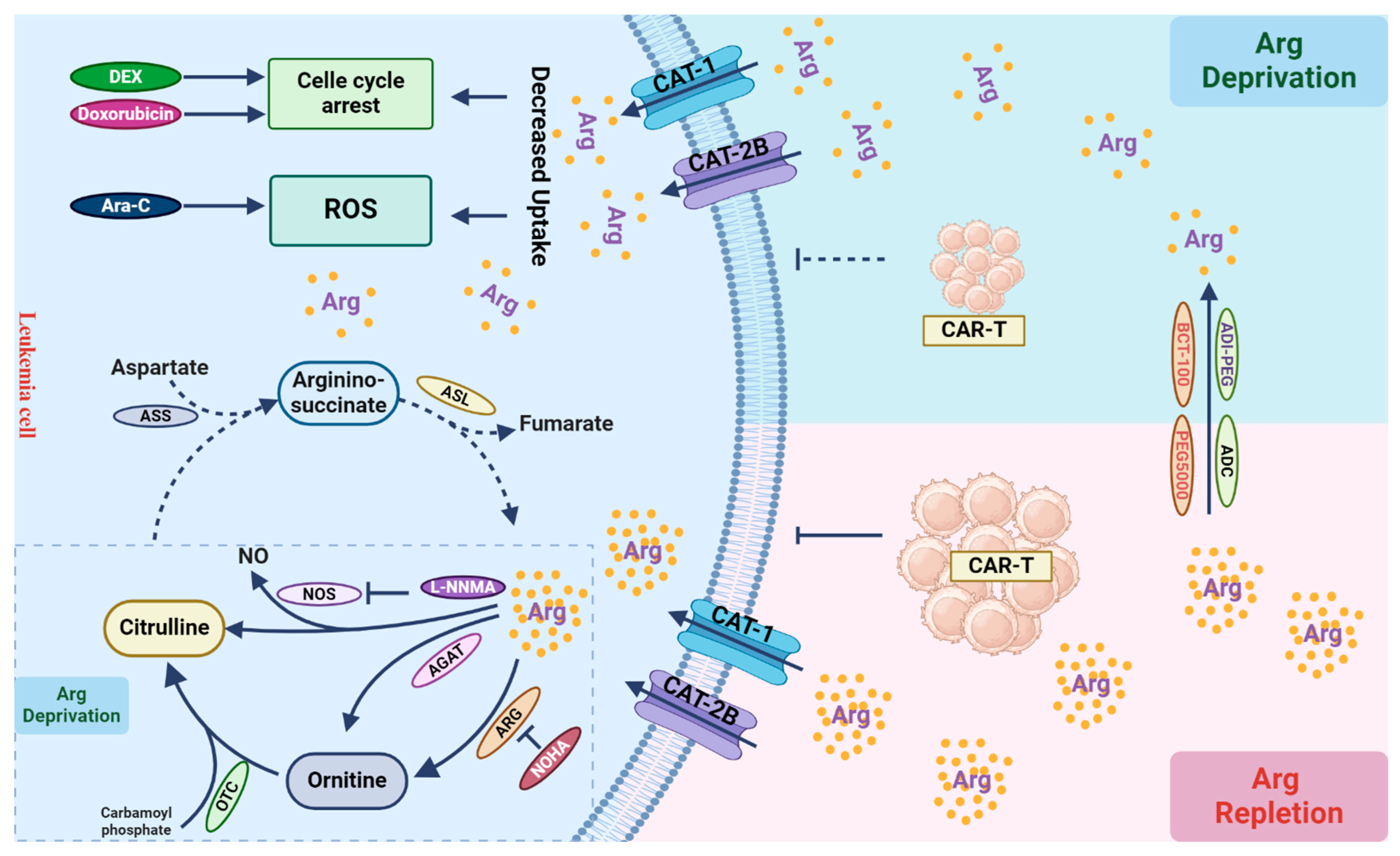
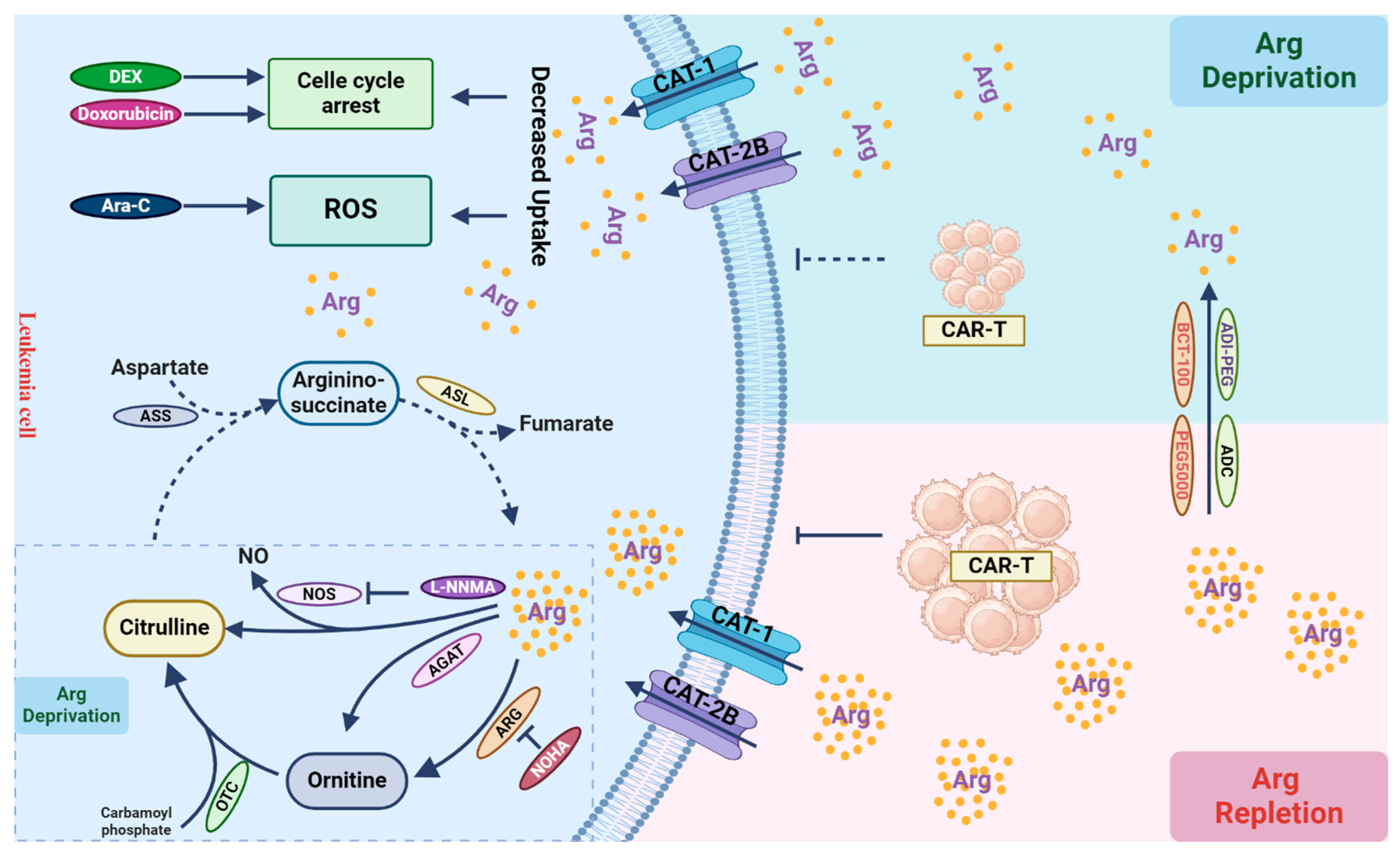
4. Arginine Deprivation: Anti-Leukemic or Immuno-Suppressive?
4.1. Arginine Metabolism and Its Depletion-Based Therapy in AML
4.2. Arginine Supports the Anti-Tumor Effect of T Cells
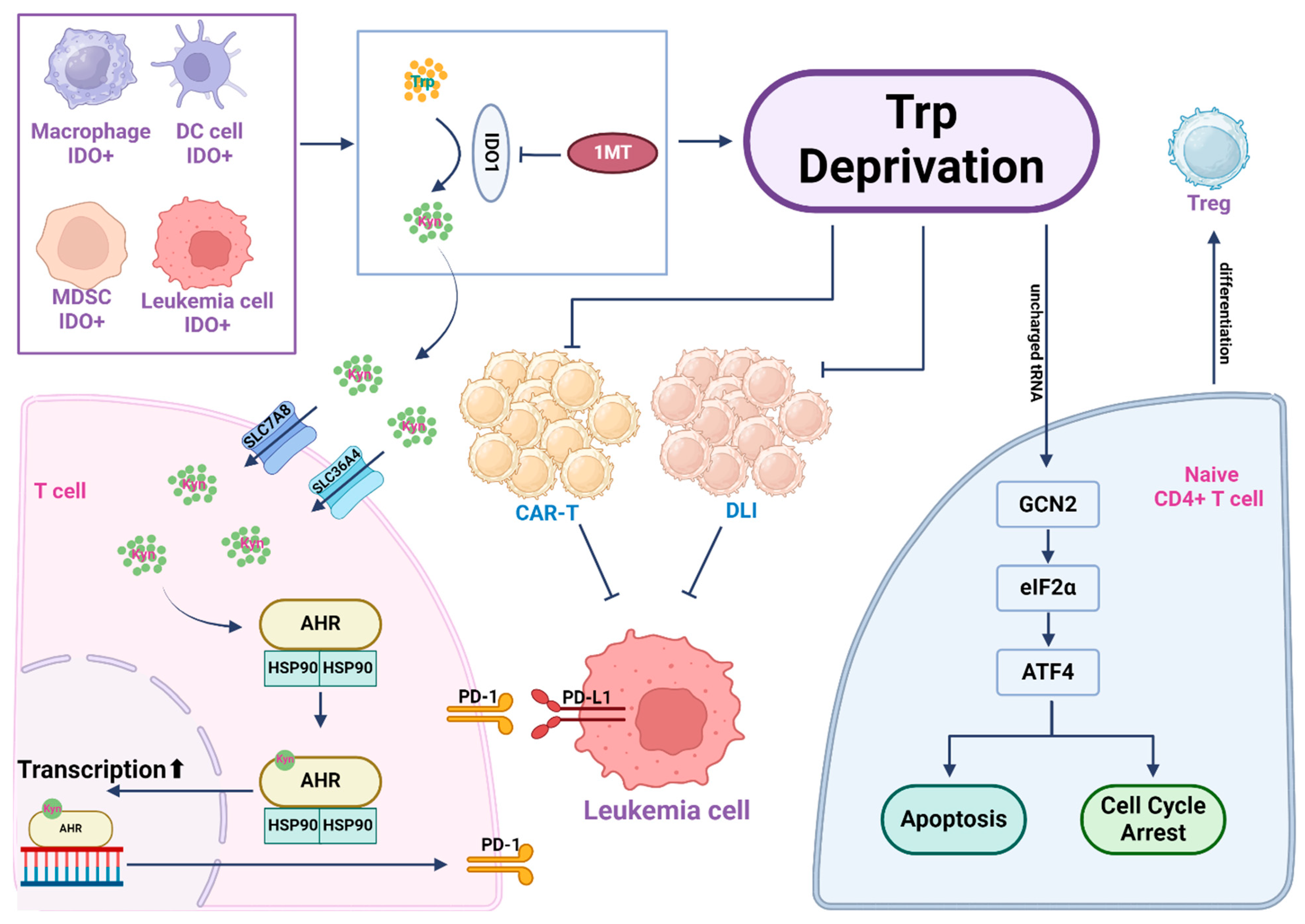
5. Tryptophan, an Immune Modulator
5.1. Tryptophan Catabolic Enzymes and Their Roles in Immune Cells in the Tumor Microenvironment
5.2. Pre-Clinical and Clinical Applications of IDO Inhibitors in Leukemia
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Kelly, B.; Pearce, E.L. Amino Assets: How Amino Acids Support Immunity. Cell Metab. 2020, 32, 154–175. [Google Scholar] [CrossRef] [PubMed]
- Safrhansova, L.; Hlozkova, K.; Starkova, J. Targeting amino acid metabolism in cancer. Int. Rev. Cell Mol. Biol 2022, 373, 37–79. [Google Scholar] [PubMed]
- Tabe, Y.; Lorenzi, P.L.; Konopleva, M. Amino acid metabolism in hematologic malignancies and the era of targeted therapy. Blood 2019, 134, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cui, L.; Lu, S.; Xu, S. Amino acid metabolism in tumor biology and therapy. Cell Death Dis. 2024, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.; Taurino, G.; Bianchi, M.G.; Bussolati, O. The role of amino acids in the crosstalk between mesenchymal stromal cells and neoplastic cells in the hematopoietic Niche. Front. Cell Dev. Biol. 2021, 9, 714755. [Google Scholar] [CrossRef]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino Acid Depletion Therapies: Starving Cancer Cells to Death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Oburoglu, L.; Tardito, S.; Fritz, V.; de Barros, S.C.; Merida, P.; Craveiro, M.; Mamede, J.; Cretenet, G.; Mongellaz, C.; An, X.; et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell 2014, 15, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutte, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.W.; Cerione, R.A. Glutaminase: A hot spot for regulation of cancer cell metabolism? Oncotarget 2010, 1, 734–740. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Katt, W.P.; Cerione, R.A. Alone and together: Current approaches to targeting glutaminase enzymes as part of anti-cancer therapies. Future Drug. Discov. 2023, 4, FDD79. [Google Scholar] [CrossRef] [PubMed]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Miwa, H.; Shikami, M. Importance of glutamine metabolism in leukemia cells by energy production through TCA cycle and by redox homeostasis. Cancer Investig. 2014, 32, 241–247. [Google Scholar] [CrossRef]
- Pei, S.; Minhajuddin, M.; Callahan, K.P.; Balys, M.; Ashton, J.M.; Neering, S.J.; Lagadinou, E.D.; Corbett, C.; Ye, H.; Liesveld, J.L.; et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J. Biol. Chem. 2013, 288, 33542–33558. [Google Scholar] [CrossRef]
- van Gastel, N.; Spinelli, J.B.; Sharda, A.; Schajnovitz, A.; Baryawno, N.; Rhee, C.; Oki, T.; Grace, E.; Soled, H.J.; Milosevic, J.; et al. Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cell Metab. 2020, 32, 391–403.e396. [Google Scholar] [CrossRef]
- Amaya, M.; Inguva, A.; Pei, S. The STAT3-MYC axis promotes survival of leukemia stem cells by regulating SLC1A5 and oxidative phosphorylation. Blood 2021, 139, 584–596. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; D’Alessandro, A.; Alvarez-Calderon, F.; Kim, J.; Nemkov, T.; Adane, B.; Rozhok, A.I.; Kumar, A.; Kumar, V.; Pollyea, D.A.; et al. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, E6669–E6678. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Nemkov, T.; Reisz, J.A.; Zaberezhnyy, V.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp. Hematol. 2018, 58, 52–58. [Google Scholar] [CrossRef]
- Gallipoli, P.; Giotopoulos, G.; Tzelepis, K.; Costa, A.S.H.; Vohra, S.; Medina-Perez, P.; Basheer, F.; Marando, L.; Di Lisio, L.; Dias, J.M.L.; et al. Glutaminolysis is a metabolic dependency in FLT3(ITD) acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 2018, 131, 1639–1653. [Google Scholar] [CrossRef]
- Saxena, K.; Konopleva, M.; Bhagat, T.D.; Guerra, V.A.; Maduike, R.; Tiziani, S.; Borthakur, G.; Jabbour, E.; Pemmaraju, N.; Kadia, T.M.; et al. AZA + Glutaminase Inhibition with Telaglenastat (CB-839) for Advanced MDS: An Updated Interim Analysis. Blood 2020, 136, 31–32. [Google Scholar] [CrossRef]
- Emadi, A.; Duong, V.H.; Pantin, J.; Imran, M.; Koka, R.; Singh, Z.; Sausville, E.A.; Law, J.Y.; Lee, S.T.; Shi, H.; et al. Indoximod Combined with Standard Induction Chemotherapy Is Well Tolerated and Induces a High Rate of Complete Remission with MRD-Negativity in Patients with Newly Diagnosed AML: Results from a Phase 1 Trial. Blood 2018, 132, 332. [Google Scholar] [CrossRef]
- Rousselot, P.; Coude, M.M.; Gokbuget, N.; Gambacorti Passerini, C.; Hayette, S.; Cayuela, J.M.; Huguet, F.; Leguay, T.; Chevallier, P.; Salanoubat, C.; et al. Dasatinib and low-intensity chemotherapy in elderly patients with Philadelphia chromosome-positive ALL. Blood 2016, 128, 774–782. [Google Scholar] [CrossRef]
- Patel, B.; Kirkwood, A.A.; Dey, A.; Marks, D.I.; McMillan, A.K.; Menne, T.F.; Micklewright, L.; Patrick, P.; Purnell, S.; Rowntree, C.J.; et al. Pegylated-asparaginase during induction therapy for adult acute lymphoblastic leukaemia: Toxicity data from the UKALL14 trial. Leukemia 2017, 31, 58–64. [Google Scholar] [CrossRef]
- Avramis, V.I. Asparaginases: Biochemical pharmacology and modes of drug resistance. Anticancer. Res. 2012, 32, 2423–2437. [Google Scholar] [PubMed]
- Emadi, A.; Law, J.Y.; Strovel, E.T.; Lapidus, R.G.; Jeng, L.J.B.; Lee, M.; Blitzer, M.G.; Carter-Cooper, B.A.; Sewell, D.; Van Der Merwe, I.; et al. Asparaginase Erwinia chrysanthemi effectively depletes plasma glutamine in adult patients with relapsed/refractory acute myeloid leukemia. Cancer Chemother. Pharmacol. 2018, 81, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A.; Kapadia, B.; Bollino, D.; Bhandary, B.; Baer, M.R.; Niyongere, S.; Strovel, E.T.; Kaizer, H.; Chang, E.; Choi, E.Y.; et al. Venetoclax and pegcrisantaspase for complex karyotype acute myeloid leukemia. Leukemia 2021, 35, 1907–1924. [Google Scholar] [CrossRef] [PubMed]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832. [Google Scholar] [CrossRef]
- Ni, F.; Yu, W.M.; Li, Z.; Graham, D.K.; Jin, L.; Kang, S.; Rossi, M.R.; Li, S.; Broxmeyer, H.E.; Qu, C.K. Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat. Metab. 2019, 1, 390–403. [Google Scholar] [CrossRef]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Jiang, J.; Batra, S.; Zhang, J. Asparagine: A Metabolite to Be Targeted in Cancers. Metabolites 2021, 11, 402. [Google Scholar] [CrossRef]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef]
- Meng, D.; Yang, Q.; Wang, H.; Melick, C.H.; Navlani, R.; Frank, A.R.; Jewell, J.L. Glutamine and asparagine activate mTORC1 independently of Rag GTPases. J. Biol. Chem. 2020, 295, 2890–2899. [Google Scholar] [CrossRef]
- Chiu, M.; Taurino, G.; Bianchi, M.G.; Kilberg, M.S.; Bussolati, O. Asparagine Synthetase in Cancer: Beyond Acute Lymphoblastic Leukemia. Front Oncol. 2019, 9, 1480. [Google Scholar] [CrossRef] [PubMed]
- Kaspers, G.J.L. Acute myeloid leukaemia niche regulates response to L-asparaginase. Br. J. Haematol. 2019, 186, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Fung, M.K.L.; Chan, G.C. Drug-induced amino acid deprivation as strategy for cancer therapy. J. Hematol. Oncol. 2017, 10, 144. [Google Scholar] [CrossRef]
- Pokrovsky, V.S.; Chepikova, O.E.; Davydov, D.Z.; Zamyatnin, A.A., Jr.; Lukashev, A.N.; Lukasheva, E.V. Amino Acid Degrading Enzymes and their Application in Cancer Therapy. Curr. Med. Chem. 2019, 26, 446–464. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, T.A.; Costa, I.M.; Biasoto, H.P.; Lima, G.M.; Silva, C.; Pessoa, A.; Monteiro, G. Critical overview of the main features and techniques used for the evaluation of the clinical applicability of L-asparaginase as a biopharmaceutical to treat blood cancer. Blood Rev. 2020, 43, 100651. [Google Scholar] [CrossRef]
- Song, P.; Ye, L.; Fan, J.; Li, Y.; Zeng, X.; Wang, Z.; Wang, S.; Zhang, G.; Yang, P.; Cao, Z.; et al. Asparaginase induces apoptosis and cytoprotective autophagy in chronic myeloid leukemia cells. Oncotarget 2015, 6, 3861–3873. [Google Scholar] [CrossRef]
- Juluri, K.R.; Siu, C.; Cassaday, R.D. Asparaginase in the Treatment of Acute Lymphoblastic Leukemia in Adults: Current Evidence and Place in Therapy. Blood Lymphat Cancer 2022, 12, 55–79. [Google Scholar] [CrossRef]
- Jiang, J.; Srivastava, S.; Seim, G.; Pavlova, N.N.; King, B.; Zou, L.; Zhang, C.; Zhong, M.; Feng, H.; Kapur, R. Promoter demethylation of the asparagine synthetase gene is required for ATF4-dependent adaptation to asparagine depletion. J. Biol. Chem. 2019, 294, 18674–18684. [Google Scholar] [CrossRef]
- Nakamura, A.; Nambu, T.; Ebara, S.; Hasegawa, Y.; Toyoshima, K.; Tsuchiya, Y.; Tomita, D.; Fujimoto, J.; Kurasawa, O.; Takahara, C.; et al. Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc. Natl. Acad. Sci. USA 2018, 115, E7776–E7785. [Google Scholar] [CrossRef]
- Stams, W.A.; den Boer, M.L.; Beverloo, H.B.; Meijerink, J.P.; Stigter, R.L.; van Wering, E.R.; Janka-Schaub, G.E.; Slater, R.; Pieters, R. Sensitivity to L-asparaginase is not associated with expression levels of asparagine synthetase in t(12;21)+ pediatric ALL. Blood 2003, 101, 2743–2747. [Google Scholar] [CrossRef]
- Touzart, A.; Lengline, E.; Latiri, M.; Belhocine, M.; Smith, C.; Thomas, X.; Spicuglia, S.; Puthier, D.; Pflumio, F.; Leguay, T.; et al. Epigenetic silencing affects L-asparaginase sensitivity and predicts outcome in T-ALL. Clin. Cancer Res. 2019, 25, 2483–2493. [Google Scholar] [CrossRef]
- Ren, Y.; Roy, S.; Ding, Y.; Iqbal, J.; Broome, J.D. Methylation of the asparagine synthetase promoter in human leukemic cell lines is associated with a specific methyl binding protein. Oncogene 2004, 23, 3953–3961. [Google Scholar] [CrossRef]
- Worton, K.S.; Kerbel, R.S.; Andrulis, I.L. Hypomethylation and reactivation of the asparagine synthetase gene induced by L-asparaginase and ethyl methanesulfonate. Cancer Res. 1991, 51, 985–989. [Google Scholar]
- Serravalle, S.; Bertuccio, S.N.; Astolfi, A.; Melchionda, F.; Pession, A. Synergistic Cytotoxic Effect of L-Asparaginase Combined with Decitabine as a Demethylating Agent in Pediatric T-ALL, with Specific Epigenetic Signature. Biomed. Res. Int. 2016, 2016, 1985750. [Google Scholar] [CrossRef]
- Acebron, S.P.; Karaulanov, E.; Berger, B.S.; Huang, Y.L.; Niehrs, C. Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol. Cell 2014, 54, 663–674. [Google Scholar] [CrossRef]
- Hinze, L.; Pfirrmann, M.; Karim, S.; Degar, J.; McGuckin, C.; Vinjamur, D.; Sacher, J.; Stevenson, K.E.; Neuberg, D.S.; Orellana, E.; et al. Synthetic Lethality of Wnt Pathway Activation and Asparaginase in Drug-Resistant Acute Leukemias. Cancer Cell 2019, 35, 664–676.e667. [Google Scholar] [CrossRef]
- Hinze, L.; Schreek, S.; Zeug, A.; Ibrahim, N.K.; Fehlhaber, B.; Loxha, L.; Cinar, B.; Ponimaskin, E.; Degar, J.; McGuckin, C.; et al. Supramolecular assembly of GSK3alpha as a cellular response to amino acid starvation. Mol. Cell 2022, 82, 2858–2870.e2858. [Google Scholar] [CrossRef]
- Tameire, F.; Verginadis, L., II; Leli, N.M.; Polte, C.; Conn, C.S.; Ojha, R.; Salas Salinas, C.; Chinga, F.; Monroy, A.M.; Fu, W.; et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 2019, 21, 889–899, published correction appears in Nat Cell Biol. 2019, 21, 1052. [Google Scholar] [CrossRef]
- Butler, M.; van Ingen Schenau, D.S.; Yu, J.; Jenni, S.; Dobay, M.P.; Hagelaar, R.; Vervoort, B.M.T.; Tee, T.M.; Hoff, F.W.; Meijerink, J.P.; et al. BTK inhibition sensitizes acute lymphoblastic leukemia to asparaginase by suppressing the amino acid response pathway. Blood 2021, 138, 2383–2395. [Google Scholar] [CrossRef]
- Srivastava, S.; Jiang, J.; Misra, J.; Seim, G.; Staschke, K.A.; Zhong, M.; Zhou, L.; Liu, Y.; Chen, C.; Dave, U.; et al. Asparagine bioavailability regulates the translation of MYC oncogene. Oncogene 2022, 41, 4855–4865. [Google Scholar] [CrossRef]
- Rosilio, C.; Nebout, M.; Imbert, V.; Griessinger, E.; Neffati, Z.; Benadiba, J.; Hagenbeek, T.; Spits, H.; Reverso, J.; Ambrosetti, D.; et al. L-type amino-acid transporter 1 (LAT1): A therapeutic target supporting growth and survival of T-cell lymphoblastic lymphoma/T-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 1253–1266. [Google Scholar] [CrossRef]
- Yanagida, O.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Segawa, H.; Nii, T.; Cha, S.H.; Matsuo, H.; Fukushima, J.; Fukasawa, Y.; et al. Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta 2001, 1514, 291–302. [Google Scholar] [CrossRef]
- Heraudet, L.; Galtier, J.; Favre, S.; Peyraud, F.; Cazaubiel, T.; Leroy, H.; Mottal, N.; Gros, F.X.; Forcade, E.; Clement, L.; et al. VANDA regimen followed by blinatumomab leads to favourable outcome in patients with Philadelphia chromosome-negative B-precursor acute lymphoblastic leukaemia in first relapse. Br. J. Haematol. 2022, 198, 523–527. [Google Scholar] [CrossRef]
- Fischer, D.; Toenges, R.; Kiil, K.; Michalik, S.; Thalhammer, A.; Bug, G.; Gokbuget, N.; Lang, F. Liver failure after treatment with inotuzumab and polychemotherapy including PEG-asparaginase in a patient with relapsed Philadelphia chromosome-negative acute lymphoblastic leukemia. Ann. Hematol. 2024, 103, 489–498. [Google Scholar] [CrossRef]
- Earl, M. Incidence and management of asparaginase-associated adverse events in patients with acute lymphoblastic leukemia. Clin. Adv. Hematol. Oncol. 2009, 7, 600–606. [Google Scholar]
- Wilson, G.J.; Bunpo, P.; Cundiff, J.K.; Wek, R.C.; Anthony, T.G. The eukaryotic initiation factor 2 kinase GCN2 protects against hepatotoxicity during asparaginase treatment. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1124–E1133. [Google Scholar] [CrossRef]
- Wilson, G.J.; Lennox, B.A.; She, P.; Mirek, E.T.; Al Baghdadi, R.J.; Fusakio, M.E.; Dixon, J.L.; Henderson, G.C.; Wek, R.C.; Anthony, T.G. GCN2 is required to increase fibroblast growth factor 21 and maintain hepatic triglyceride homeostasis during asparaginase treatment. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E283–E293. [Google Scholar] [CrossRef]
- Phillipson-Weiner, L.; Mirek, E.T.; Wang, Y.; McAuliffe, W.G.; Wek, R.C.; Anthony, T.G. General control nonderepressible 2 deletion predisposes to asparaginase-associated pancreatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G1061–G1070. [Google Scholar] [CrossRef]
- Morris, S.M., Jr. Arginine Metabolism Revisited. J. Nutr. 2016, 146, 2579S–2586S. [Google Scholar] [CrossRef]
- Closs, E.I.; Simon, A.; Vekony, N.; Rotmann, A. Plasma membrane transporters for arginine. J. Nutr. 2004, 134, 2752S–2759S; discussion 2765S–2767S. [Google Scholar] [CrossRef] [PubMed]
- Appleton, J. Arginine: Clinical potential of a semi-essential amino acid. Altern Med. Rev. 2002, 7, 512–522. [Google Scholar] [PubMed]
- Szefel, J.; Danielak, A.; Kruszewski, W.J. Metabolic pathways of L-arginine and therapeutic consequences in tumors. Adv. Med. Sci. 2019, 64, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Fenouille, N.; Bassil, C.F.; Ben-Sahra, I. The creatine kinase pathway is a metabolic vulnerability in EVI1-positive acute myeloid leukemia. Nat. Med. 2017, 23, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Santo, C.; Booth, S.; Vardon, A. The arginine metabolome in acute lymphoblastic leukemia can be targeted by the pegylated-recombinant arginase I BCT-100. Int. J. Cancer 2018, 142, 1490–1502. [Google Scholar] [CrossRef]
- Mussai, F.; Egan, S.; Higginbotham-Jones, J.; Perry, T.; Beggs, A.; Odintsova, E.; Loke, J.; Pratt, G.; U, K.P.; Lo, A.; et al. Arginine dependence of acute myeloid leukemia blast proliferation: A novel therapeutic target. Blood 2015, 125, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.P.; Morrow, K.; Lopez-Barcons, L.A.; Zabaleta, J.; Sierra, R.; Velasco, C.; Cole, J.; Rodriguez, P.C. Pegylated arginase I: A potential therapeutic approach in T-ALL. Blood 2010, 115, 5214–5221. [Google Scholar] [CrossRef]
- Patil, M.D.; Bhaumik, J.; Babykutty, S.; Banerjee, U.C.; Fukumura, D. Arginine dependence of tumor cells: Targeting a chink in cancer’s armor. Oncogene 2016, 35, 4957–4972. [Google Scholar] [CrossRef]
- Yau, T.; Cheng, P.N.; Chan, P.; Chen, L.; Yuen, J.; Pang, R.; Fan, S.T.; Wheatley, D.N.; Poon, R.T. Preliminary efficacy, safety, pharmacokinetics, pharmacodynamics and quality of life study of pegylated recombinant human arginase 1 in patients with advanced hepatocellular carcinoma. Investig. New Drugs 2015, 33, 496–504. [Google Scholar] [CrossRef]
- Major, P.P.; Egan, E.M.; Beardsley, G.P.; Minden, M.D. Lethality of human myeloblasts correlates with the incorporation of arabinofuranosylcytosine into DNA. Proc. Natl. Acad. Sci. USA 1981, 78, 3235–3239. [Google Scholar] [CrossRef]
- Glazer, E.S.; Stone, E.M.; Zhu, C.; Massey, K.L.; Hamir, A.N.; Curley, S.A. Bioengineered human arginase I with enhanced activity and stability controls hepatocellular and pancreatic carcinoma xenografts. Transl. Oncol. 2012, 4, 138–146. [Google Scholar] [CrossRef]
- Tanios, R.; Bekdash, A.; Kassab, E.; Stone, E.; Georgiou, G.; Frankel, A.E.; Abi-Habib, R.J. Human recombinant arginase I(Co)-PEG5000 [HuArgI(Co)-PEG5000]-induced arginine depletion is selectively cytotoxic to human acute myeloid leukemia cells. Leuk. Res. 2013, 37, 1565–1571. [Google Scholar] [CrossRef]
- Jones, J.B. The Effect of Arginine Deiminase on Murine Leukemic Lymphoblasts; The University of Oklahoma Health Sciences Center: Oklahoma, OK, USA, 1981. [Google Scholar]
- Takaku, H.; Matsumoto, M.; Misawa, S.; Miyazaki, K. Anti-tumor activity of arginine deiminase from Mycoplasma argini and its growth-inhibitory mechanism. Jpn J. Cancer Res. 1995, 86, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Takaku, H.; Takase, M.; Abe, S.; Hayashi, H.; Miyazaki, K. In vivo anti-tumor activity of arginine deiminase purified from Mycoplasma arginini. Int. J. Cancer 1992, 51, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.J.; Shen, W.C. Drug evaluation: ADI-PEG-20--a PEGylated arginine deiminase for arginine-auxotrophic cancers. Curr. Opin. Mol. Ther. 2006, 8, 240–248. [Google Scholar]
- Gong, H.; Zolzer, F.; von Recklinghausen, G.; Havers, W.; Schweigerer, L. Arginine deiminase inhibits proliferation of human leukemia cells more potently than asparaginase by inducing cell cycle arrest and apoptosis. Leukemia 2000, 14, 826–829. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Scala, S.; Castello, G.; Daponte, A.; Simeone, E.; Ottaiano, A.; Beneduce, G.; De Rosa, V.; Izzo, F.; Melucci, M.T.; et al. Pegylated arginine deiminase treatment of patients with metastatic melanoma: Results from phase I and II studies. J. Clin. Oncol. 2005, 23, 7660–7668. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.J.; Jiang, S.S.; Hung, W.C.; Borthakur, G.; Lin, S.F.; Pemmaraju, N.; Jabbour, E.; Bomalaski, J.S.; Chen, Y.P.; Hsiao, H.H.; et al. A phase II study of arginine deiminase (ADI-PEG20) in relapsed/refractory or poor-risk acute myeloid leukemia patients. Sci. Rep. 2017, 7, 11253. [Google Scholar] [CrossRef]
- Miraki-Moud, F.; Ghazaly, E.; Ariza-McNaughton, L.; Hodby, K.A.; Clear, A.; Anjos-Afonso, F.; Liapis, K.; Grantham, M.; Sohrabi, F.; Cavenagh, J.; et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood 2015, 125, 4060–4068. [Google Scholar] [CrossRef]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef]
- Deng, M.; Gui, X.; Kim, J.; Xie, L.; Chen, W.; Li, Z.; He, L.; Chen, Y.; Chen, H.; Luo, W.; et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature 2018, 562, 605–609. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef]
- i Líndez, A.-A.M.; Dunand-Sauthier, I.; Conti, M.; Gobet, F.; Núñez, N.; Hannich, J.T.; Riezman, H.; Geiger, R.; Piersigilli, A.; Hahn, K. Mitochondrial arginase-2 is a cell-autonomous regulator of CD8+ T cell function and antitumor efficacy. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Mussai, F.; Wheat, R.; Sarrou, E.; Booth, S.; Stavrou, V.; Fultang, L.; Perry, T.; Kearns, P.; Cheng, P.; Keeshan, K.; et al. Targeting the arginine metabolic brake enhances immunotherapy for leukaemia. Int. J. Cancer 2019, 145, 2201–2208. [Google Scholar] [CrossRef]
- Silk, J.D.; Lakhal, S.; Laynes, R.; Vallius, L.; Karydis, I.; Marcea, C.; Boyd, C.A.; Cerundolo, V. IDO induces expression of a novel tryptophan transporter in mouse and human tumor cells. J. Immunol. 2011, 187, 1617–1625. [Google Scholar] [CrossRef]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, 9794. [Google Scholar] [CrossRef] [PubMed]
- Le Floc’h, N.; Otten, W.; Merlot, E. Tryptophan metabolism, from nutrition to potential therapeutic applications. Amino Acids 2011, 41, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Chamuleau, M.E.; van de Loosdrecht, A.A.; Hess, C.J.; Janssen, J.J.; Zevenbergen, A.; Delwel, R.; Valk, P.J.; Lowenberg, B.; Ossenkoppele, G.J. High INDO (indoleamine 2,3-dioxygenase) mRNA level in blasts of acute myeloid leukemic patients predicts poor clinical outcome. Haematologica 2008, 93, 1894–1898. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e487. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25 into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Rannug, A.; Stockinger, B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009, 30, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Neamah, W.H.; Singh, N.P.; Alghetaa, H.; Abdulla, O.A.; Chatterjee, S.; Busbee, P.B.; Nagarkatti, M.; Nagarkatti, P. AhR Activation Leads to Massive Mobilization of Myeloid-Derived Suppressor Cells with Immunosuppressive Activity through Regulation of CXCR2 and MicroRNA miR-150-5p and miR-543-3p That Target Anti-Inflammatory Genes. J. Immunol. 2019, 203, 1830–1844. [Google Scholar] [CrossRef] [PubMed]
- Sobash, P.T.; Kolhe, R.; Karim, N.A.; Guddati, A.K.; Jillella, A.; Kota, V. Role of indoleamine 2,3-dioxygenase in acute myeloid leukemia. Future Oncol. 2020, 16, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef]
- Stone, T.W.; Williams, R.O. Modulation of T cells by tryptophan metabolites in the kynurenine pathway. Trends Pharmacol. Sci. 2023, 44, 442–456. [Google Scholar] [CrossRef]
- Liu, D.; Chen, B.; Mo, Y. Redox-activated porphyrin-based liposome remote-loaded with indoleamine 2,3-dioxygenase (IDO) inhibitor for synergistic photoimmunotherapy through induction of immunogenic cell death and blockage of IDO pathway. Nano Lett. 2019, 19, 6964–6976. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Kolb, H.J.; Schattenberg, A.; Goldman, J.M.; Hertenstein, B.; Jacobsen, N.; Arcese, W.; Ljungman, P.; Ferrant, A.; Verdonck, L.; Niederwieser, D.; et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood 1995, 86, 2041–2050. [Google Scholar] [CrossRef]
- Lim, J.-Y.; Lee, S.-E.; Park, G.; Choi, E.Y.; Min, C.-K. Inhibition of indoleamine 2, 3-dioxygenase by stereoisomers of 1-methyl tryptophan in an experimental graft-versus-tumor model. Exp. Hematol. 2014, 42, 862–866.e863. [Google Scholar] [CrossRef]
- Cruz, C.R.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef]
- El Kholy, N.M.; Sallam, M.M.; Ahmed, M.B.; Sallam, R.M.; Asfour, I.A.; Hammouda, J.A.; Habib, H.Z.; Abu-Zahra, F. Expression of indoleamine 2,3-dioxygenase in acute myeloid leukemia and the effect of its inhibition on cultured leukemia blast cells. Med. Oncol. 2011, 28, 270–278. [Google Scholar] [CrossRef]
- Mellor, A.L.; Chandler, P.; Baban, B.; Hansen, A.M.; Marshall, B.; Pihkala, J.; Waldmann, H.; Cobbold, S.; Adams, E.; Munn, D.H. Specific subsets of murine dendritic cells acquire potent T cell regulatory functions following CTLA4-mediated induction of indoleamine 2, 3 dioxygenase. Int. Immunol. 2004, 16, 1391–1401. [Google Scholar] [CrossRef]
- Muller, A.J.; Sharma, M.D.; Chandler, P.R.; DuHadaway, J.B.; Everhart, M.E.; Johnson III, B.A.; Kahler, D.J.; Pihkala, J.; Soler, A.P.; Munn, D.H. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2, 3 dioxygenase. Proc. Natl. Acad. Sci. USA 2008, 105, 17073–17078. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Hou, D.; Baban, B.; Lee, J.R.; Antonia, S.J.; Messina, J.L.; Chandler, P.; Koni, P.A.; Mellor, A.L. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.; Oliver, T.; Rowe, M.; Thomas, S.; Zakharia, Y.; Gilman, P.B.; Muller, A.J.; Prendergast, G.C. Indoximod: An immunometabolic adjuvant that empowers T cell activity in cancer. Front. Oncol. 2018, 8, 370. [Google Scholar] [CrossRef]
- Metz, R.; Duhadaway, J.B.; Kamasani, U.; Laury-Kleintop, L.; Muller, A.J.; Prendergast, G.C. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007, 67, 7082–7087. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Opitz, U.; Sahm, F.; Ochs, K.; Lutz, C.; Wick, W.; Platten, M. The indoleamine-2, 3-dioxygenase (IDO) inhibitor 1-methyl-D-tryptophan upregulates IDO1 in human cancer cells. PLoS ONE 2011, 6, e19823. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, G.; Li, L.; Li, D.; Dong, Z.; Jiang, P. Asparagine enhances LCK signalling to potentiate CD8(+) T-cell activation and anti-tumour responses. Nat. Cell Biol. 2021, 23, 75–86. [Google Scholar] [CrossRef]
- Gnanaprakasam, J.N.R.; Kushwaha, B.; Liu, L.; Chen, X.; Kang, S.; Wang, T.; Cassel, T.A.; Adams, C.M.; Higashi, R.M.; Scott, D.A.; et al. Asparagine restriction enhances CD8(+) T cell metabolic fitness and antitumoral functionality through an NRF2-dependent stress response. Nat. Metab. 2023, 5, 1423–1439. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Registered Number | Disease | Phase | Group | Status | Reference |
|---|---|---|---|---|---|
| NCT02071927 | R/R leukemia | I | Single-agent CB-839/CB-839 and AZA | Complete | N.A |
| NCT03047993 | Advanced MDS | I/II | CB-839 and AZA | Complete | [29] |
| NCT04666649 | R/R AML | I | Ven-PegC | Ongoing | N.A |
| NCT02899286 | R/R AML | II | PEG-BCT-100 | Complete | N.A |
| NCT01551628 | R/R leukemia | I | Recombinant human arginase1 Peg-5000 | Terminated(slow patient recruitment) | N.A |
| NCT02732184 | R/R AML or MDS | II | Co-ArgI-PEG modified human arginase I | Complete | N.A |
| NCT01910012 | R/RAML | II | ADI-PEG 20 | Complete | [8] |
| NCT05001828 | High risk AML | I | ADI-PEG 20, Venetoclax and Azacitidine | Ongoing | N.A |
| NCT02835729 | ND-AML | I | Indoximodin, Idarubicin and Cytarabine | Complete | [30] |
| 2006-005694-21 (EWALL-PH-01) | ND-Ph+ and/or CR-ABL1+ ALL | II | Dasatinib, cytarabine, asparaginase and methotrexate | Complete | [31] |
| NCT01085617(UKALL14) | newly diagnosed ALL | III | PEG-ASP and standard induction regimen (ph + disease received continuous oral imatinib) | Complete | [32] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Zhang, J. Enhancing Leukemia Treatment: The Role of Combined Therapies Based on Amino Acid Starvation. Cancers 2024, 16, 1171. https://doi.org/10.3390/cancers16061171
Chen C, Zhang J. Enhancing Leukemia Treatment: The Role of Combined Therapies Based on Amino Acid Starvation. Cancers. 2024; 16(6):1171. https://doi.org/10.3390/cancers16061171
Chicago/Turabian StyleChen, Can, and Ji Zhang. 2024. "Enhancing Leukemia Treatment: The Role of Combined Therapies Based on Amino Acid Starvation" Cancers 16, no. 6: 1171. https://doi.org/10.3390/cancers16061171
APA StyleChen, C., & Zhang, J. (2024). Enhancing Leukemia Treatment: The Role of Combined Therapies Based on Amino Acid Starvation. Cancers, 16(6), 1171. https://doi.org/10.3390/cancers16061171