


Triple-Negative Breast Cancer and Emerging Therapeutic Strategies: ATR and CHK1/2 as Promising Targets
 , ,
, ,
Simple Summary
Abstract
1. Introduction
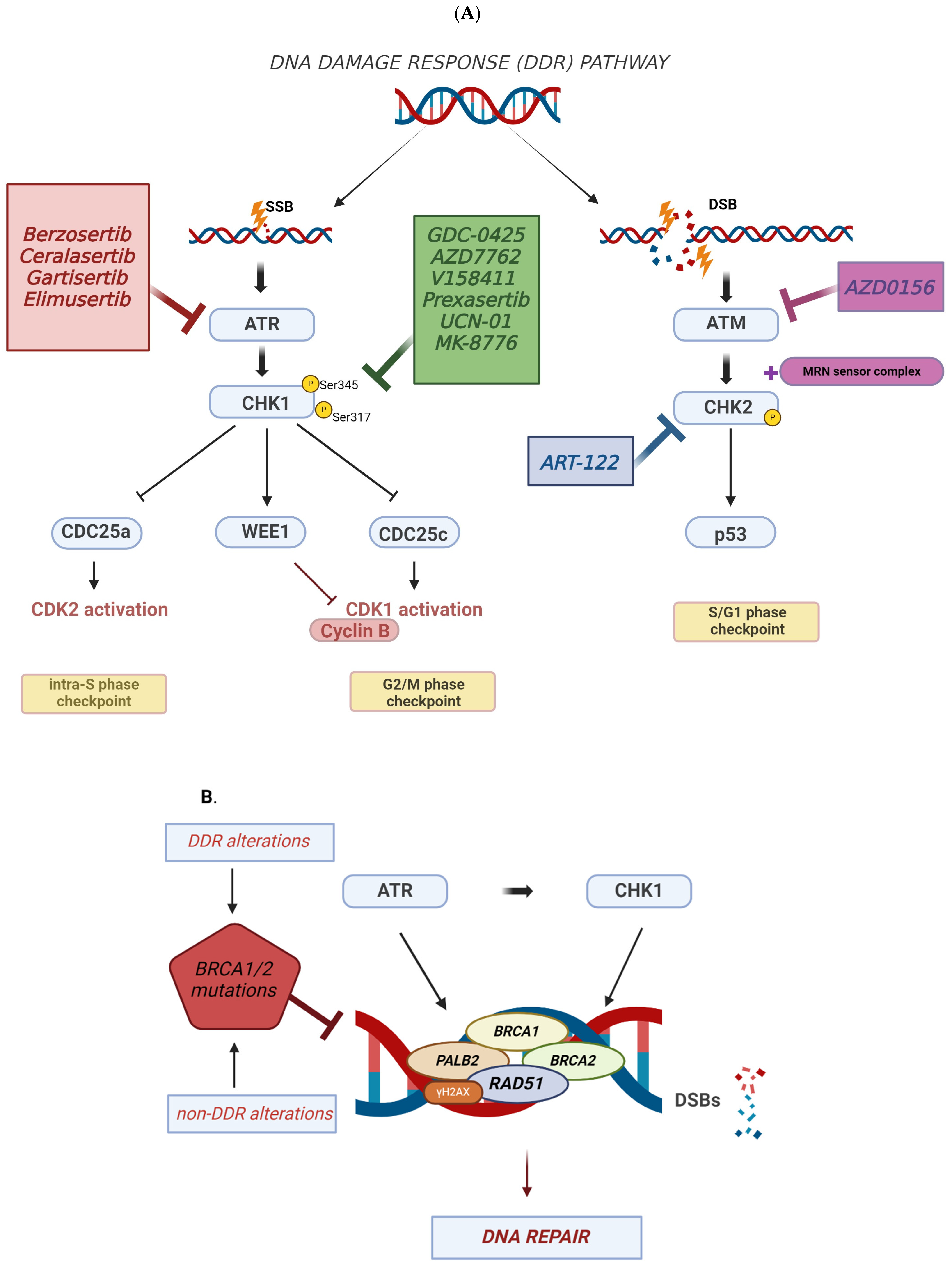
2. DNA Damage Response (DDR) and TNBC
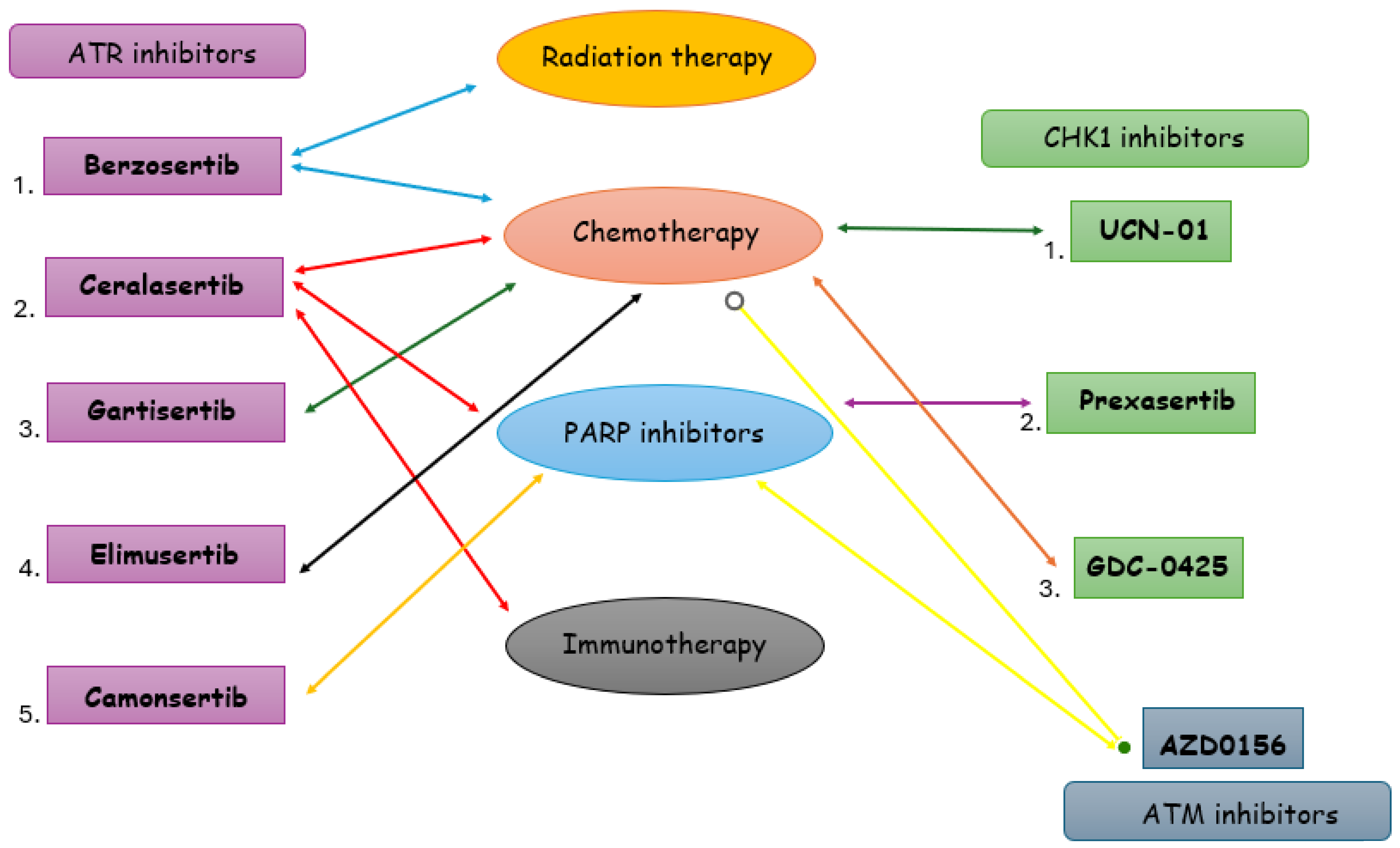
3. Targeting DDR in TNBC
3.1. ATR Inhibitors in TNBC
3.1.1. Berzosertib
3.1.2. Ceralasertib
3.1.3. Gartisertib
3.1.4. Elimusertib
3.1.5. Camonsertib
3.2. CHK1 Inhibitors in TNBC
3.2.1. GDC-0425
3.2.2. AZD7762
3.2.3. V158411
3.2.4. Prexasertib
3.2.5. UCN-01
3.2.6. MK-8776
3.3. ATM-CHK2 Inhibitors in TNBC
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Statistics Center. Available online: http://cancerstatisticscenter.cancer.org (accessed on 31 July 2023).
- Katsura, C.; Ogunmwonyi, I.; Kankam, H.K.; Saha, S. Breast Cancer: Presentation, Investigation and Management. Br. J. Hosp. Med. 2022, 83, 1–7. [Google Scholar] [CrossRef]
- Fahad Ullah, M. Breast Cancer: Current Perspectives on the Disease Status. In Breast Cancer Metastasis and Drug Resistance; Ahmad, A., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; Volume 1152, pp. 51–64. ISBN 978-3-030-20300-9. [Google Scholar]
- Alkabban, F.M.; Ferguson, T. Breast Cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- GLOBOCAN 2020: New Global Cancer Data. Available online: https://www.uicc.org/news/globocan-2020-new-global-cancer-data (accessed on 24 November 2021).
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanisławek, A. Breast Cancer—Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies—An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef]
- Miller, K.D.; Ortiz, A.P.; Pinheiro, P.S.; Bandi, P.; Minihan, A.; Fuchs, H.E.; Martinez Tyson, D.; Tortolero-Luna, G.; Fedewa, S.A.; Jemal, A.M.; et al. Cancer Statistics for the US Hispanic/Latino Population, 2021. CA Cancer J. Clin. 2021, 71, 466–487. [Google Scholar] [CrossRef] [PubMed]
- Giaquinto, A.N.; Miller, K.D.; Tossas, K.Y.; Winn, R.A.; Jemal, A.; Siegel, R.L. Cancer Statistics for African American/Black People 2022. CA Cancer J. Clin. 2022, 72, 202–229. [Google Scholar] [CrossRef] [PubMed]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast Cancer: Biology, Biomarkers, and Treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.M.; Eun Kwon, J.; Ali, T.; Jang, J.; Ullah, I.; Lee, Y.-G.; Park, D.W.; Park, J.; Jeang, J.W.; Kang, S.C. Triple-Negative Breast Cancer: Epidemiology, Molecular Mechanisms, and Modern Vaccine-Based Treatment Strategies. Biochem. Pharmacol. 2023, 212, 115545. [Google Scholar] [CrossRef]
- Shirman, Y.; Lubovsky, S.; Shai, A. HER2-Low Breast Cancer: Current Landscape and Future Prospects. Breast Cancer Dove Med. Press 2023, 15, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Aggarwal, R. An Overview of Triple-Negative Breast Cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients with Triple-Negative Breast Cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Irshad, S.; Ellis, P.; Tutt, A. Molecular Heterogeneity of Triple-Negative Breast Cancer and Its Clinical Implications. Curr. Opin. Oncol. 2011, 23, 566–577. [Google Scholar] [CrossRef]
- Metzger-Filho, O.; Tutt, A.; De Azambuja, E.; Saini, K.S.; Viale, G.; Loi, S.; Bradbury, I.; Bliss, J.M.; Azim, H.A.; Ellis, P.; et al. Dissecting the Heterogeneity of Triple-Negative Breast Cancer. J. Clin. Oncol. 2012, 30, 1879–1887. [Google Scholar] [CrossRef]
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast Cancer: Epidemiology and Etiology. Cell Biochem. Biophys. 2015, 72, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Eliyatkin, N.; Yalcin, E.; Zengel, B.; Aktaş, S.; Vardar, E. Molecular Classification of Breast Carcinoma: From Traditional, Old-Fashioned Way to A New Age, and A New Way. J. Breast Health 2015, 11, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Oakman, C.; Viale, G.; Di Leo, A. Management of Triple Negative Breast Cancer. Breast 2010, 19, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Brenton, J.D.; Carey, L.A.; Ahmed, A.A.; Caldas, C. Molecular Classification and Molecular Forecasting of Breast Cancer: Ready for Clinical Application? J. Clin. Oncol. 2005, 23, 7350–7360. [Google Scholar] [CrossRef]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz Van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Chaudhary, L.N.; Wilkinson, K.H.; Kong, A. Triple-Negative Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 141–153. [Google Scholar] [CrossRef]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment Landscape of Triple-Negative Breast Cancer—Expanded Options, Evolving Needs. Nat. Rev. Clin. Oncol. 2022, 19, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia Telangiectasia and Rad3-Related Inhibitors and Cancer Therapy: Where We Stand. J. Hematol. Oncol. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 Inhibitors and Cancer Therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the Biology and Clinical Potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A. DNA Damage in Cancer Therapeutics: A Boon or a Curse? Cancer Res. 2015, 75, 2133–2138. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA Repair Dysregulation from Cancer Driver to Therapeutic Target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA Damage Response. Cold Spring Harb. Perspect. Biol. 2011, 3, a000745. [Google Scholar] [CrossRef]
- Tian, H.; Gao, Z.; Li, H.; Zhang, B.; Wang, G.; Zhang, Q.; Pei, D.; Zheng, J. DNA Damage Response—A Double-Edged Sword in Cancer Prevention and Cancer Therapy. Cancer Lett. 2015, 358, 8–16. [Google Scholar] [CrossRef]
- Jin, J.; Tao, Z.; Cao, J.; Li, T.; Hu, X. DNA Damage Response Inhibitors: An Avenue for TNBC Treatment. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188521. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. ATR: An Essential Regulator of Genome Integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef]
- Mailand, N.; Falck, J.; Lukas, C.; Syljuåsen, R.G.; Welcker, M.; Bartek, J.; Lukas, J. Rapid Destruction of Human Cdc25A in Response to DNA Damage. Science 2000, 288, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 Phosphatases in Cancer Cells: Key Players? Good Targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef]
- Xiao, Z.; Chen, Z.; Gunasekera, A.H.; Sowin, T.J.; Rosenberg, S.H.; Fesik, S.; Zhang, H. Chk1 Mediates S and G2 Arrests through Cdc25A Degradation in Response to DNA-Damaging Agents. J. Biol. Chem. 2003, 278, 21767–21773. [Google Scholar] [CrossRef]
- Kalous, J.; Jansová, D.; Šušor, A. Role of Cyclin-Dependent Kinase 1 in Translational Regulation in the M-Phase. Cells 2020, 9, 1568. [Google Scholar] [CrossRef] [PubMed]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef]
- Ronco, C.; Martin, A.R.; Demange, L.; Benhida, R. ATM, ATR, CHK1, CHK2 and WEE1 Inhibitors in Cancer and Cancer Stem Cells. MedChemComm 2017, 8, 295–319. [Google Scholar] [CrossRef]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell Cycle Checkpoint in Cancer: A Therapeutically Targetable Double-Edged Sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef]
- Boudny, M.; Trbusek, M. ATR-CHK1 Pathway as a Therapeutic Target for Acute and Chronic Leukemias. Cancer Treat. Rev. 2020, 88, 102026. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A Genome-Wide Homologous Recombination Screen Identifies the RNA-Binding Protein RBMX as a Component of the DNA-Damage Response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.; Powell, S.N.; Iliakis, G.; Wang, Y. ATR Affecting Cell Radiosensitivity Is Dependent on Homologous Recombination Repair but Independent of Nonhomologous End Joining. Cancer Res. 2004, 64, 7139–7143. [Google Scholar] [CrossRef]
- Jachimowicz, R.D.; Goergens, J.; Reinhardt, H.C. DNA Double-Strand Break Repair Pathway Choice—from Basic Biology to Clinical Exploitation. Cell Cycle 2019, 18, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Jackson, S.P. Human CtIP Mediates Cell Cycle Control of DNA End Resection and Double Strand Break Repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef]
- Huertas, P.; Cortés-Ledesma, F.; Sartori, A.A.; Aguilera, A.; Jackson, S.P. CDK Targets Sae2 to Control DNA-End Resection and Homologous Recombination. Nature 2008, 455, 689–692. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.M.A.; Middleton, F.K.; Arora, A.; Agarwal, D.; Chen, T.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.; Green, A.R.; et al. Untangling the ATR-CHEK1 Network for Prognostication, Prediction and Therapeutic Target Validation in Breast Cancer. Mol. Oncol. 2015, 9, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Rundle, S.; Bradbury, A.; Drew, Y.; Curtin, N. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers 2017, 9, 41. [Google Scholar] [CrossRef]
- Orthwein, A.; Noordermeer, S.M.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A Mechanism for the Suppression of Homologous Recombination in G1 Cells. Nature 2015, 528, 422–426. [Google Scholar] [CrossRef]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 Is an Integral Component of the BRCA Complex Required for Homologous Recombination Repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef]
- Thorslund, T.; McIlwraith, M.J.; Compton, S.A.; Lekomtsev, S.; Petronczki, M.; Griffith, J.D.; West, S.C. The Breast Cancer Tumor Suppressor BRCA2 Promotes the Specific Targeting of RAD51 to Single-Stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1263–1265. [Google Scholar] [CrossRef]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.-D. Human BRCA2 Protein Promotes RAD51 Filament Formation on RPA-Covered Single-Stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified Human BRCA2 Stimulates RAD51-Mediated Recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef]
- Buisson, R.; Dion-Côté, A.-M.; Coulombe, Y.; Launay, H.; Cai, H.; Stasiak, A.Z.; Stasiak, A.; Xia, B.; Masson, J.-Y. Cooperation of Breast Cancer Proteins PALB2 and Piccolo BRCA2 in Stimulating Homologous Recombination. Nat. Struct. Mol. Biol. 2010, 17, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.; Krishnamoorthy, A.; Dungrawala, H.; Garcin, E.B.; Modesti, M.; Cortez, D. RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep. 2018, 24, 538–545. [Google Scholar] [CrossRef]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010, 2010, 920161. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.-X. Gamma-H2AX—A Novel Biomarker for DNA Double-Strand Breaks. In Vivo Athens Greece 2008, 22, 305–309. [Google Scholar] [PubMed]
- Garcia-Canton, C.; Anadón, A.; Meredith, C. γH2AX as a Novel Endpoint to Detect DNA Damage: Applications for the Assessment of the in Vitro Genotoxicity of Cigarette Smoke. Toxicol. In Vitro 2012, 26, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Moretti, E.; Siclari, O.; Migliaccio, I.; Santarpia, L.; D’Incalci, M.; Piccolo, S.; Veronesi, A.; Zambelli, A.; Del Sal, G.; et al. Targeting Triple Negative Breast Cancer: Is P53 the Answer? Cancer Treat. Rev. 2013, 39, 541–550. [Google Scholar] [CrossRef]
- Herschkowitz, J.I.; He, X.; Fan, C.; Perou, C.M. The Functional Loss of the Retinoblastoma Tumour Suppressor Is a Common Event in Basal-like and Luminal B Breast Carcinomas. Breast Cancer Res. 2008, 10, R75. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The Clonal and Mutational Evolution Spectrum of Primary Triple-Negative Breast Cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A Cell-Based Screen Identifies ATR Inhibitors with Synthetic Lethal Properties for Cancer-Associated Mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective Killing of ATM- or P53-Deficient Cancer Cells through Inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating “BRCAness”. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen Receptor Inhibitor-Induced “BRCAness” and PARP Inhibition Are Synthetically Lethal for Castration-Resistant Prostate Cancer. Sci. Signal. 2017, 10, eaam7479. [Google Scholar] [CrossRef] [PubMed]
- Karanika, S.; Karantanos, T.; Li, L.; Wang, J.; Park, S.; Yang, G.; Zuo, X.; Song, J.H.; Maity, S.N.; Manyam, G.C.; et al. Targeting DNA Damage Response in Prostate Cancer by Inhibiting Androgen Receptor-CDC6-ATR-Chk1 Signaling. Cell Rep. 2017, 18, 1970–1981. [Google Scholar] [CrossRef]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic Lethality between Androgen Receptor Signalling and the PARP Pathway in Prostate Cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Milton, S.; Murphy, C.E.; et al. Potentiation of Tumor Responses to DNA Damaging Therapy by the Selective ATR Inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in Vivo Using the Novel Inhibitor VE-822 Results in Selective Sensitization of Pancreatic Tumors to Radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef]
- Gorecki, L.; Andrs, M.; Rezacova, M.; Korabecny, J. Discovery of ATR Kinase Inhibitor Berzosertib (VX-970, M6620): Clinical Candidate for Cancer Therapy. Pharmacol. Ther. 2020, 210, 107518. [Google Scholar] [CrossRef]
- Jossé, R.; Martin, S.E.; Guha, R.; Ormanoglu, P.; Pfister, T.D.; Reaper, P.M.; Barnes, C.S.; Jones, J.; Charlton, P.; Pollard, J.R.; et al. ATR Inhibitors VE-821 and VX-970 Sensitize Cancer Cells to Topoisomerase i Inhibitors by Disabling DNA Replication Initiation and Fork Elongation Responses. Cancer Res. 2014, 74, 6968–6979. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.K.; Pollard, J.R.; Curtin, N.J. The Impact of P53 Dysfunction in ATR Inhibitor Cytotoxicity and Chemo- and Radiosensitisation. Cancers 2018, 10, 275. [Google Scholar] [CrossRef]
- Jo, U.; Senatorov, I.S.; Zimmermann, A.; Saha, L.K.; Murai, Y.; Kim, S.H.; Rajapakse, V.N.; Elloumi, F.; Takahashi, N.; Schultz, C.W.; et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells with Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol. Cancer Ther. 2021, 20, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Fontana, E.; Lee, E.K.; Spigel, D.R.; Højgaard, M.; Lheureux, S.; Mettu, N.B.; Carneiro, B.A.; Carter, L.; Plummer, R.; et al. Camonsertib in DNA Damage Response-Deficient Advanced Solid Tumors: Phase 1 Trial Results. Nat. Med. 2023, 29, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. NCI Dictionary of Cancer Terms. 2023. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms (accessed on 14 August 2023).
- Tu, X.; Kahila, M.M.; Zhou, Q.; Yu, J.; Kalari, K.R.; Wang, L.; Harmsen, W.S.; Yuan, J.; Boughey, J.C.; Goetz, M.P.; et al. ATR Inhibition Is a Promising Radiosensitizing Strategy for Triple-Negative Breast Cancer. Mol. Cancer Ther. 2018, 17, 2462–2472. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 Study of the ATR Inhibitor Berzosertib in Combination with Cisplatin in Patients with Advanced Solid Tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef]
- Plummer, E.R.; Dean, E.J.; Evans, T.R.J.; Greystoke, A.; Herbschleb, K.; Ranson, M.; Brown, J.; Zhang, Y.; Karan, S.; Pollard, J.; et al. Phase I Trial of First-in-Class ATR Inhibitor VX-970 in Combination with Gemcitabine (Gem) in Advanced Solid Tumors (NCT02157792). J. Clin. Oncol. 2016, 34, 2513. [Google Scholar] [CrossRef]
- Telli, M.L.; Tolaney, S.M.; Shapiro, G.I.; Middleton, M.; Lord, S.R.; Arkenau, H.T.; Tutt, A.; Abramson, V.; Dean, E.; Haddad, T.C.; et al. Phase 1b Study of Berzosertib and Cisplatin in Patients with Advanced Triple-Negative Breast Cancer. NPJ Breast Cancer 2022, 8, 45. [Google Scholar] [CrossRef]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; De Miguel Luken, M.J.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination with Carboplatin in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Jin, J.; Fang, H.; Yang, F.; Ji, W.; Guan, N.; Sun, Z.; Shi, Y.; Zhou, G.; Guan, X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia 2018, 20, 478–488. [Google Scholar] [CrossRef]
- Wilson, Z.; Odedra, R.; Wallez, Y.; Wijnhoven, P.W.G.; Hughes, A.M.; Gerrard, J.; Jones, G.N.; Bargh-Dawson, H.; Brown, E.; Young, L.A.; et al. ATR Inhibitor AZD6738 (Ceralasertib) Exerts Antitumor Activity as a Monotherapy and in Combination with Chemotherapy and the PARP Inhibitor Olaparib. Cancer Res. 2022, 82, 1140–1152. [Google Scholar] [CrossRef]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; El-Khouiery, A.; Soria, J.-C.; Lopez, J.; Berges, A.; Cheung, S.Y.A.; Irurzun-Arana, I.; Goldwin, A.; et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2021, 27, 5213–5224. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Nowecki, Z.; Szoszkiewicz, R.; Im, S.-A.; Arkenau, H.-T.; Armstrong, A.; Jacot, W.; Kim, J.H.; Webster, M.; Balmana, J.; et al. 161O VIOLETTE: Randomised Phase II Study of Olaparib (Ola) + Ceralasertib (Cer) or Adavosertib (Ada) vs Ola Alone in Patients (Pts) with Metastatic Triple-Negative Breast Cancer (mTNBC). Ann. Oncol. 2022, 33, S194–S195. [Google Scholar] [CrossRef]
- Kirk, R. Cancer Risk Reduction in BRCA Mutation Carriers. Nat. Rev. Clin. Oncol. 2010, 7, 609. [Google Scholar] [CrossRef]
- Byrski, T.; Gronwald, J.; Huzarski, T.; Grzybowska, E.; Budryk, M.; Stawicka, M.; Mierzwa, T.; Szwiec, M.; Wiśniowski, R.; Siolek, M.; et al. Response to Neo-Adjuvant Chemotherapy in Women with BRCA1-Positive Breast Cancers. Breast Cancer Res. Treat. 2008, 108, 289–296. [Google Scholar] [CrossRef]
- Musolino, A.; Bella, M.A.; Bortesi, B.; Michiara, M.; Naldi, N.; Zanelli, P.; Capelletti, M.; Pezzuolo, D.; Camisa, R.; Savi, M.; et al. BRCA Mutations, Molecular Markers, and Clinical Variables in Early-Onset Breast Cancer: A Population-Based Study. Breast 2007, 16, 280–292. [Google Scholar] [CrossRef]
- Zenke, F.T.; Zimmermann, A.; Dahmen, H.; Elenbaas, B.; Pollard, J.; Reaper, P.; Bagrodia, S.; Spilker, M.E.; Amendt, C.; Blaukat, A. Abstract 369: Antitumor Activity of M4344, a Potent and Selective ATR Inhibitor, in Monotherapy and Combination Therapy. Cancer Res. 2019, 79, 369. [Google Scholar] [CrossRef]
- Villette, C.C.; Brightman, F.; Dupuy, N.; Zimmermann, A.; Lignet, F.; Zenke, F.T.; Terranova, N.; Bolleddula, J.; Bawab, S.E.; Chassagnole, C. Abstract 5699: Development and Validation of a Quantitative Systems Pharmacology Model for Prediction of Preclinical Efficacy of PARP Inhibitors Rucaparib and Talazoparib Combined with the ATR Inhibitor Gartisertib (M4344). Cancer Res. 2023, 83, 5699. [Google Scholar] [CrossRef]
- Burris, H.A.; Berlin, J.; Arkenau, T.; Cote, G.M.; Lolkema, M.P.; Ferrer-Playan, J.; Kalapur, A.; Bolleddula, J.; Locatelli, G.; Goddemeier, T.; et al. A Phase I Study of ATR Inhibitor Gartisertib (M4344) as a Single Agent and in Combination with Carboplatin in Patients with Advanced Solid Tumours. Br. J. Cancer 2024. Online ahead of print. [Google Scholar] [CrossRef]
- Luecking, U.T.; Lefranc, J.; Wengner, A.; Wortmann, L.; Schick, H.; Briem, H.; Siemeister, G.; Lienau, P.; Schatz, C.; Bader, B.; et al. Abstract 983: Identification of Potent, Highly Selective and Orally Available ATR Inhibitor BAY 1895344 with Favorable PK Properties and Promising Efficacy in Monotherapy and Combination in Preclinical Tumor Models. Cancer Res. 2017, 77, 983. [Google Scholar] [CrossRef]
- Wengner, A.M.; Siemeister, G.; Luecking, U.; Lefranc, J.; Lienau, P.; Deeg, G.; Lagkadinou, E.; Liu, L.; Golfier, S.; Schatz, C.; et al. Abstract 836: ATR Inhibitor BAY 1895344 Shows Potent Anti-Tumor Efficacy in Monotherapy and Strong Combination Potential with the Targeted Alpha Therapy Radium-223 Dichloride in Preclinical Tumor Models. Cancer Res. 2017, 77, 836. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Madoz-Gúrpide, J.; Cañamero, M.; Sanchez, L.; Solano, J.; Alfonso, P.; Casal, J.I. A Proteomics Analysis of Cell Signaling Alterations in Colorectal Cancer. Mol. Cell. Proteom. 2007, 6, 2150–2164. [Google Scholar] [CrossRef] [PubMed]
- Verlinden, L.; Vanden Bempt, I.; Eelen, G.; Drijkoningen, M.; Verlinden, I.; Marchal, K.; De Wolf-Peeters, C.; Christiaens, M.-R.; Michiels, L.; Bouillon, R.; et al. The E2F-Regulated Gene Chk1 Is Highly Expressed in Triple-Negative Estrogen Receptor−/Progesterone Receptor−/HER-2− Breast Carcinomas. Cancer Res. 2007, 67, 6574–6581. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Yang, Z.; Li, Y. Expression of Checkpoint Kinase 1 and Polo-like Kinase 1 and Its Clinicopathological Significance in Benign and Malignant Lesions of the Stomach. J. Cent. South Univ. Med. Sci. 2010, 35, 1080–1084. [Google Scholar] [CrossRef]
- Xu, J.; Li, Y.; Wang, F.; Wang, X.; Cheng, B.; Ye, F.; Xie, X.; Zhou, C.; Lu, W. Suppressed miR-424 Expression via Upregulation of Target Gene Chk1 Contributes to the Progression of Cervical Cancer. Oncogene 2013, 32, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Hu, K.; Yuan, Y.; Sang, Y.; Bu, Q.; Chen, G.; Yang, L.; Li, B.; Huang, P.; Chen, D.; et al. CHK1 Targets Spleen Tyrosine Kinase (L) for Proteolysis in Hepatocellular Carcinoma. J. Clin. Investig. 2012, 122, 2165–2175. [Google Scholar] [CrossRef]
- Albiges, L.; Goubar, A.; Scott, V.; Vicier, C.; Lefèbvre, C.; Alsafadi, S.; Commo, F.; Saghatchian, M.; Lazar, V.; Dessen, P.; et al. Chk1 as a New Therapeutic Target in Triple-Negative Breast Cancer. Breast 2014, 23, 250–258. [Google Scholar] [CrossRef]
- Kim, H.-J.; Seo, B.-G.; Seo, E.-C.; Lee, K.-M.; Hwangbo, C. Checkpoint Kinase 1 (CHK1) Functions as Both a Diagnostic Marker and a Regulator of Epithelial-to-Mesenchymal Transition (EMT) in Triple-Negative Breast Cancer. Curr. Issues Mol. Biol. 2022, 44, 5848–5865. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Minami, K.; Chen, A.; Wan, Q.; Yin, Y.; Gan, L.; Xu, A.; Matsuura, N.; Koizumi, M.; et al. Inhibiting Checkpoint Kinase 1 Protects Bone from Bone Resorption by Mammary Tumor in a Mouse Model. Oncotarget 2018, 9, 9364–9378. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Svensson, S.; Romania, P.; Dattilo, R.; Patrizii, M.; Signore, M.; Navarra, S.; Lotti, F.; Biffoni, M.; Pilozzi, E.; et al. Therapeutic Targeting of Chk1 in NSCLC Stem Cells during Chemotherapy. Cell Death Differ. 2012, 19, 768–778. [Google Scholar] [CrossRef]
- Meyer, F.; Becker, S.; Classen, S.; Parplys, A.C.; Mansour, W.Y.; Riepen, B.; Timm, S.; Ruebe, C.; Jasin, M.; Wikman, H.; et al. Prevention of DNA Replication Stress by CHK1 Leads to Chemoresistance Despite a DNA Repair Defect in Homologous Recombination in Breast Cancer. Cells 2020, 9, 238. [Google Scholar] [CrossRef]
- Bryant, C.; Stokes, S.; Massey, A.J. Abstract 4458: Chk1 Inhibition as a Novel Therapeutic Strategy for Treating Triple Negative Breast and Ovarian Cancers. Cancer Res. 2011, 71, 4458. [Google Scholar] [CrossRef]
- Bennett, C.N.; Tomlinson, C.C.; Michalowski, A.M.; Chu, I.M.; Luger, D.; Mittereder, L.R.; Aprelikova, O.; Shou, J.; Piwinica-Worms, H.; Caplen, N.J.; et al. Cross-Species Genomic and Functional Analyses Identify a Combination Therapy Using a CHK1 Inhibitor and a Ribonucleotide Reductase Inhibitor to Treat Triple-Negative Breast Cancer. Breast Cancer Res. 2012, 14, R109. [Google Scholar] [CrossRef]
- Ding, X.; Chen, Y.; Sahasranaman, S.; Shi, Y.; McKnight, J.; Dean, B. A Supported Liquid Extraction LC-MS/MS Method for Determination of Concentrations of GDC-0425, a Small Molecule Checkpoint Kinase 1 Inhibitor, in Human Plasma: Determination of GDC-0425 in Human Plasma by SLE LC-MS/M. Biomed. Chromatogr. 2016, 30, 1984–1991. [Google Scholar] [CrossRef]
- Infante, J.R.; Hollebecque, A.; Postel-Vinay, S.; Bauer, T.M.; Blackwood, E.M.; Evangelista, M.; Mahrus, S.; Peale, F.V.; Lu, X.; Sahasranaman, S.; et al. Phase I Study of GDC-0425, a Checkpoint Kinase 1 Inhibitor, in Combination with Gemcitabine in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2017, 23, 2423–2432. [Google Scholar] [CrossRef] [PubMed]
- Gazzard, L.; Appleton, B.; Chapman, K.; Chen, H.; Clark, K.; Drobnick, J.; Goodacre, S.; Halladay, J.; Lyssikatos, J.; Schmidt, S.; et al. Discovery of the 1,7-Diazacarbazole Class of Inhibitors of Checkpoint Kinase 1. Bioorg. Med. Chem. Lett. 2014, 24, 5704–5709. [Google Scholar] [CrossRef]
- Osborne, J.D.; Matthews, T.P.; McHardy, T.; Proisy, N.; Cheung, K.-M.J.; Lainchbury, M.; Brown, N.; Walton, M.I.; Eve, P.D.; Boxall, K.J.; et al. Multiparameter Lead Optimization to Give an Oral Checkpoint Kinase 1 (CHK1) Inhibitor Clinical Candidate: (R)-5-((4-((Morpholin-2-Ylmethyl)Amino)-5-(Trifluoromethyl)Pyridin-2-Yl)Amino)Pyrazine-2-Carbonitrile (CCT245737). J. Med. Chem. 2016, 59, 5221–5237. [Google Scholar] [CrossRef]
- Zabludoff, S.D.; Deng, C.; Grondine, M.R.; Sheehy, A.M.; Ashwell, S.; Caleb, B.L.; Green, S.; Haye, H.R.; Horn, C.L.; Janetka, J.W.; et al. AZD7762, a Novel Checkpoint Kinase Inhibitor, Drives Checkpoint Abrogation and Potentiates DNA-Targeted Therapies. Mol. Cancer Ther. 2008, 7, 2955–2966. [Google Scholar] [CrossRef]
- Min, D.-J.; He, S.; Green, J.E. Birinapant (TL32711) Improves Responses to GEM/AZD7762 Combination Therapy in Triple-Negative Breast Cancer Cell Lines. Anticancer Res. 2016, 36, 2649–2657. [Google Scholar] [PubMed]
- Witkiewicz, A.K.; Chung, S.; Brough, R.; Vail, P.; Franco, J.; Lord, C.J.; Knudsen, E.S. Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cancer. Cell Rep. 2018, 22, 1185–1199. [Google Scholar] [CrossRef]
- Schwartz, G. Neoadjuvant Induction Chemotherapy. Minerva Ginecol. 2005, 57, 327–348. [Google Scholar] [PubMed]
- Stover, D.G.; Winer, E.P. Tailoring Adjuvant Chemotherapy Regimens for Patients with Triple Negative Breast Cancer. Breast 2015, 24, S132–S135. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.M.; Shalaby, A.; O’Loughlin, M.; Keane, N.; Webber, M.J.; Kerin, M.J.; Keane, M.M.; Glynn, S.A.; Callagy, G.M. Outcome for Triple Negative Breast Cancer in a Retrospective Cohort with an Emphasis on Response to Platinum-Based Neoadjuvant Therapy. Breast Cancer Res. Treat. 2019, 174, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Rao, Z.; Yuan, S.; You, J.; Hong, C.; He, Q.; Yang, B.; Du, C.; Cao, J. One Therapeutic Approach for Triple-Negative Breast Cancer: Checkpoint Kinase 1 Inhibitor AZD7762 Combination with Neoadjuvant Carboplatin. Eur. J. Pharmacol. 2021, 908, 174366. [Google Scholar] [CrossRef]
- Sausville, E.; LoRusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I Dose-Escalation Study of AZD7762, a Checkpoint Kinase Inhibitor, in Combination with Gemcitabine in US Patients with Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef]
- Wayne, J.; Brooks, T.; Massey, A.J. Inhibition of Chk1 with the Small Molecule Inhibitor V158411 Induces DNA Damage and Cell Death in an Unperturbed S-Phase. Oncotarget 2016, 7, 85033–85048. [Google Scholar] [CrossRef]
- Bryant, C.; Rawlinson, R.; Massey, A.J. Chk1 Inhibition as a Novel Therapeutic Strategy for Treating Triple-Negative Breast and Ovarian Cancers. BMC Cancer 2014, 14, 570. [Google Scholar] [CrossRef]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef]
- Wu, W.; King, C.; Donoho, G.; Iversen, P.; Capen, A.; Castanares, M.; Stephens, J.; Ding, Y.; Pratt, S.; Martinez, R.; et al. Abstract 336: Anti-Tumor Activity of the Chk1 Inhibitor Prexasertib (LY2606368) as a Single Agent in Triple Negative Breast Cancer Models. Cancer Res. 2018, 78, 336. [Google Scholar] [CrossRef]
- Lee, J.-M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.-J.; et al. Prexasertib, a Cell Cycle Checkpoint Kinase 1 and 2 Inhibitor, in BRCA Wild-Type Recurrent High-Grade Serous Ovarian Cancer: A First-in-Class Proof-of-Concept Phase 2 Study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, J.-B.; Parmar, K.; Shapiro, G.I.; D’Andrea, A.D. Abstract 2729: Estrogen Receptor-Negative Breast Cancer Cell Lines Exhibit Hypersensitivity to the CHK1 Inhibitor LY2606368. Cancer Res. 2016, 76, 2729. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.-J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, 1013-e1824. [Google Scholar] [CrossRef] [PubMed]
- Mani, C.; Jonnalagadda, S.; Lingareddy, J.; Awasthi, S.; Gmeiner, W.H.; Palle, K. Prexasertib Treatment Induces Homologous Recombination Deficiency and Synergizes with Olaparib in Triple-Negative Breast Cancer Cells. Breast Cancer Res. 2019, 21, 104. [Google Scholar] [CrossRef]
- Frey, M.K.; Pothuri, B. Homologous Recombination Deficiency (HRD) Testing in Ovarian Cancer Clinical Practice: A Review of the Literature. Gynecol. Oncol. Res. Pract. 2017, 4, 4. [Google Scholar] [CrossRef]
- Rubinstein, M.M.; Hyman, D.M.; Caird, I.; Won, H.; Soldan, K.; Seier, K.; Iasonos, A.; Tew, W.P.; O’Cearbhaill, R.E.; Grisham, R.N.; et al. Phase 2 Study of LY3023414 in Patients with Advanced Endometrial Cancer Harboring Activating Mutations in the PI3K Pathway. Cancer 2020, 126, 1274–1282. [Google Scholar] [CrossRef]
- Hong, D.S.; Moore, K.N.; Bendell, J.C.; Karp, D.D.; Wang, J.S.; Ulahannan, S.V.; Jones, S.; Wu, W.; Donoho, G.P.; Ding, Y.; et al. Preclinical Evaluation and Phase Ib Study of Prexasertib, a CHK1 Inhibitor, and Samotolisib (LY3023414), a Dual PI3K/mTOR Inhibitor. Clin. Cancer Res. 2021, 27, 1864–1874. [Google Scholar] [CrossRef]
- Ma, C.X.; Janetka, J.W.; Piwnica-Worms, H. Death by Releasing the Breaks: CHK1 Inhibitors as Cancer Therapeutics. Trends Mol. Med. 2011, 17, 88–96. [Google Scholar] [CrossRef]
- Busby, E.C.; Leistritz, D.F.; Abraham, R.T.; Karnitz, L.M.; Sarkaria, J.N. The Radiosensitizing Agent 7-Hydroxystaurosporine (UCN-01) Inhibits the DNA Damage Checkpoint Kinase hChk1. Cancer Res. 2000, 60, 2108–2112. [Google Scholar]
- Graves, P.R.; Yu, L.; Schwarz, J.K.; Gales, J.; Sausville, E.A.; O’Connor, P.M.; Piwnica-Worms, H. The Chk1 Protein Kinase and the Cdc25C Regulatory Pathways Are Targets of the Anticancer Agent UCN-01. J. Biol. Chem. 2000, 275, 5600–5605. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Cai, S.; Li, S.; Ryan, C.E.; Guo, Z.; Schaiff, W.T.; Lin, L.; Hoog, J.; Goiffon, R.J.; Prat, A.; et al. Targeting Chk1 in P53-Deficient Triple-Negative Breast Cancer Is Therapeutically Beneficial in Human-in-Mouse Tumor Models. J. Clin. Investig. 2012, 122, 1541–1552. [Google Scholar] [CrossRef]
- Fracasso, P.M.; Williams, K.J.; Chen, R.C.; Picus, J.; Ma, C.X.; Ellis, M.J.; Tan, B.R.; Pluard, T.J.; Adkins, D.R.; Naughton, M.J.; et al. A Phase 1 Study of UCN-01 in Combination with Irinotecan in Patients with Resistant Solid Tumor Malignancies. Cancer Chemother. Pharmacol. 2011, 67, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Rudek, M.A.; Purcell, T.; Laheru, D.A.; Messersmith, W.A.; Dancey, J.; Carducci, M.A.; Baker, S.D.; Hidalgo, M.; Donehower, R.C. Phase I and Pharmacokinetic Study of UCN-01 in Combination with Irinotecan in Patients with Solid Tumors. Cancer Chemother. Pharmacol. 2008, 61, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Sausville, E.A.; Arbuck, S.G.; Messmann, R.; Headlee, D.; Bauer, K.S.; Lush, R.M.; Murgo, A.; Figg, W.D.; Lahusen, T.; Jaken, S.; et al. Phase I Trial of 72-Hour Continuous Infusion UCN-01 in Patients with Refractory Neoplasms. J. Clin. Oncol. 2001, 19, 2319–2333. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.P.; Lewis, L.D.; Beelen, A.P.; Olszanski, A.J.; Johnston, N.; Rhodes, C.H.; Beaulieu, B.; Ernstoff, M.S.; Eastman, A. Modulation of Cell Cycle Progression in Human Tumors: A Pharmacokinetic and Tumor Molecular Pharmacodynamic Study of Cisplatin Plus the Chk1 Inhibitor UCN-01 (NSC 638850). Clin. Cancer Res. 2006, 12, 7079–7085. [Google Scholar] [CrossRef]
- Zhou, Z.; Yang, Z.; Wang, S.; Zhang, L.; Luo, J.; Feng, Y.; Yu, X.; Chen, X.; Guo, X. The Chk1 Inhibitor MK-8776 Increases the Radiosensitivity of Human Triple-Negative Breast Cancer by Inhibiting Autophagy. Acta Pharmacol. Sin. 2017, 38, 513–523. [Google Scholar] [CrossRef]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; Bang, Y.J.; El-Khoueiry, A.; Abida, W.; Harrington, K.; Sundar, R.; Carter, L.; Castanon-Alvarez, E.; et al. Phase I Modular Study of AZD6738, a Novel Oral, Potent and Selective Ataxia Telangiectasia Rad3-Related (ATR) Inhibitor in Combination (Combo) with Carboplatin, Olaparib or Durvalumab in Patients (Pts) with Advanced Cancers. Eur. J. Cancer 2016, 69, S2. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Suzuki, K.; Kodama, S.; Watanabe, M. Recruitment of ATM Protein to Double Strand DNA Irradiated with Ionizing Radiation. J. Biol. Chem. 1999, 274, 25571–25575. [Google Scholar] [CrossRef]
- Dart, D.A.; Adams, K.E.; Akerman, I.; Lakin, N.D. Recruitment of the Cell Cycle Checkpoint Kinase ATR to Chromatin during S-Phase. J. Biol. Chem. 2004, 279, 16433–16440. [Google Scholar] [CrossRef] [PubMed]
- Lupardus, P.J.; Byun, T.; Yee, M.; Hekmat-Nejad, M.; Cimprich, K.A. A Requirement for Replication in Activation of the ATR-Dependent DNA Damage Checkpoint. Genes Dev. 2002, 16, 2327–2332. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Zhang, J.; Xie, Y. Prevalence and Characterization of ATM Germline Mutations in Chinese BRCA1/2-Negative Breast Cancer Patients. Breast Cancer Res. Treat. 2019, 174, 639–647. [Google Scholar] [CrossRef]
- Tommiska, J.; Bartkova, J.; Heinonen, M.; Hautala, L.; Kilpivaara, O.; Eerola, H.; Aittomäki, K.; Hofstetter, B.; Lukas, J.; Von Smitten, K.; et al. The DNA Damage Signalling Kinase ATM Is Aberrantly Reduced or Lost in BRCA1/BRCA2-Deficient and ER/PR/ERBB2-Triple-Negative Breast Cancer. Oncogene 2008, 27, 2501–2506. [Google Scholar] [CrossRef]
- Baik, T.; Park, C.; Kang, Y.W.; Joh, D.; Lee, H.-Y.; Park, S.; Hwang, S.; Kim, I.K.; Kim, J.O. Abstract 2608: Pre-Clinical Evaluation of Small Molecule CHK2 Inhibitor, ART-122 Combined with Olaparib in BRCA Wild Type Tumors. Cancer Res. 2022, 82, 2608. [Google Scholar] [CrossRef]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer Ther. 2020, 19, 13–25. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR Inhibition Disrupts Rewired Homologous Recombination and Fork Protection Pathways in PARP Inhibitor-Resistant BRCA-Deficient Cancer Cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef]
- Martorana, F.; Da Silva, L.A.; Sessa, C.; Colombo, I. Everything Comes with a Price: The Toxicity Profile of DNA-Damage Response Targeting Agents. Cancers 2022, 14, 953. [Google Scholar] [CrossRef]
- Lee, K.J.; Wright, G.; Bryant, H.; Wiggins, L.A.; Schuler, M.; Gassman, N.R. EGFR Signaling Promotes Resistance to CHK1 Inhibitor Prexasertib in Triple Negative Breast Cancer. Cancer Drug Resist. 2021, 3, 980–991. [Google Scholar] [CrossRef]
- Stefanski, C.D.; Prosperi, J.R. Combating CHK1 Resistance in Triple Negative Breast Cancer: EGFR Inhibition as Potential Combinational Therapy. Cancer Drug Resist. 2022, 5, 229–232. [Google Scholar] [CrossRef]
- Dunlop, C.R.; Wallez, Y.; Johnson, T.I.; Bernaldo De Quirós Fernández, S.; Durant, S.T.; Cadogan, E.B.; Lau, A.; Richards, F.M.; Jodrell, D.I. Complete Loss of ATM Function Augments Replication Catastrophe Induced by ATR Inhibition and Gemcitabine in Pancreatic Cancer Models. Br. J. Cancer 2020, 123, 1424–1436. [Google Scholar] [CrossRef]
- Menezes, D.L.; Holt, J.; Tang, Y.; Feng, J.; Barsanti, P.; Pan, Y.; Ghoddusi, M.; Zhang, W.; Thomas, G.; Holash, J.; et al. A Synthetic Lethal Screen Reveals Enhanced Sensitivity to ATR Inhibitor Treatment in Mantle Cell Lymphoma with ATM Loss-of-Function. Mol. Cancer Res. MCR 2015, 13, 120–129. [Google Scholar] [CrossRef]
- Rafiei, S.; Fitzpatrick, K.; Liu, D.; Cai, M.-Y.; Elmarakeby, H.A.; Park, J.; Ricker, C.; Kochupurakkal, B.S.; Choudhury, A.D.; Hahn, W.C.; et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition Than PARP Inhibition in Prostate Cancer. Cancer Res. 2020, 80, 2094–2100. [Google Scholar] [CrossRef]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR Inhibitors as a Synthetic Lethal Therapy for Tumours Deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Matsumoto, S.; Wakatsuki, K.; Kunishige, T.; Nakade, H.; Miyao, S.; Sho, M. RNF126 as a Marker of Prognosis and Proliferation of Gastric Cancer. Anticancer Res. 2020, 40, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Z.; Shi, R.; Zhu, X.; Zhou, J.; Peng, B.; Xu, X. RNF126 Quenches RNF168 Function in the DNA Damage Response. Genom. Proteom. Bioinform. 2018, 16, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Fa, P.; Qiu, Z.; Wang, Q.-E.; Yan, C.; Zhang, J. A Novel Role for RNF126 in the Promotion of G2 Arrest via Interaction with 14-3-3σ. Int. J. Radiat. Oncol. 2022, 112, 542–553. [Google Scholar] [CrossRef] [PubMed]
- He, M.Y.; Rancoule, C.; Rehailia-Blanchard, A.; Espenel, S.; Trone, J.-C.; Bernichon, E.; Guillaume, E.; Vallard, A.; Magné, N. Radiotherapy in Triple-Negative Breast Cancer: Current Situation and Upcoming Strategies. Crit. Rev. Oncol. Hematol. 2018, 131, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zheng, M.; Zhang, R.; Jiang, Q.; Du, G.; Wu, Y.; Yang, C.; Li, F.; Li, W.; Wang, L.; et al. RNF126-Mediated MRE11 Ubiquitination Activates the DNA Damage Response and Confers Resistance of Triple-Negative Breast Cancer to Radiotherapy. Adv. Sci. 2023, 10, 2203884. [Google Scholar] [CrossRef] [PubMed]
- Jo, U.; Murai, Y.; Agama, K.K.; Sun, Y.; Saha, L.K.; Yang, X.; Arakawa, Y.; Gayle, S.; Jones, K.; Paralkar, V.; et al. TOP1-DNA Trapping by Exatecan and Combination Therapy with ATR Inhibitor. Mol. Cancer Ther. 2022, 21, 1090–1102. [Google Scholar] [CrossRef]
- Takimoto, R.; Kamigaki, T.; Okada, S.; Ibe, H.; Oguma, E.; Goto, S. Prognostic Factors for Advanced/Recurrent Breast Cancer Treated with Immune-Cell Therapy. Anticancer Res. 2021, 41, 4133–4141. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| DDRi | Agent | NCT ID Number | Status | Disease | Phase | Patient Number | Treatment | Safety | Result |
|---|---|---|---|---|---|---|---|---|---|
| ATRi | VX-970/Berzosertib | NCT02157792 | Completed | BC | I | 4 (cohort 1) | berzosertib iv (140 mg/m2; D2, D9), +cisplatin (75 mg/m2; D1) q21 days | Most common grade ≥ 3 TRAEs: neutropenia (20.0%) and anemia (16.7%). | 1 TNBC patient achieved PR but progressed after 17 months [82] |
| Completed | mTNBC | I | 47 (cohort 2) | berzosertib iv (140 mg/m2; D2, D9), +cisplatin (75 mg/m2; D1) q21 days | Grade ≥ 3 TEAEs: neutropenia (38.3%), anemia (25.5%), thrombocytopenia (12.8%), and vomiting (12.8%). | ORR: 11/47 (23.4%) patients [84] | |||
| Completed | Refractory Solid Tumors | I | 2 (cohort 3) | berzosertib iv 90 mg/m2 (D2, D9) + carboplatin AUC 5 (D1) q21 days | Grade ≥ 3 AEs: neutropenia (21.7%), anemia (4.3%), and thrombocytopenia (4.3%). | 1 mBC patient experienced dose-limiting febrile neutropenia (berzosertib 90 mg/m2 once weekly + carboplatin) [85] | |||
| AZD6738/Ceralasertib | NCT02264678 | Recruiting | Advanced Solid Malignancies | I/Ib | Still recruiting | ceralasertib (in escalating doses 60 mg OD to 240 mg BD D5–D14) + carboplatin, Olaparib, or durvalumab | Frequent TRAEs: thrombocytopenia, anemia, neutropenia, fatigue, decreased appetite, nausea, vomiting, constipation, diarrhea, cough. | FIRST RESULTS: 2 BRCA mutant TNBC patients achieved PRs [143] | |
| VIOLETTE (NCT03330847) | Active, Not Recruiting | mTNBC | II | 226 | olaparib (300 mg BID q28 days) + ceralasertib (160 mg D1–D7) q28 days vs. olaparib (300 mg BID q28 days) monotherapy | Most common TRAEs: nausea and anemia (47% of patients). | No significant difference in ORR or PFS with the combination [89] | ||
| VX-803/M4344/Gartisertib | NCT02278250 | Completed | Advanced Solid Tumors | I | 97 | Gartisertib (250 mg qd) monotherapy or in combination with carboplatin | Significant liver toxicity [95]. | n/a | |
| RP-3500/Camonsertib | NCT04497116 | Active, Not Recruiting | Advanced Solid Tumors harboring DDR mutations | I | 120 | Camonsertib orally (160 mg D1–D3 qw) monotherapy | Anemia was the most common drug-related toxicity (32% grade 3). | Upon camonsertib treatment in 1 patient with BRCA1-mutated TNBC, all BRCA1 mutations declined in blood but then rebounded before progression of non-target lesions at 29 weeks of treatment. Overall, anemia was the most common drug-related toxicity (32% Grade 3) [79] | |
| CHK1i | GDC-0425 | NCT01359696 | Completed | Refractory Solid Tumors | I | 5 | GDC-0425 orally (60 mg). +gemcitabine iv (1000 mg/m2) | DLTs: thrombocytopenia (n = 5), neutropenia (n = 4), dyspnea, nausea, pyrexia, syncope, and increased. alanine aminotransferase (n = 1 each). Common TRAEs: nausea (48%); anemia, neutropenia, vomiting (45% each); fatigue (43%); pyrexia (40%); and thrombocytopenia (35%). | 2 PRs in TP53 mutated TNBC [112] |
| LY2606368/Prexasertib | NCT02203513 | Terminated | BRCAwt patients | II | 9 | Prexasertib iv 105 mg/m2 q2w monotherapy | Grade 3/4 TRAEs: afebrile neutropenia (n = 8; 88.9%), anemia (n = 3; 33.3%), and thrombocytopenia (n = 1; 11.1%). | 1 PR and 4 SD [129] | |
| NCT02873975 | Completed | Advanced Solid Tumors with specific mutations | II | n/a | n/a | n/a | n/a | ||
| NCT03057145 | Completed | Advanced Solid Tumors | I | n/a | Prexasertib + Olaparib | n/a | n/a | ||
| NCT02124148 | Completed | Advanced and/or Metastatic Solid Tumors | Ib | 53 | Prexasertib iv (105 mg/m2 q14 days) + Samotolisib orally (200 mg twice daily) | Common TRAEs: leukopenia/neutropenia (94.3%), thrombocytopenia (62.3%), and nausea (52.8%). | ORR for TNBC: 25% [133] | ||
| UCN-01 | NCT00031681 | Completed | Resistant Solid Tumor Malignancies | I | 5 | UCN-01 iv (70 mg/m2 D2, 35 mg/m2 D23) +irinotecan iv (125 mg/m2 D1, D8, D15, D22) | DLTs: grade 3 diarrhea/dehydration and dyspnea. | 2 out of 5 PRs in p53 mutated TNBC [138] | |
| n/a | Completed | mTNBC | II | 25 | UCN-01 + irinotecan | n/a | 1 PR [138,139,140,141] |
| DDRi | Agent | NCT ID Number | Disease | Phase | Treatment |
|---|---|---|---|---|---|
| ATRi | VX-970/Berzosertib | NCT04052555 | Chemotherapy resistant TNBC/HR+, HER2-BC | Ib | Berzosertib + Radiation Therapy |
| AZD6738/Ceralasertib | NCT04090567 | Germline BRCA-mutated BC advanced or metastatic | II | Olaparib + Cediranib or AZD6738 | |
| PHOENIX (NCT03740893) | Neoadjuvant chemotherapy-resistant residual TNBC | IIa | short exposure to DDRi and/or anti-PD-L1 immunotherapy preoperatively | ||
| NCT03801369 | mTNBC | II | Olaparib + durvalumab, selumetinib, or capivasertib or ceralasertib alone | ||
| BAY-1895344/Elimusertib | NCT04491942 | Solid Tumors | I | Elimusertib + chemotherapy | |
| RP-3500/Camonsertib | NCT04972110 | Advanced Solid Tumors | I/II | Camonsertib + Niraparib/Olaparib | |
| CHKi | LY2606368/Prexasertib | NCT04032080 | mTNBC | II | Prexasertib + Samotolisib |
| ATMi | AZD0156 | NCT02588105 | Metastatic disease | I | AZD0156 as monotherapy or +olaparib or FOLRIRI |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sofianidi, A.; Dumbrava, E.E.; Syrigos, K.N.; Nasrazadani, A. Triple-Negative Breast Cancer and Emerging Therapeutic Strategies: ATR and CHK1/2 as Promising Targets. Cancers 2024, 16, 1139. https://doi.org/10.3390/cancers16061139
Sofianidi A, Dumbrava EE, Syrigos KN, Nasrazadani A. Triple-Negative Breast Cancer and Emerging Therapeutic Strategies: ATR and CHK1/2 as Promising Targets. Cancers. 2024; 16(6):1139. https://doi.org/10.3390/cancers16061139
Chicago/Turabian StyleSofianidi, Amalia, Ecaterina E. Dumbrava, Konstantinos N. Syrigos, and Azadeh Nasrazadani. 2024. "Triple-Negative Breast Cancer and Emerging Therapeutic Strategies: ATR and CHK1/2 as Promising Targets" Cancers 16, no. 6: 1139. https://doi.org/10.3390/cancers16061139
APA StyleSofianidi, A., Dumbrava, E. E., Syrigos, K. N., & Nasrazadani, A. (2024). Triple-Negative Breast Cancer and Emerging Therapeutic Strategies: ATR and CHK1/2 as Promising Targets. Cancers, 16(6), 1139. https://doi.org/10.3390/cancers16061139