
The Diagnostic Value of ACSL1, ACSL4, and ACSL5 and the Clinical Potential of an ACSL Inhibitor in Non-Small-Cell Lung Cancer
 ,
,  , ,
, ,  , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. RNA Extraction, Reverse Transcription (RT), and Quantitative PCR (qPCR)
2.3. Protein Isolation and Western Blotting (WB)
2.4. Measurement of Total ACSLs Activity Using the Substrate [3H]-Palmitic Acid (PA)
2.5. Multiplex Immunofluorescence (mIF)
2.6. Immunohistochemistry (IHC)
2.7. Drug Treatment and Transfection
2.8. Cell Viability Assay
2.9. Data Analysis
3. Results
3.1. Expression and Enzymatic Activity of ACSL Isoforms in Normal Human Bronchial Epithelium (NHBE) Cells and Lung Cancer Cell Lines
3.2. Tissue Distribution of ACSL Isoforms in Normal Human Lung Tissues
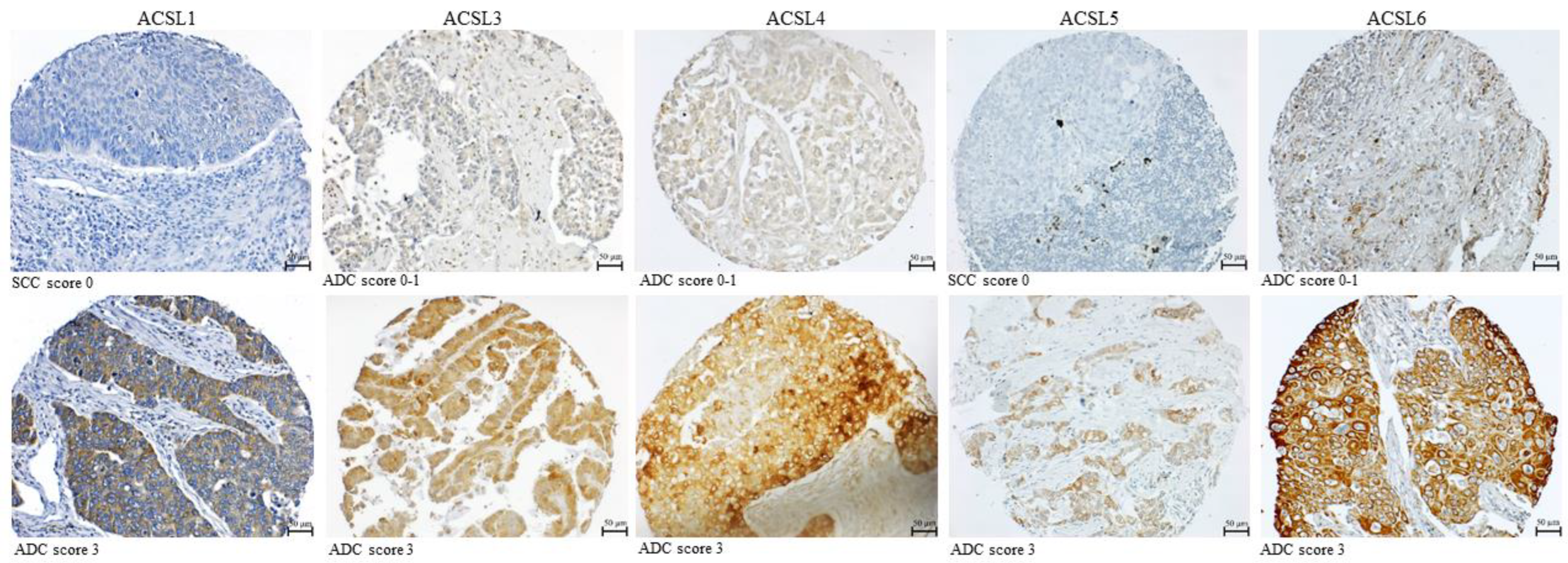
3.3. The Correlation between the Expression of ACSL Isoforms and the Clinicopathological Parameters in Primary Lung Tumor Tissues
3.4. ACSL Enzymatic Activity after ACSL Inhibitor (Triacsin C) Treatment
3.5. The Effect of Triacsin C Combined with Chemotherapy/Targeted Therapeutic Drugs
3.6. The Effects of ACSL1, ACSL4, and ACSL5 Knockdown on Cell Viability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Zahan, T.; Das, P.K.; Akter, S.F.; Habib, R.; Rahman, M.H.; Karim, M.R.; Islam, F. Therapy Resistance in Cancers: Phenotypic, Metabolic, Epigenetic and Tumour Microenvironmental Perspectives. Anti-Cancer Agents Med. Chem. 2020, 20, 2190–2206. [Google Scholar] [CrossRef]
- Wang, Y.; Patti, G.J. The Warburg effect: A signature of mitochondrial overload. Trends Cell Biol. 2023, 33, 1014–1020. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Kaur, H.; Jung, K.H.; Yang, S.; Tripathi, S.; Galbraith, M.; Deng, Y.; Jolly, M.K.; Kaipparettu, B.A.; et al. Towards decoding the coupled decision-making of metabolism and epithelial-to-mesenchymal transition in cancer. Br. J. Cancer 2021, 124, 1902–1911. [Google Scholar] [CrossRef]
- Ma, Y.; Nenkov, M.; Chen, Y.; Press, A.T.; Kaemmerer, E.; Gassler, N. Fatty acid metabolism and acyl-CoA synthetases in the liver-gut axis. World J. Hepatol. 2021, 13, 1512–1533. [Google Scholar] [CrossRef]
- Gassler, N.; Roth, W.; Funke, B.; Schneider, A.; Herzog, F.; Tischendorf, J.J.; Grund, K.; Penzel, R.; Bravo, I.G.; Mariadason, J.; et al. Regulation of enterocyte apoptosis by acyl-CoA synthetase 5 splicing. Gastroenterology 2007, 133, 587–598. [Google Scholar] [CrossRef]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.H.; Luu, T.T.; Kim, D.; An, Y.J.; Park, S.; Park, H.J.; Lee, S.K. BMP4 Upregulation Is Associated with Acquired Drug Resistance and Fatty Acid Metabolism in EGFR-Mutant Non-Small-Cell Lung Cancer Cells. Mol. Ther. Nucleic Acids 2018, 12, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Klett, E.L.; Chen, S.; Yechoor, A.; Lih, F.B.; Coleman, R.A. Long-chain acyl-CoA synthetase isoforms differ in preferences for eicosanoid species and long-chain fatty acids. J. Lipid Res. 2017, 58, 884–894. [Google Scholar] [CrossRef] [PubMed]
- TNMplot. Available online: https://tnmplot.com (accessed on 18 January 2024).
- Bartha, Á.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2262. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Deng, Y.; Yang, Y.; Xu, L.; Fu, J. FATP2 regulates non-small cell lung cancer by mediating lipid metabolism through ACSL1. Tissue Cell 2023, 82, 102105. [Google Scholar] [CrossRef]
- Chen, W.C.; Wang, C.Y.; Hung, Y.H.; Weng, T.Y.; Yen, M.C.; Lai, M.D. Systematic Analysis of Gene Expression Alterations and Clinical Outcomes for Long-Chain Acyl-Coenzyme A Synthetase Family in Cancer. PLoS ONE 2016, 11, e0155660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, X.; Liu, Y.; Yan, F.; Zeng, Y.; Song, Y.; Fang, H.; Song, D.; Wang, X. Lysophosphatidylcholine inhibits lung cancer cell proliferation by regulating fatty acid metabolism enzyme long-chain acyl-coenzyme A synthase 5. Clin. Transl. Med. 2023, 13, e1180. [Google Scholar] [CrossRef]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Too complex to fail? Targeting fatty acid metabolism for cancer therapy. Prog. Lipid Res. 2022, 85, 101143. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lewin, T.M.; Coleman, R.A. Expression and characterization of recombinant rat Acyl-CoA synthetases 1, 4, and 5. Selective inhibition by triacsin C and thiazolidinediones. J. Biol. Chem. 2001, 276, 24667–24673. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, C.G.; Caviglia, J.M.; Li, L.O.; Wang, S.; Granger, D.A.; Coleman, R.A. Characterization of recombinant long-chain rat acyl-CoA synthetase isoforms 3 and 6: Identification of a novel variant of isoform 6. Biochemistry 2005, 44, 1635–1642. [Google Scholar] [CrossRef]
- Saraswathi, V.; Hasty, A.H. Inhibition of long-chain acyl coenzyme A synthetases during fatty acid loading induces lipotoxicity in macrophages. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1937–1943. [Google Scholar] [CrossRef]
- Dechandt, C.R.P.; Zuccolotto-Dos-Reis, F.H.; Teodoro, B.G.; Fernandes, A.; Eberlin, M.N.; Kettelhut, I.C.; Curti, C.; Alberici, L.C. Triacsin C reduces lipid droplet formation and induces mitochondrial biogenesis in primary rat hepatocytes. J. Bioenerg. Biomembr. 2017, 49, 399–411. [Google Scholar] [CrossRef]
- Mashima, T.; Oh-hara, T.; Sato, S.; Mochizuki, M.; Sugimoto, Y.; Yamazaki, K.; Hamada, J.; Tada, M.; Moriuchi, T.; Ishikawa, Y.; et al. p53-defective tumors with a functional apoptosome-mediated pathway: A new therapeutic target. J. Natl. Cancer Inst. 2005, 97, 765–777. [Google Scholar] [CrossRef]
- Zou, J.; Ganji, S.; Pass, I.; Ardecky, R.; Peddibhotla, M.; Loribelle, M.; Heynen-Genel, S.; Sauer, M.; Pass, I.; Vasile, S.; et al. Potent Inhibitors of Lipid Droplet Formation. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Mashima, T.; Sato, S.; Okabe, S.; Miyata, S.; Matsuura, M.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Acyl-CoA synthetase as a cancer survival factor: Its inhibition enhances the efficacy of etoposide. Cancer Sci. 2009, 100, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Polonio-Alcalá, E.; Porta, R.; Ruiz-Martínez, S.; Vásquez-Dongo, C.; Relat, J.; Bosch-Barrera, J.; Ciurana, J.; Puig, T. AZ12756122, a novel fatty acid synthase inhibitor, decreases resistance features in EGFR-TKI resistant EGFR-mutated NSCLC cell models. Biomed. Pharmacother. 2022, 156, 113942. [Google Scholar] [CrossRef] [PubMed]
- Füllekrug, J.; Poppelreuther, M. Measurement of Long-Chain Fatty Acyl-CoA Synthetase Activity. Methods Mol. Biol. 2016, 1376, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Meier Plotter. Available online: https://kmplot.com/analysis/index.php?p=service (accessed on 18 January 2024).
- Zhao, B.X.; Wang, J.; Song, B.; Wei, H.; Lv, W.P.; Tian, L.M.; Li, M.; Lv, S. Establishment and biological characteristics of acquired gefitinib resistance in cell line NCI-H1975/gefinitib-resistant with epidermal growth factor receptor T790M mutation. Mol. Med. Rep. 2015, 11, 2767–2774. [Google Scholar] [CrossRef]
- Choi, Y.J.; Rho, J.K.; Jeon, B.S.; Choi, S.J.; Park, S.C.; Lee, S.S.; Kim, H.R.; Kim, C.H.; Lee, J.C. Combined inhibition of IGFR enhances the effects of gefitinib in H1650: A lung cancer cell line with EGFR mutation and primary resistance to EGFR-TK inhibitors. Cancer Chemother. Pharmacol. 2010, 66, 381–388. [Google Scholar] [CrossRef]
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr. 2014, 34, 1–30. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.; Li, F.; Lv, C.; Yang, Q.K. High-fat diet impairs ferroptosis and promotes cancer invasiveness via downregulating tumor suppressor ACSL4 in lung adenocarcinoma. Biol. Direct 2021, 16, 10. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 18 January 2024).
- Agudelo, C.W.; Samaha, G.; Garcia-Arcos, I. Alveolar lipids in pulmonary disease. A review. Lipids Health Dis. 2020, 19, 122. [Google Scholar] [CrossRef]
- Surkova, S.; Sokolkova, A.; Kozlov, K.; Nuzhdin, S.V.; Samsonova, M. Quantitative analysis reveals genotype- and domain-specific differences between mRNA and protein expression of segmentation genes in Drosophila. Dev. Biol. 2019, 448, 48–58. [Google Scholar] [CrossRef]
- Jia, P.; Zhao, Z. Impacts of somatic mutations on gene expression: An association perspective. Brief. Bioinform. 2017, 18, 413–425. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Sanford, J.D.; Jin, A.; Grois, G.A.; Zhang, Y. A role of cytoplasmic p53 in the regulation of metabolism shown by bat-mimicking p53 NLS mutant mice. Cell Rep. 2023, 42, 111920. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Marques, M.; Cunha, I.; Reis-Henriques, M.A.; Santos, M.M.; Castro, L.F. Diversity and history of the long-chain acyl-CoA synthetase (Acsl) gene family in vertebrates. BMC Evol. Biol. 2013, 13, 271. [Google Scholar] [CrossRef]
- Brinkman, B.M. Splice variants as cancer biomarkers. Clin. Biochem. 2004, 37, 584–594. [Google Scholar] [CrossRef]
- Soupene, E.; Kuypers, F.A. Multiple erythroid isoforms of human long-chain acyl-CoA synthetases are produced by switch of the fatty acid gate domains. BMC Mol. Biol. 2006, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yu, Y.; Zhang, L.; Liu, Y.; Zheng, K.; Wang, S.; Jin, H.; Liu, L.; Cao, Y. Transcript variants of long-chain acyl-CoA synthase 1 have distinct roles in sheep lipid metabolism. Front. Genet. 2022, 13, 1021103. [Google Scholar] [CrossRef]
- Yang, G.; Wang, Y.; Feng, J.; Liu, Y.; Wang, T.; Zhao, M.; Ye, L.; Zhang, X. Aspirin suppresses the abnormal lipid metabolism in liver cancer cells via disrupting an NFκB-ACSL1 signaling. Biochem. Biophys. Res. Commun. 2017, 486, 827–832. [Google Scholar] [CrossRef]
- Sánchez-Martínez, R.; Cruz-Gil, S.; García-Álvarez, M.S.; Reglero, G.; Ramírez de Molina, A. Complementary ACSL isoforms contribute to a non-Warburg advantageous energetic status characterizing invasive colon cancer cells. Sci. Rep. 2017, 7, 11143. [Google Scholar] [CrossRef]
- Ma, Y.; Zha, J.; Yang, X.; Li, Q.; Zhang, Q.; Yin, A.; Beharry, Z.; Huang, H.; Huang, J.; Bartlett, M.; et al. Long-chain fatty acyl-CoA synthetase 1 promotes prostate cancer progression by elevation of lipogenesis and fatty acid beta-oxidation. Oncogene 2021, 40, 1806–1820. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, X.; Zhang, S.; Cui, M.; Liu, F.; Sun, B.; Zhang, W.; Zhang, X.; Ye, L. HBXIP up-regulates ACSL1 through activating transcriptional factor Sp1 in breast cancer. Biochem. Biophys. Res. Commun. 2017, 484, 565–571. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhou, W.; Yu, S.; Ju, Y.; To, S.K.Y.; Wong, A.S.T.; Jiao, Y.; Poon, T.C.W.; Tam, K.Y.; Lee, L.T.O. Metabolic reprogramming of ovarian cancer involves ACSL1-mediated metastasis stimulation through upregulated protein myristoylation. Oncogene 2021, 40, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Y.; Liu, F.; Cao, Y.; Peng, S.L.; Pan, H.W.; Hong, X.Q.; Zheng, P.F. ACSL1, CH25H, GPCPD1, and PLA2G12A as the potential lipid-related diagnostic biomarkers of acute myocardial infarction. Aging 2023, 15, 1394–1411. [Google Scholar] [CrossRef]
- Roelands, J.; Garand, M.; Hinchcliff, E.; Ma, Y.; Shah, P.; Toufiq, M.; Alfaki, M.; Hendrickx, W.; Boughorbel, S.; Rinchai, D.; et al. Long-Chain Acyl-CoA Synthetase 1 Role in Sepsis and Immunity: Perspectives From a Parallel Review of Public Transcriptome Datasets and of the Literature. Front. Immunol. 2019, 10, 2410. [Google Scholar] [CrossRef] [PubMed]
- Matesanz, F.; Fedetz, M.; Barrionuevo, C.; Karaky, M.; Catalá-Rabasa, A.; Potenciano, V.; Bello-Morales, R.; López-Guerrero, J.A.; Alcina, A. A splice variant in the ACSL5 gene relates migraine with fatty acid activation in mitochondria. Eur. J. Hum. Genet. 2016, 24, 1572–1577. [Google Scholar] [CrossRef]
- Rajkumar, A.; Liaghati, A.; Chan, J.; Lamothe, G.; Dent, R.; Doucet, É.; Rabasa-Lhoret, R.; Prud’homme, D.; Harper, M.E.; Tesson, F. ACSL5 genotype influence on fatty acid metabolism: A cellular, tissue, and whole-body study. Metabolism 2018, 83, 271–279. [Google Scholar] [CrossRef]
- Pérez-Núñez, I.; Karaky, M.; Fedetz, M.; Barrionuevo, C.; Izquierdo, G.; Matesanz, F.; Alcina, A. Splice-site variant in ACSL5: A marker promoting opposing effect on cell viability and protein expression. Eur. J. Hum. Genet. 2019, 27, 1836–1844. [Google Scholar] [CrossRef]
- Teng, A.C.; Adamo, K.; Tesson, F.; Stewart, A.F. Functional characterization of a promoter polymorphism that drives ACSL5 gene expression in skeletal muscle and associates with diet-induced weight loss. FASEB J. 2009, 23, 1705–1709. [Google Scholar] [CrossRef]
- Reinartz, A.; Ehling, J.; Leue, A.; Liedtke, C.; Schneider, U.; Kopitz, J.; Weiss, T.; Hellerbrand, C.; Weiskirchen, R.; Knüchel, R.; et al. Lipid-induced up-regulation of human acyl-CoA synthetase 5 promotes hepatocellular apoptosis. Biochim. Biophys. Acta 2010, 1801, 1025–1035. [Google Scholar] [CrossRef]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef]
- Klaus, C.; Schneider, U.; Hedberg, C.; Schütz, A.K.; Bernhagen, J.; Waldmann, H.; Gassler, N.; Kaemmerer, E. Modulating effects of acyl-CoA synthetase 5-derived mitochondrial Wnt2B palmitoylation on intestinal Wnt activity. World J. Gastroenterol. 2014, 20, 14855–14864. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Sato, S.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Promotion of glioma cell survival by acyl-CoA synthetase 5 under extracellular acidosis conditions. Oncogene 2009, 28, 9–19. [Google Scholar] [CrossRef]
- Belkaid, A.; Ouellette, R.J.; Surette, M.E. 17β-estradiol-induced ACSL4 protein expression promotes an invasive phenotype in estrogen receptor positive mammary carcinoma cells. Carcinogenesis 2017, 38, 402–410. [Google Scholar] [CrossRef]
- Wu, M.; Liu, H.; Chen, W.; Fujimoto, Y.; Liu, J. Hepatic expression of long-chain acyl-CoA synthetase 3 is upregulated in hyperlipidemic hamsters. Lipids 2009, 44, 989–998. [Google Scholar] [CrossRef]
- Ansari, I.H.; Longacre, M.J.; Stoker, S.W.; Kendrick, M.A.; O’Neill, L.M.; Zitur, L.J.; Fernandez, L.A.; Ntambi, J.M.; MacDonald, M.J. Characterization of Acyl-CoA synthetase isoforms in pancreatic beta cells: Gene silencing shows participation of ACSL3 and ACSL4 in insulin secretion. Arch. Biochem. Biophys. 2017, 618, 32–43. [Google Scholar] [CrossRef]
- Kuwata, H.; Hara, S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat. 2019, 144, 106363. [Google Scholar] [CrossRef]
- Kang, M.J.; Fujino, T.; Sasano, H.; Minekura, H.; Yabuki, N.; Nagura, H.; Iijima, H.; Yamamoto, T.T. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc. Natl. Acad. Sci. USA 1997, 94, 2880–2884. [Google Scholar] [CrossRef]
- Shimbara-Matsubayashi, S.; Kuwata, H.; Tanaka, N.; Kato, M.; Hara, S. Analysis on the Substrate Specificity of Recombinant Human Acyl-CoA Synthetase ACSL4 Variants. Biol. Pharm. Bull. 2019, 42, 850–855. [Google Scholar] [CrossRef]
- Sánchez-Martínez, R.; Cruz-Gil, S.; Gómez de Cedrón, M.; Álvarez-Fernández, M.; Vargas, T.; Molina, S.; García, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719–38736. [Google Scholar] [CrossRef]
- Marszalek, J.R.; Kitidis, C.; Dirusso, C.C.; Lodish, H.F. Long-chain acyl-CoA synthetase 6 preferentially promotes DHA metabolism. J. Biol. Chem. 2005, 280, 10817–10826. [Google Scholar] [CrossRef] [PubMed]
- Orlando, U.D.; Castillo, A.F.; Medrano, M.A.R.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Acyl-CoA synthetase-4 is implicated in drug resistance in breast cancer cell lines involving the regulation of energy-dependent transporter expression. Biochem. Pharmacol. 2019, 159, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Nenkov, M.; Ma, Y.; Gaßler, N.; Chen, Y. Metabolic Reprogramming of Colorectal Cancer Cells and the Microenvironment: Implication for Therapy. Int. J. Mol. Sci. 2021, 22, 6262. [Google Scholar] [CrossRef]
- Cao, A.; Li, H.; Zhou, Y.; Wu, M.; Liu, J. Long chain acyl-CoA synthetase-3 is a molecular target for peroxisome proliferator-activated receptor delta in HepG2 hepatoma cells. J. Biol. Chem. 2010, 285, 16664–16674. [Google Scholar] [CrossRef]
- Kim, Y.; George, D.; Prior, A.M.; Prasain, K.; Hao, S.; Le, D.D.; Hua, D.H.; Chang, K.O. Novel triacsin C analogs as potential antivirals against rotavirus infections. Eur. J. Med. Chem. 2012, 50, 311–318. [Google Scholar] [CrossRef]
- Li, Y.J.; Fahrmann, J.F.; Aftabizadeh, M.; Zhao, Q.; Tripathi, S.C.; Zhang, C.; Yuan, Y.; Ann, D.; Hanash, S.; Yu, H. Fatty acid oxidation protects cancer cells from apoptosis by increasing mitochondrial membrane lipids. Cell Rep. 2022, 39, 111044. [Google Scholar] [CrossRef]
- Xia, H.; Zhang, Z.; You, F. Inhibiting ACSL1-Related Ferroptosis Restrains Murine Coronavirus Infection. Viruses 2021, 13, 2383. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e429. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Number | ACSL1 | p-Value | Sample Number | ACSL3 | p-Value | Sample Number | ACSL4 | p-Value | Sample Number | ACSL5 | p-Value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1–3 | 0–1 | 2–3 | 0–1 | 2–3 | 0 | 1–3 | ||||||||||
| Type | ADC | 85 | 4 | 43 | 0.011 | 44 | 13 | 12 | 0.213 | 69 | 30 | 11 | 0.021 | 88 | 27 | 22 | 0 |
| SCC | 12 | 26 | 14 | 5 | 27 | 1 | 38 | 1 | |||||||||
| Gender | Male | 97 | 18 | 57 | 0.387 | 53 | 29 | 12 | 0.09 | 78 | 48 | 12 | 0.501 | 101 | 62 | 16 | 0.069 |
| Female | 3 | 19 | 5 | 7 | 16 | 2 | 14 | 9 | |||||||||
| Age | <60 | 97 | 4 | 29 | 0.124 | 53 | 7 | 9 | 0.042 | 78 | 18 | 8 | 0.037 | 101 | 24 | 9 | 0.683 |
| ≥60 | 17 | 47 | 27 | 10 | 46 | 6 | 52 | 16 | |||||||||
| pT | 1–2 | 93 | 18 | 64 | 1 | 49 | 24 | 16 | 0.454 | 75 | 56 | 10 | 0.345 | 97 | 61 | 23 | 0.504 |
| 3–4 | 2 | 9 | 7 | 2 | 6 | 3 | 11 | 2 | |||||||||
| pN | 0–1 | 91 | 12 | 51 | 0.519 | 49 | 20 | 15 | 0.202 | 73 | 45 | 7 | 0.127 | 95 | 45 | 19 | 0.210 |
| 2–4 | 7 | 21 | 11 | 3 | 15 | 6 | 26 | 5 | |||||||||
| pM | 0 | 97 | 18 | 69 | 0.685 | 53 | 30 | 17 | 1 | 78 | 58 | 12 | 0.629 | 101 | 70 | 22 | 0.686 |
| 1–3 | 3 | 7 | 4 | 2 | 6 | 2 | 6 | 3 | |||||||||
| Grade | 1–2 | 97 | 6 | 43 | 0.023 | 53 | 14 | 13 | 0.057 | 78 | 39 | 3 | 0.009 | 101 | 32 | 17 | 0.025 |
| 3–4 | 15 | 33 | 20 | 6 | 25 | 11 | 44 | 8 | |||||||||
| Type | Sample Number | Grade | ACSL1 | p-Value | Sample Number | Grade | ACSL3 | p-Value | Sample Number | Grade | ACSL4 | p-Value | Sample Number | Grade | ACSL5 | p-Value | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1–3 | 0–1 | 2–3 | 0–1 | 2–3 | 0 | 1–3 | |||||||||||||
| ADC | 47 | 1–2 | 0 | 25 | 0.026 | 25 | 1–2 | 4 | 9 | 0.027 | 41 | 1–2 | 20 | 3 | 0.024 | 49 | 1–2 | 10 | 15 | 0.03 |
| 3–4 | 4 | 18 | 3–4 | 9 | 3 | 3–4 | 10 | 8 | 3–4 | 17 | 7 | |||||||||
| SCC | 38 | 1–2 | 6 | 16 | 0.503 | 19 | 1–2 | 10 | 3 | 1 | 28 | 1–2 | 17 | 0 | 0.393 | 39 | 1–2 | 21 | 1 | 1 |
| 3–4 | 6 | 10 | 3–4 | 4 | 2 | 3–4 | 10 | 1 | 3–4 | 17 | 0 | |||||||||
| 50 Samples | ACSL1 | ACSL3 | ACSL4 | ACSL5 | ACSL6 |
|---|---|---|---|---|---|
| ACSL1 | 1 | r = 0.260, p = 0.069 | r = 0.039, p = 0.788 | r = 0.292, p = 0.04 | r = −0.123, p = 0.394 |
| ACSL3 | 1 | r = −0.078, p = 0.589 | r = 0.246, p = 0.085 | r = −0.280, p = 0.048 | |
| ACSL4 | 1 | r = −0.005, p = 0.972 | r = −0.064, p = 0.659 | ||
| ACSL5 | 1 | r = −0.314, p = 0.026 | |||
| ACSL6 | 1 |
| ACSL1 vs. Ki67 | ACSL3 vs. Ki67 | ACSL4 vs. Ki67 | ACSL5 vs. Ki67 | ACSL6 vs. Ki67 | |
|---|---|---|---|---|---|
| Sample number | 88 | 51 | 72 | 86 | 85 |
| Correlation coefficient (r Value) | 0.079 | 0.076 | 0.07 | −0.15 | −0.077 |
| Sig. (two-tailed) p | 0.458 | 0.585 | 0.551 | 0.164 | 0.477 |
| Grade 1–2 | 44 | 26 | 38 | 42 | 44 |
| −0.122 | −0.243 | −0.293 | −0.36 | 0.079 | |
| 0.42 | 0.234 | 0.071 | 0.02 | 0.598 | |
| Grade 3–4 | 44 | 25 | 34 | 44 | 41 |
| 0.232 | 0.397 | 0.063 | 0.102 | −0.241 | |
| 0.124 | 0.047 | 0.714 | 0.498 | 0.123 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Nenkov, M.; Berndt, A.; Abubrig, M.; Schmidt, M.; Sandhaus, T.; Huber, O.; Clement, J.H.; Lang, S.M.; Chen, Y.; et al. The Diagnostic Value of ACSL1, ACSL4, and ACSL5 and the Clinical Potential of an ACSL Inhibitor in Non-Small-Cell Lung Cancer. Cancers 2024, 16, 1170. https://doi.org/10.3390/cancers16061170
Ma Y, Nenkov M, Berndt A, Abubrig M, Schmidt M, Sandhaus T, Huber O, Clement JH, Lang SM, Chen Y, et al. The Diagnostic Value of ACSL1, ACSL4, and ACSL5 and the Clinical Potential of an ACSL Inhibitor in Non-Small-Cell Lung Cancer. Cancers. 2024; 16(6):1170. https://doi.org/10.3390/cancers16061170
Chicago/Turabian StyleMa, Yunxia, Miljana Nenkov, Alexander Berndt, Mohamed Abubrig, Martin Schmidt, Tim Sandhaus, Otmar Huber, Joachim H. Clement, Susanne M. Lang, Yuan Chen, and et al. 2024. "The Diagnostic Value of ACSL1, ACSL4, and ACSL5 and the Clinical Potential of an ACSL Inhibitor in Non-Small-Cell Lung Cancer" Cancers 16, no. 6: 1170. https://doi.org/10.3390/cancers16061170
APA StyleMa, Y., Nenkov, M., Berndt, A., Abubrig, M., Schmidt, M., Sandhaus, T., Huber, O., Clement, J. H., Lang, S. M., Chen, Y., & Gaßler, N. (2024). The Diagnostic Value of ACSL1, ACSL4, and ACSL5 and the Clinical Potential of an ACSL Inhibitor in Non-Small-Cell Lung Cancer. Cancers, 16(6), 1170. https://doi.org/10.3390/cancers16061170