
T Cell Features in Glioblastoma May Guide Therapeutic Strategies to Overcome Microenvironment Immunosuppression

 , , , , ,
, , , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Gliomas Immune Landscape
2.1. Microglia and Macrophages
2.2. Neutrophils
2.3. Dendritic Cells
2.4. T Cells
2.4.1. Conventional T Cells
2.4.2. T Regulatory Cells
2.5. Tumor Microenvironment: Specific Changes in Recurrent GBM
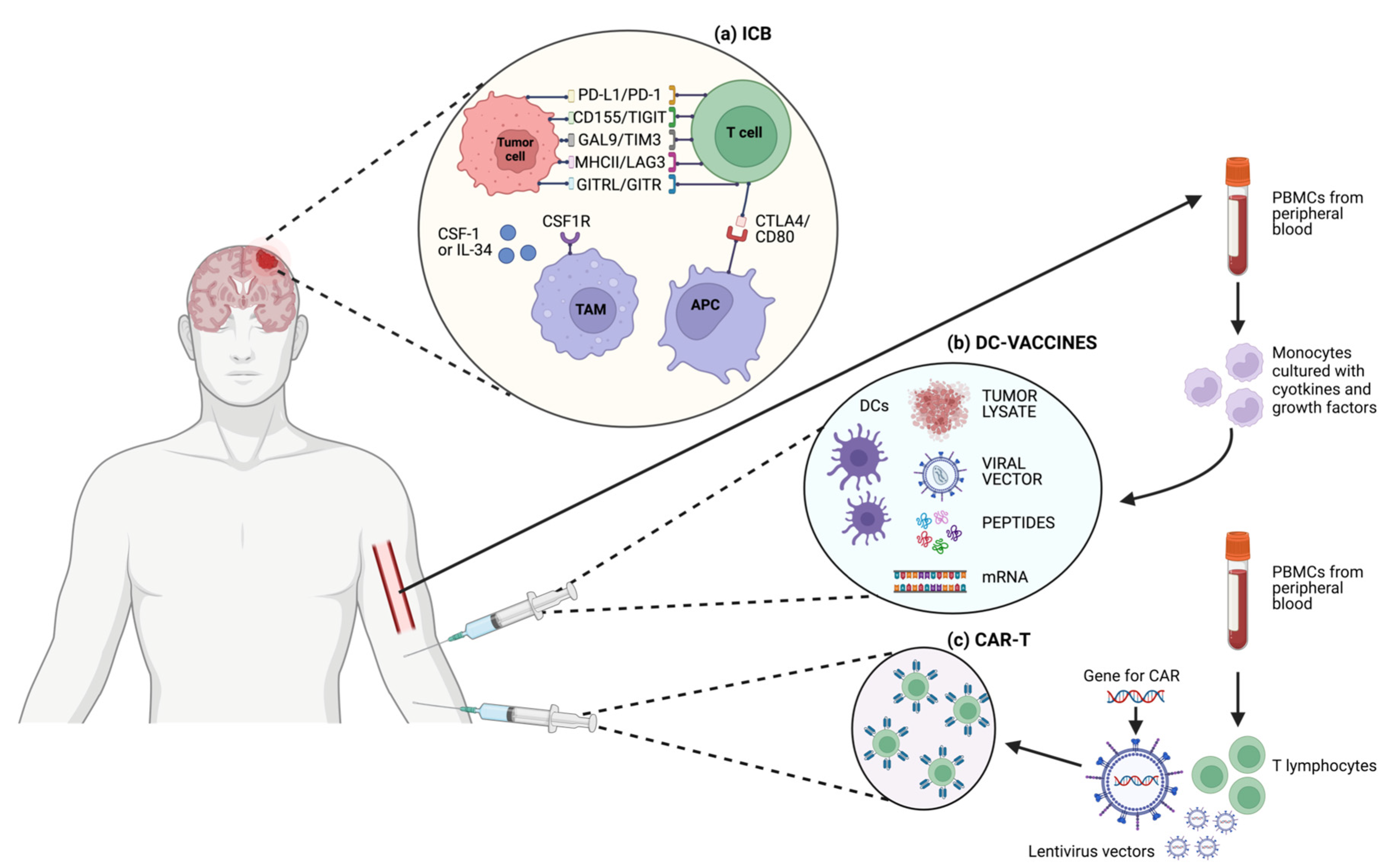
3. Immunotherapy
3.1. Targeting the PD-1/PD-L1 Axis, Alone or in Combination with Anti-CTLA4
3.2. Other Co-Stimulatory and Co-Inhibitory Pathways
3.3. Vaccines
3.4. Adoptive Cell Therapy and Chimeric Antigen Receptor T Cell Therapy
4. How to Overcome Immunoresistance: Future Perspectives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro Oncol. 2022, 24 (Suppl. S5), v1–v95. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Sun, W.; Hussain, S.F.; Dey, M.; Crutcher, L.; Aldape, K.; Gilbert, M.; Hassenbusch, S.J.; Sawaya, R.; Schmittling, B.; et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: Case study. Neuro Oncol. 2008, 10, 98–103. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I.; et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Vidyarthi, A.; Agnihotri, T.; Khan, N.; Singh, S.; Tewari, M.K.; Radotra, B.D.; Chatterjee, D.; Agrewala, J.N. Predominance of M2 macrophages in gliomas leads to the suppression of local and systemic immunity. Cancer Immunol. Immunother. 2019, 68, 1995–2004. [Google Scholar] [CrossRef]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef]
- Hor, W.-S.; Huang, W.-L.; Lin, Y.-S.; Yang, B.-C. Cross-talk between tumor cells and neutrophils through the Fas (APO-1, CD95)/FasL system: Human glioma cells enhance cell viability and stimulate cytokine production in neutrophils. J. Leukoc. Biol. 2003, 73, 363–368. [Google Scholar] [CrossRef]
- Chio, C.-C.; Wang, Y.-S.; Chen, Y.-L.; Lin, S.-J.; Yang, B.-C. Down-regulation of Fas-L in glioma cells by ribozyme reduces cell apoptosis, tumour-infiltrating cells, and liver damage but accelerates tumour formation in nude mice. Br. J. Cancer 2001, 85, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; De Groot, J.F. Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef]
- Maas, R.R.; Soukup, K.; Fournier, N.; Massara, M.; Galland, S.; Kornete, M.; Wischnewski, V.; Lourenco, J.; Croci, D.; Álvarez-Prado, Á.F.; et al. The local microenvironment drives activation of neutrophils in human brain tumors. Cell 2023, 186, 4546–4566. [Google Scholar] [CrossRef]
- Bambury, R.M.; Teo, M.Y.; Power, D.G.; Yusuf, A.; Murray, S.; Battley, J.E.; Drake, C.; O’Dea, P.; Bermingham, N.; Keohane, C.; et al. The association of pre-treatment neutrophil to lymphocyte ratio with overall survival in patients with glioblastoma multiforme. J. Neurooncol. 2013, 114, 149–154. [Google Scholar] [CrossRef]
- McNamara, M.G.; Lwin, Z.; Jiang, H.; Chung, C.; Millar, B.-A.; Sahgal, A.; Laperriere, N.; Mason, W.P. Conditional probability of survival and post-progression survival in patients with glioblastoma in the temozolomide treatment era. J. Neurooncol. 2014, 117, 153–160. [Google Scholar] [CrossRef]
- Mason, M.; Maurice, C.; McNamara, M.G.; Tieu, M.T.; Lwin, Z.; Millar, B.-A.; Menard, C.; Laperriere, N.; Milosevic, M.; Atenafu, E.G.; et al. Neutrophil-lymphocyte ratio dynamics during concurrent chemo-radiotherapy for glioblastoma is an independent predictor for overall survival. J. Neurooncol. 2017, 132, 463–471. [Google Scholar] [CrossRef]
- Wang, P.-F.; Song, H.-W.; Cai, H.-Q.; Kong, L.-W.; Yao, K.; Jiang, T.; Li, S.-W.; Yan, C.-X. Preoperative inflammation markers and IDH mutation status predict glioblastoma patient survival. Oncotarget 2017, 8, 50117–50123. [Google Scholar] [CrossRef]
- Bertaut, A.; Truntzer, C.; Madkouri, R.; Kaderbhai, C.G.; Derangère, V.; Vincent, J.; Chauffert, B.; Aubriot-Lorton, M.H.; Farah, W.; Mourier, K.L. Blood baseline neutrophil count predicts bevacizumab efficacy in glioblastoma. Oncotarget 2016, 7, 70948–70958. [Google Scholar] [CrossRef]
- Della Bella, S.; Clerici, M.; Villa, M.L. Disarming dendritic cells: A tumor strategy to escape from immune control? Expert Rev. Clin. Immunol. 2007, 3, 411–422. [Google Scholar] [CrossRef]
- Marland, G.; Bakker, A.B.; Adema, G.J.; Figdor, C.G. Dendritic cells in immune response induction. Stem Cells 1996, 14, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Villadangos, J.A.; Young, L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity 2008, 29, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Irla, M.; Küpfer, N.; Suter, T.; Lissilaa, R.; Benkhoucha, M.; Skupsky, J.; Lalive, P.H.; Fontana, A.; Reith, W.; Hugues, S. MHC class II-restricted antigen presentation by plasmacytoid dendritic cells inhibits T cell-mediated autoimmunity. J. Exp. Med. 2010, 207, 1891–1905. [Google Scholar] [CrossRef]
- Conrad, C.; Gregorio, J.; Wang, Y.-H.; Ito, T.; Meller, S.; Hanabuchi, S.; Anderson, S.; Atkinson, N.; Ramirez, P.T.; Liu, Y.-J. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3(+) T-regulatory cells. Cancer Res. 2012, 72, 5240–5249. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, G.; Garcia, S.; Escriou, N.; Freitas, A.A.; Leclerc, C.; Dadaglio, G. Murine plasmacytoid dendritic cells induce effector/memory CD8+ T-cell responses in vivo after viral stimulation. Blood 2004, 104, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Salio, M.; Palmowski, M.J.; Atzberger, A.; Hermans, I.F.; Cerundolo, V. CpG-matured murine plasmacytoid dendritic cells are capable of in vivo priming of functional CD8 T cell responses to endogenous but not exogenous antigens. J. Exp. Med. 2004, 199, 567–579. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Stichel, D.; Hielscher, T.; Sievers, P.; Berghoff, A.S.; Schrimpf, D.; Sill, M.; Euskirchen, P.; Blume, C.; Patel, A.; et al. Integrated Molecular-Morphologic Meningioma Classification: A Multicenter Retrospective Analysis, Retrospectively and Prospectively Validated. J. Clin. Oncol. 2021, 39, 3839–3852. [Google Scholar] [CrossRef]
- Wischnewski, V.; Maas, R.R.; Aruffo, P.G.; Soukup, K.; Galletti, G.; Kornete, M.; Galland, S.; Fournier, N.; Lilja, J.; Wirapati, P.; et al. Phenotypic diversity of T cells in human primary and metastatic brain tumors revealed by multiomic interrogation. Nat. Cancer 2023, 4, 908–924. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef]
- Davidson, T.B.; Lee, A.; Hsu, M.; Sedighim, S.; Orpilla, J.; Treger, J.; Mastall, M.; Roesch, S.; Rapp, C.; Galvez, M. Expression of PD-1 by T Cells in Malignant Glioma Patients Reflects Exhaustion and Activation. Clin. Cancer Res. 2019, 25, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; De Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724. [Google Scholar] [CrossRef] [PubMed]
- Losurdo, A.; Scirgolea, C.; Alvisi, G.; Brummelman, J.; Errico, V.; Di Tommaso, L.; Pilipow, K.; Colombo, F.S.; Fernandes, B.; Peano, C.; et al. Author Correction: Single-cell profiling defines the prognostic benefit of CD39(high) tissue resident memory CD8+ T cells in luminal-like breast cancer. Commun. Biol. 2021, 4, 1252. [Google Scholar] [CrossRef]
- Abdelfattah, N.; Kumar, P.; Wang, C.; Leu, J.-S.; Flynn, W.F.; Gao, R.; Baskin, D.S.; Pichumani, K.; Ijare, O.B.; Wood, S.L.; et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat. Commun. 2022, 13, 767. [Google Scholar] [CrossRef] [PubMed]
- Ambartsumian, N.; Klingelhofer, J.; Grigorian, M. The Multifaceted S100A4 Protein in Cancer and Inflammation. Methods Mol. Biol. 2019, 1929, 339–365. [Google Scholar]
- Ravi, V.M.; Will, P.; Kueckelhaus, J.; Sun, N.; Joseph, K.; Salié, H.; Vollmer, L.; Kuliesiute, U.; Von Ehr, J.; Benotmane, J.K.; et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 2022, 40, 639–655. [Google Scholar] [CrossRef]
- Fornara, O.; Odeberg, J.; Wolmer Solberg, N.; Tammik, C.; Skarman, P.; Peredo, I.; Stragliotto, G.; Rahbar, A.; Söderberg-Nauclér, C. Poor survival in glioblastoma patients is associated with early signs of immunosenescence in the CD4 T-cell compartment after surgery. Oncoimmunology 2015, 4, e1036211. [Google Scholar] [CrossRef]
- El Andaloussi, A.; Lesniak, M.S. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol. 2006, 8, 234–243. [Google Scholar] [CrossRef]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E.; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.-S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef] [PubMed]
- Campos, B.; Olsen, L.R.; Urup, T.; Poulsen, H.S. A comprehensive profile of recurrent glioblastoma. Oncogene 2016, 35, 5819–5825. [Google Scholar] [CrossRef]
- De Groot, J.; Penas-Prado, M.; Alfaro-Munoz, K.; Hunter, K.; Pei, B.L.; O’Brien, B.; Weathers, S.-P.; Loghin, M.; Kamiya Matsouka, C.; Yung, W.K.A.; et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. 2020, 22, 539–549. [Google Scholar] [CrossRef]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol. 2013, 15, 1079–1087. [Google Scholar] [CrossRef]
- Hoogstrate, Y.; Draaisma, K.; Ghisai, S.A.; Van Hijfte, L.; Barin, N.; De Heer, I.; Coppieters, W.; Van Den Bosch, T.P.P.; Bolleboom, A.; Gao, Z.; et al. Transcriptome analysis reveals tumor microenvironment changes in glioblastoma. Cancer Cell. 2023, 41, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Van Hooren, L.; Handgraaf, S.M.; Kloosterman, D.J.; Karimi, E.; Van Mil, L.W.H.G.; Gassama, A.A.; Solsona, B.G.; De Groot, M.H.P.; Brandsma, D.; Quail, D.F.; et al. CD103(+) regulatory T cells underlie resistance to radio-immunotherapy and impair CD8(+) T cell activation in glioblastoma. Nat. Cancer 2023, 4, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Ochiai, H.; Mitchell, D.A.; Grossi, P.M.; Sweeney, A.E.; Archer, G.E.; Cummings, T.; Allison, J.P.; Bigner, D.D.; Sampson, J.H. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin. Cancer Res. 2007, 13, 2158–2167. [Google Scholar] [CrossRef]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; Van Den Bent, M.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. 2023, 25, 123–134. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022, 24, 1935–1949. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Nayak, L.; Standifer, N.; Dietrich, J.; Clarke, J.L.; Dunn, G.P.; Lim, M.; Cloughesy, T.; Gan, H.K.; Flagg, E.; George, E.; et al. Circulating Immune Cell and Outcome Analysis from the Phase II Study of PD-L1 Blockade with Durvalumab for Newly Diagnosed and Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 2567–2578. [Google Scholar] [CrossRef]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; De Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: A phase I clinical trial. J. Immunother. Cancer 2021, 9, e002296. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Gelb, A.B.; Chen, C.C.; Rao, G.; Reardon, D.A.; Wen, P.Y.; Bi, W.L.; Peruzzi, P.; Amidei, C.; Triggs, D.; et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial. Neuro Oncol. 2022, 24, 951–963. [Google Scholar] [CrossRef]
- Webb, M.; Sener, U.; Burns, T.; Twohy, E.; Kizilbash, S.H.; Ruff, M.; Uhm, J.; Galanis, E.; D’Andre, S.; Riviere-Cazaux, C.; et al. Efficacy and safety study of neoadjuvant efineptakin alfa (NT-I7) and pembrolizumab in recurrent glioblastoma. J. Clin. Oncol. 2023, 41 (Suppl. S16). [Google Scholar] [CrossRef]
- Brown, N.F.; Ng, S.M.; Brooks, C.; Coutts, T.; Holmes, J.; Roberts, C.; Elhussein, L.; Hoskin, P.; Maughan, T.; Blagden, S.; et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: The Ipi-Glio trial protocol. BMC Cancer 2020, 20, 198. [Google Scholar] [CrossRef] [PubMed]
- Pouessel, D.; Ken, S.; Gouaze-Andersson, V.; Piram, L.; Mervoyer, A.; Larrieu-Ciron, D.; Cabarrou, B.; Lusque, A.; Robert, M.; Frenel, J.-S.; et al. Hypofractionated Stereotactic Re-irradiation and Anti-PDL1 Durvalumab Combination in Recurrent Glioblastoma: STERIMGLI Phase I Results. Oncologist 2023, 28, e817–e825. [Google Scholar] [CrossRef]
- Ahluwalia, M.; Peereboom, D.; Rauf, Y.; Schilero, C.; Ciolfi, M.; Ciesielski, M.; Fenstermaker, R. Phase II study of pembrolizumab plus SurVaxM for glioblastoma at first recurrence. J. Clin. Oncol. 2020, 38 (Suppl. S15), TPS2581. [Google Scholar] [CrossRef]
- Ahluwalia, M.; Peereboom, D.; Rauf, Y.; Schilero, C.; Ciolfi, M.; Ciesielski, M.; Fenstermaker, R. Avelumab in newly diagnosed glioblastoma. Neurooncol. Adv. 2021, 3, vdab118. [Google Scholar]
- Hormigo, A.; Chiu, D.; Hahn, M.; Qi, J.; Lee, B.; Mandeli, J.; Ghatan, S.; Hadjipanayis, C.; Yong, R.; Germano, I.; et al. CTIM-09. Phase I study of PD-L1 inhibition with avelumab and laser interstitial thermal therapy in patients with recurrent glioblastoma. Neuro-Oncology 2021, 23 (Suppl. S6), vi51. [Google Scholar] [CrossRef]
- Raphael, I.; Kumar, R.; McCarl, L.H.; Shoger, K.; Wang, L.; Sandlesh, P.; Sneiderman, C.T.; Allen, J.; Zhai, S.; Campagna, M.L.; et al. TIGIT and PD-1 Immune Checkpoint Pathways Are Associated With Patient Outcome and Anti-Tumor Immunity in Glioblastoma. Front. Immunol. 2021, 12, 637146. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Medikonda, R.; Saleh, L.; Kim, T.; Pant, A.; Srivastava, S.; Kim, Y.-H.; Jackson, C.; Tong, L.; Routkevitch, D.; et al. Combination checkpoint therapy with anti-PD-1 and anti-BTLA results in a synergistic therapeutic effect against murine glioblastoma. Oncoimmunology 2021, 10, 1956142. [Google Scholar] [CrossRef]
- Kim, S.T.; Klempner, S.J.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, K.-M.; Lee, J. Correlating programmed death ligand 1 (PD-L1) expression, mismatch repair deficiency, and outcomes across tumor types: Implications for immunotherapy. Oncotarget 2017, 8, 77415–77423. [Google Scholar] [CrossRef]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef]
- Lim, M.; Ye, X.; Nabors, L.B.; Piotrowski, A.; Ahluwalia, M.; Desai, A.; Walbert, T.; Fisher, J.; Desideri, S.; Sims, M.; et al. Updated phase I trial of anti-LAG-3 or anti-CD137 alone and in combination with anti-PD-1 in patients with recurrent GBM. J. Clin. Oncol. 2019, 37 (Suppl. S15), 2017. [Google Scholar] [CrossRef]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef]
- Davar, D.; Zappasodi, R. Targeting GITR in cancer immunotherapy—There is no perfect knowledge. Oncotarget 2023, 14, 614–621. [Google Scholar] [CrossRef]
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235. [Google Scholar] [CrossRef]
- Lu, L.; Xu, X.; Zhang, B.; Zhang, R.; Ji, H.; Wang, X. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J. Transl. Med. 2014, 12, 36. [Google Scholar] [CrossRef]
- Schoenhals, J.E.; Cushman, T.R.; Barsoumian, H.B.; Li, A.; Cadena, A.P.; Niknam, S.; Younes, A.I.; Caetano, M.D.S.; Cortez, M.A.; Welsh, J.W. Anti-glucocorticoid-induced Tumor Necrosis Factor-Related Protein (GITR) Therapy Overcomes Radiation-Induced Treg Immunosuppression and Drives Abscopal Effects. Front. Immunol. 2018, 9, 2170. [Google Scholar] [CrossRef]
- Pouessel, D.; Ken, S.; Gouaze-Andersson, V.; Piram, L.; Mervoyer, A.; Larrieu-Ciron, D.; Cabarrou, B.; Lusque, A.; Robert, M.; Frenel, J.-S.; et al. PD1 inhibition and GITR agonism in combination with fractionated stereotactic radiotherapy for the treatment of recurrent glioblastoma: A phase 2, multi-arm study. J. Clin. Oncol. 2023, 41 (Suppl. S16), 2004. [Google Scholar]
- Mathewson, N.D.; Ashenberg, O.; Tirosh, I.; Gritsch, S.; Perez, E.M.; Marx, S.; Jerby-Arnon, L.; Chanoch-Myers, R.; Hara, T.; Richman, A.R.; et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 2021, 184, 1281–1298. [Google Scholar] [CrossRef]
- Saxena, M.; Van Der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, D.M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2023, 41, 1453–1465. [Google Scholar] [CrossRef]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef]
- Chometon, T.Q.; Siqueira, M.D.S.; Sant´anna, J.C.; Almeida, M.R.; Gandini, M.; Martins De Almeida Nogueira, A.C.; Antas, P.R.Z. A protocol for rapid monocyte isolation and generation of singular human monocyte-derived dendritic cells. PLoS ONE 2020, 15, e0231132. [Google Scholar] [CrossRef]
- Hotchkiss, K.M.; Batich, K.A.; Mohan, A.; Rahman, R.; Piantadosi, S.; Khasraw, M. Dendritic cell vaccine trials in gliomas: Untangling the lines. Neuro Oncol. 2023, 25, 1752–1762. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; van den Bent, M.J. Autologous tumor lysate-loaded dendritic cell vaccination (DCVax-L) in glioblastoma: Breakthrough or fata morgana? Neuro Oncol. 2023, 25, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Taylor, T.H.; Piccioni, D.E.; Duma, C.M.; LaRocca, R.V.; Kesari, S.; Carrillo, J.A.; Abedi, M.; Aiken, R.D.; Hsu, F.P.K.; et al. Phase 2 study of AV-GBM-1 (a tumor-initiating cell targeted dendritic cell vaccine) in newly diagnosed Glioblastoma patients: Safety and efficacy assessment. J. Exp. Clin. Cancer Res. 2022, 41, 344. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.-J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef]
- Hu, J.L.; Omofoye, O.A.; Rudnick, J.D.; Kim, S.; Tighiouart, M.; Phuphanich, S.; Wang, H.; Mazer, M.; Ganaway, T.; Chu, R.M.; et al. A Phase I Study of Autologous Dendritic Cell Vaccine Pulsed with Allogeneic Stem-like Cell Line Lysate in Patients with Newly Diagnosed or Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 689–696. [Google Scholar] [CrossRef]
- Rudnick, J.D.; Sarmiento, J.M.; Uy, B.; Nuno, M.; Wheeler, C.J.; Mazer, M.J.; Wang, H.; Hu, J.L.; Chu, R.M.; Phuphanich, S.; et al. A phase I trial of surgical resection with Gliadel Wafer placement followed by vaccination with dendritic cells pulsed with tumor lysate for patients with malignant glioma. J. Clin. Neurosci. 2020, 74, 187–193. [Google Scholar] [CrossRef]
- Rahman, M.; Dastmalchi, F.; Karachi, A.; Mitchell, D. The role of CMV in glioblastoma and implications for immunotherapeutic strategies. Oncoimmunology 2019, 8, e1514921. [Google Scholar] [CrossRef]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H. Reproducibility of outcomes in sequential trials using CMV-targeted dendritic cell vaccination for glioblastoma. J. Clin. Oncol. 2022, 40 (Suppl. S16), 2005. [Google Scholar]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E.; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef]
- Miller, A.; Kosaloglu-Yalcin, Z.; Westernberg, L.; Montero, L.; Bahmanof, M.; Frentzen, A.; Logandha Ramamoorthy Premlal, A.; Greenbaum, J.; Seumois, G.; Habbaba, R.; et al. A phase 1b study of personalized neoantigen vaccine plus pembrolizumab in adults with advanced cancer. J. Clin. Oncol. 2021, 39 (Suppl. S15), 2615. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Lee, D.W.; Yates, B.; Yuan, C.M.; Shalabi, H.; Martin, S.; Wolters, P.L.; Steinberg, S.M.; Baker, E.H.; Delbrook, C.P.; et al. Long-Term Follow-Up of CD19-CAR T-Cell Therapy in Children and Young Adults With B-ALL. J. Clin. Oncol. 2021, 39, 1650–1659. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef]
- Katz, S.C.; Burga, R.A.; McCormack, E.; Wang, L.J.; Mooring, W.; Point, G.R.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor-Modified T-cell Therapy for CEA+ Liver Metastases. Clin. Cancer Res. 2015, 21, 3149–3159. [Google Scholar] [CrossRef]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef]
- Learn, C.A.; Hartzell, T.L.; Wikstrand, C.J.; Archer, G.E.; Rich, J.N.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin. Cancer Res. 2004, 10, 3216–3224. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Mineo, J.-F.; Bordron, A.; Baroncini, M.; Maurage, C.-A.; Ramirez, C.; Siminski, R.-M.; Berthou, C.; Dam Hieu, P. Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. J. Neurooncol. 2007, 85, 281–287. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. Pilot study of T cells redirected to EGFRvIII with a chimeric antigen receptor in patients with EGFRvIII+ glioblastoma. J. Clin. Oncol. 2016, 34 (Suppl. S15), 2067. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Xuan, W.; Khan, F.; James, C.D.; Heimberger, A.B.; Lesniak, M.S.; Chen, P. Circadian regulation of cancer cell and tumor microenvironment crosstalk. Trends Cell Biol. 2021, 31, 940–950. [Google Scholar] [CrossRef]
- Weller, M.; Van Den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Di Ianni, N.; Maffezzini, M.; Eoli, M.; Pellegatta, S. Revisiting the Immunological Aspects of Temozolomide Considering the Genetic Landscape and the Immune Microenvironment Composition of Glioblastoma. Front. Oncol. 2021, 11, 747690. [Google Scholar] [CrossRef]
- De Martino, M.; Padilla, O.; Daviaud, C.; Wu, C.-C.; Gartrell, R.D.; Vanpouille-Box, C. Exploiting Radiation Therapy to Restore Immune Reactivity of Glioblastoma. Front. Oncol. 2021, 11, 671044. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, Q.; Tan, L.; Wu, X.; Huang, R.; Zuo, Y.; Chen, L.; Yang, J.; Zhang, Z.-X.; Ruan, W.; et al. Neutralizing IL-8 potentiates immune checkpoint blockade efficacy for glioma. Cancer Cell 2023, 41, 693–710. [Google Scholar] [CrossRef]
- Forrester, J.V.; McMenamin, P.G.; Dando, S.J. CNS infection and immune privilege. Nat. Rev. Neurosci. 2018, 19, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642. [Google Scholar] [CrossRef]
- Tawbi, H.A.-H.; Amaria, R.N.; Glitza, I.C.; Milton, D.; Hwu, W.-J.; Patel, S.P.; Wong, M.K.K.; Yee, C.; Woodman, S.E.; McQuade, J.L. Safety and preliminary activity data from a single center phase II study of triplet combination of nivolumab (N) with dabrafenib (D) and trametinib (T) [trident] in patients (Pts) with BRAF-mutated metastatic melanoma (MM). J. Clin. Oncol. 2018, 36 (Suppl. S15), 9560. [Google Scholar] [CrossRef]
- Goldberg, S.B.; Schalper, K.A.; Gettinger, S.N.; Mahajan, A.; Herbst, R.S.; Chiang, A.C.; Lilenbaum, R.; Wilson, F.H.; Omay, S.B.; Yu, J.B.; et al. Pembrolizumab for management of patients with NSCLC and brain metastases: Long-term results and biomarker analysis from a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2020, 21, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.J.; Burgess-Brown, N.A.; Jiang, S. Beyond Just Peptide Antigens: The Complex World of Peptide-Based Cancer Vaccines. Front. Immunol. 2021, 12, 696791. [Google Scholar] [CrossRef] [PubMed]
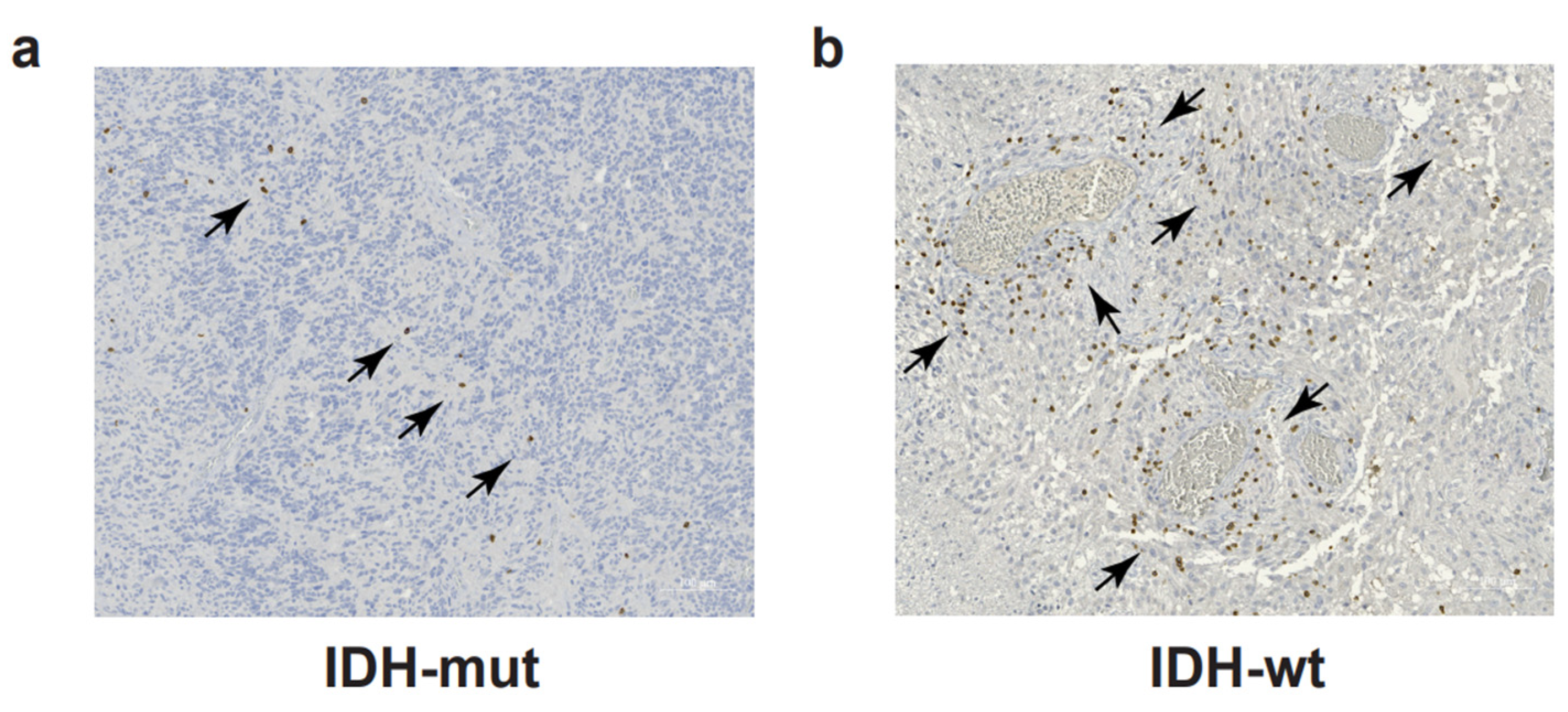

{kind=link}
{kind=link}
| Title | Identifier | Study Design | Treatment | Population | Main Results | Main TRAEs |
|---|---|---|---|---|---|---|
| A Randomized Phase 3 Open Label Study of Nivolumab vs. Temozolomide in Combination with Radiation Therapy in Newly Diagnosed Adult Subjects with Unmethylated MGMT Glioblastoma (CheckMate 498) [64] | NCT02617589 | Phase 3 | A: nivolumab + radiotherapy B: temozolomide + radiotherapy | Newly diagnosed methylated GBM | A: 13.0 months B: 14.2 months | A: 21.9% G3-4 TRAEs B: 25.1% G3-4 TRAEs |
| Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter (CheckMate548) [65] | NCT02667587 | Phase 3 | A: nivolumab + temozolomide + radiotherapy B: placebo + temozolomide + radiotherapy | Newly diagnosed methylated GBM | A: mPFS 10.6 months; mOS 31.3 months B: mPFS 10.3 months; mOS 33 months | A: 52.4% G3-4 TRAEs B: 33.6% G3-4 TRAEs |
| Effect of Nivolumab vs. Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial [63] | NCT02017717 | Phase 3 | A: nivolumab B: bevacizumab | Recurrent GBM | A: mOS: 9.8 months B: mOS 10 months | A: 18.1% G3-4 TRAEs B: 15.2% G3-4 TRAEs |
| Circulating Immune Cells and Outcome Analysis from the Phase II Study of PD-L1 Blockade with Durvalumab for Newly Diagnosed and Recurrent Glioblastoma [68] | NCT02336165 | Phase 2 | A: RT + durvalumab B: durvalumab B2: durvalumab + bevacizumab B3: bevacizumab C: durvalumab + bevacizumab | Newly diagnosed unmethylated GBM Recurrent GBM | A: mOS 15.1 months B: mOS 6.7 months B2: mOS 8.7 months B3: mOS 9.3 months C: mOS 4.5 months | Fatigue G>2: 18% Hyperlypase G>2: 8.8% Hyperamilase G>2: 5.6% Hypertension G>2: 5.6% |
| Tremelimumab and Durvalumab in Combination or Alone in Treating Patients with Recurrent Malignant Glioma | NCT02794883 | Phase 2 | A: tremelimumab B: durvalumab C: tremelimumab + durvalumab | Recurrent GBM | A: mOS 7.2 months B: mOS 11.7 months C: mOS 7.7 months | NR |
| Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma [69] | NCT02337491 | Phase 2 | A: pembrolizumab + bevacizumab B: pembrolizumab + placebo | Recurrent GBM | A: mOS 8.8 months B: mOS 10.3 months | A: 20% Hypertension G3 B: 10% Headache G3 |
| Nivolumab with DC Vaccines for Recurrent Brain Tumors | NCT02529072 | Phase 1 | A: pre-surgery nivolumab B: pre-surgery nivolumab + DC vaccine | Grade III and Grade IV brain tumors | A: mOS 8 months B: mOS 15.3 months | NR |
| Study of Cabiralizumab in Combination with Nivolumab in Patients With Selected Advanced Cancers | NCT02526017 | Phase 1 | cabiralizumab + nivolumab | GBM Other advanced solid tumors | mOS 8 months | NR |
| Intracerebral administration of CTLA-4 and PD-1 immune checkpoint-blocking monoclonal antibodies in patients with recurrent glioblastoma: a phase I clinical trial [70] | NCT03233152 | Phase 1 | nivolumab (iv) + ipilimumab (intratumoral) | Recurrent GBM | mOS 37 weeks mPFS 11.7 weeks | NR |
| Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial [71] | NCT04006119 | Phase 1 | Cemiplimab-rwlc + Ad-RTS-hIL-12 (intratumoral) + Veledimex (oral) | Recurrent GBM | mOS 8.9 months | NR |
| Title | Identifier | Study Design | Treatment | Population | Recruitment Status |
|---|---|---|---|---|---|
| Neoadjuvant PD-1 Antibody Alone or Combined with Autologous Glioblastoma Stem-like Cell Antigen-Primed DC Vaccines (GSC-DCV) for Patients With Recurrent Glioblastoma: A Phase II, Randomized Controlled, Double Blind Clinical Trial | NCT04888611 | Phase 2 | A: camrelizumab + GSC-DCV B: camrelizumab + placebo | Recurrent GBM | Recruiting |
| Testing the Use of the Immunotherapy Drugs Ipilimumab and Nivolumab Plus Radiation Therapy Compared to the Usual Treatment (Temozolomide and Radiation Therapy) for Newly Diagnosed MGMT Unmethylated Glioblastoma | NCT04396860 | Phase 2/3 | A: TMZ+RT B: TMZ+RT + ipilimumab + nivolumab | Newly diagnosed unmethylated GBM Gliosarcoma | Active, not recruiting |
| Efficacy and Safety Study of Neoadjuvant Efineptakin Alfa (NT-I7) and Pembrolizumab in Recurrent Glioblastoma [72] | NCT05465954 | Phase 2 | Efineptakin Alfa + pembrolizumab | Recurrent GBM | Recruiting |
| A phase II open-label, randomized study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: the Ipi-Glio trial protocol [73] | ISRCTN84434175 | Phase 2 | A: temozolomide + ipilimumab B: temozolomide | Newly diagnosed GBM | Active, not recruiting |
| A Phase I/II Multicenter Trial Evaluating the Association of Hypofractionated Stereotactic Radiation Therapy and the Anti-Programmed Death-ligand 1 (PD-L1) Durvalumab (Medi4736) for Patients With Recurrent Glioblastoma (STERIMGLI) [74] | NCT02866747 | Phase 1/2 | A: hFSRT B: durvalumab + hFSRT | Recurrent GBM | Active, not recruiting |
| Phase Ib/II Trial of Anti-PD-1 Immunotherapy and Stereotactic Radiation in Patients with Recurrent Glioblastoma | NCT04977375 | Phase 1/2 | pembrolizumab + stereotactic RT | Recurrent GBM | Recruiting |
| Pembrolizumab and Vorinostat Combined With Temozolomide for Newly Diagnosed Glioblastoma | NCT03426891 | Phase 1 | TMZ+RT + pembrolizumab + vorinostat | Newly diagnosed GBM | Active, not recruiting |
| A Phase II Study of Checkpoint Blockade Immunotherapy in Patients with Somatically Hypermutated Recurrent WHO Grade 4 Glioma | NCT04145115 | Phase 2 | nivolumab + ipilimumab | Recurrent Somatically Hypermutated GBM | Recruiting |
| Phase II Study of Pembrolizumab Plus SurVaxM for Glioblastoma at First Recurrence [75] | NCT04013672 | Phase 2 | A: SurVaxM + pembrolizumab (no prior anti-PD1 therapy) B: SurVaxM + pembrolizumab (prior anti-PD1 therapy) | Recurrent GBM | Active, not recruiting |
| Phase II Study of Pembrolizumab (MK-3475) in Combination with Standard Therapy for Newly Diagnosed Glioblastoma | NCT03197506 | Phase 2 | A: neoadj and adj pembrolizumab (TMZ+RT) B: adj pembrolizumab (TMZ+RT) | Newly diagnosed GBM | Recruiting |
| Pembrolizumab for Newly Diagnosed Glioblastoma (PERGOLA) | NCT03899857 | Phase 2 | TMZ+RT + pembrolizumab | Newly diagnosed GBM | Active, not recruiting |
| Avelumab in Patients With Newly Diagnosed Glioblastoma Multiforme (SEJ) [76] | NCT03047473 | Phase 2 | TMZ+RT + avelumab | Newly diagnosed GBM | Completed |
| Phase I/II Study to Evaluate the Safety and Clinical Efficacy of Atezolizumab (Anti-PD-L1) in Combination With Cabozantinib in Patients with Recurrent Glioblastoma (rGBM) | NCT05039281 | Phase 1/2 | cabozantinib + atezolizumab | Recurrent GBM | Recruiting |
| Combination of Checkpoint Inhibition and IDO1 Inhibition Together With Standard Radiotherapy or Chemoradiotherapy in Newly Diagnosed Glioblastoma. A Phase 1 Clinical and Translational Trial | NCT04047706 | Phase 1 | A (methylated): BMS-986205 + nivolumab (TMZ+RT) B (unmethylated): BMS-986205 + nivolumab (RT) | Newly diagnosed GBM | Active, not recruiting |
| ASP8374 + Cemiplimab in Recurrent Glioma | NCT04826393 | Phase 1 | ASP8374 + cemiplimab | Recurrent GBM | Active, not recruiting |
| Pembrolizumab and a Vaccine (ATL-DC) for the Treatment of Surgically Accessible Recurrent Glioblastoma | NCT04201873 | Phase 1 | A: DC Tumor Cell Lysate Vaccine + pembrolizumab B: DC Tumor Cell Lysate Vaccine + placebo | Recurrent GBM | Recruiting |
| Pharmacodynamic Study of Pembrolizumab in Patients With Recurrent Glioblastoma | NCT02337686 | Phase 2 | Pembrolizumab | Recurrent GBM | Active, not recruiting |
| An Open-Label, Multi-Center Trial of INO-5401 and INO-9012 Delivered by Electroporation (EP) in Combination With REGN2810 in Subjects With Newly-Diagnosed Glioblastoma (GBM) | NCT03491683 | Phase 1\2 | A (unmethylated): INO-5401 + INO-9012 + cemiplimab (TMZ+RT) B (methylated): INO-5401 + INO-9012 + cemiplimab + nivolumab (RT) | Newly Diagnosed GBM | Active, not recruiting |
| A Phase 1 Study to Evaluate IL13Rα2-Targeted Chimeric Antigen Receptor (CAR) T Cells Combined with Checkpoint Inhibition for Patients With Resectable Recurrent Glioblastoma | NCT04003649 | Phase 1 | A: IL13Rα2-CAR-T + ipilimumab + nivolumab B: IL13Rα2-CAR-T + nivolumab C: IL13Rα2-CAR-T | Recurrent GBM | Recruiting |
| Phase I Study of PD-L1 Inhibition With Avelumab and Laser Interstitial Thermal Therapy (LITT) in Patients with Recurrent Glioblastoma [77] | NCT03341806 | Phase 1 | A: avelumab B: avelumab + MRI-guided LITT | Recurrent GBM | Completed |
| Title | Identifier | Study Design | Vaccine Production | Population | Main Results | Treatment Related Adverse Events (TRAEs) |
|---|---|---|---|---|---|---|
| A Phase III Clinical Trial Evaluating DCVax®-L, Autologous Dendritic Cells Pulsed With Tumor Lysate Antigen For The Treatment Of Glioblastoma Multiforme (GBM) [96] | NCT00045968 | Phase 3 | Tumor lysate | Newly diagnosed GBM Recurrent GBM | nGBM mOS: 19.3 months rGBM mOS: 13.2 months | G 3\4 TRAEs: 2 cases of intracranial edema (G3), 1 case of nausea (G3), and 1 case of lymph node infection (G3) |
| Phase II Trial of Autologous Dendritic Cells Loaded With Autologous Tumor Associated Antigens (AV-GBM-1) as an Adjunctive Therapy Following Primary Surgery Plus Concurrent Chemoradiation in Patients With Newly Diagnosed Glioblastoma [98] | NCT03400917 | Phase 2 | Lysate of irradiated autologous tumor-initiating cells | Newly diagnosed GBM | mOS: 16 months 2-years OS: 27% mPFS: 10.4 months | G 3\4 TRAEs: none |
| A Randomized, Double-blind, Controlled Phase IIb Study of the Safety and Efficacy of ICT-107 in Newly Diagnosed Patients With Glioblastoma Multiforme (GBM) Following Resection and Chemoradiation [99] | NCT01280552 | Phase 2 | Synthetic peptide epitopes targeting GBM associated antigens MAGE-1, HER-2, AIM-2, TRP-2, gp100, and IL13Rα2 | Newly diagnosed GBM | mOS: 17 months (vs. 15 months, NS) mPFS: 11.2 months (vs. 9 months, p = 0.011) | G 3\4 TRAEs: no difference from the control group (G3 nervous system disorders, fatigue, WBC decrease, and infections) |
| A Phase II Feasibility Study of Adjuvant Intra-Nodal Autologous Dendritic Cell Vaccination for Newly Diagnosed Glioblastoma Multiforme | NCT00323115 | Phase 2 | Tumor lysate | Newly diagnosed GBM | mOS: 28 (15-44) months mPFS: 9.5 (5-41) months | N.A. |
| Phase I Trial of Vaccination With Autologous Dendritic Cells Pulsed With Lysate Derived From an Allogeneic Glioblastoma Stem-like Cell Line for Patients With Newly Diagnosed or Recurrent Glioblastoma [100] | NCT02010606 | Phase 1 | Lysate from an allogeneic stem-like cell line | Newly diagnosed GBM Recurrent GBM | nGBM mOS: 20.36 months mPFS: 8.75 months rGBM mOS: 11.97 months mPFS: 3.23 months | G 3\4 TRAEs: none |
| A phase I trial of surgical resection with Gliadel Wafer placement followed by vaccination with dendritic cells pulsed with tumor lysate for patients with malignant glioma [101] | N.A. | Phase 1 | Tumor lysate | Newly diagnosed GBM Recurrent GBM | nGBM mOS: 27.7 months mPFS: 4.8 months rGBM mOS: 10.9 months mPFS: 1.9 months | G 3\4 TRAEs: none |
| Anti-Tumor Immunotherapy Targeted Against Cytomegalovirus in Patients With Newly-Diagnosed Glioblastoma Multiforme During Recovery From Therapeutic Temozolomide-induced Lymphopenia | NCT00639639 | Phase 1 | CMV pp65 | Newly diagnosed GBM | ATTAC-GM mOS: 37.7 months ATTAC-Td mOS: 38.3 months | N.A. |
| Evaluation of Overcoming Limited Migration and Enhancing Cytomegalovirus-specific Dendritic Cell Vaccines With Adjuvant TEtanus Pre-conditioning in Patients With Newly-diagnosed Glioblastoma | NCT02366728 | Phase 2 | CMV pp65 | Newly diagnosed GBM | Arm1 (unpulsed DC): mOS 16 months Arm2 (Td pre-conditioning): mOS 20 months Arm3 (Basiliximab + Td pre-conditioning): mOS 19 months | SAEs: Arm 1: 1\27 pts (lung infection) Arm 2: 4\28 pts (urinary infection, nervous system, and psychiatric disorders) Arm 3: 2\9 pts (colon perforation, nervous system disorders) |
| Title | Identifier | Study Design | Target Antigen | Status | Main Results | Main TRAEs |
|---|---|---|---|---|---|---|
| Administration of HER2 Chimeric Antigen Receptor Expressing CMV-Specific Cytotoxic T Cells In Patients With Glioblastoma Multiforme (HERT-GBM) | NCT01109095 | Phase 1 | HER2 | Completed | 16 evaluable pts (9 adults and 7 children) 1 PR (>9 months); 7 SD (8 weeks to 29 months); 8 PD mOS 11.1 months (95% CI, 4.1–27.2 months) | No DLTs G2 seizures and/or headaches |
| Pilot Study of Autologous T Cells Redirected to EGFRVIII with a Chimeric Antigen Receptor in Patients With EGFRVIII+ Glioblastoma [121] | NCT02209376 | Phase 1 | EGFRvIII | Terminated | mOS: 251 days (~8 months) (1 patient alive at 18 months) mPFS: not evaluable | No dose-limiting toxicities AEs: neurological events (seizures and neurologic decline), fatigue, fever, muscle weakness, skin and subcutaneous tissue disorders |
| EGFRvIII Chimeric Antigen Receptor (CAR) Gene-Modified T Cells for Patients with Newly-Diagnosed GBM During Lymphopenia [115] | NCT02664363 | Phase 1 | EGFRvIII | Terminated | This study terminated before a MTD could be determined | SAEs: Generalized muscle weakness and confusion Other AEs: Anemia, nausea, vomiting, WBC and PLT count decrease, hyperglycemia, hypocalcemia, cognitive disturbance, hypertension |
| A Phase I/II Study of the Safety and Feasibility of Administering T Cells Expressing Anti-EGFRvIII Chimeric Antigen Receptor to Patients With Malignant Gliomas Expressing EGFRvIII [122] | NCT01454596 | Phase 1 | EGFRvIII | Completed | No clinical responses were observed The protocol was closed rather than proceed into the Ph II portion | SAEs: 1 death due to multi-organ failure and G3/4 dyspnea Other TRAEs: febrile neutropenia, arrhythmia, diarrhea, WBC and platelet count decrease, transaminitis, creatinine increase, electrolyte disorders, confusion/dizziness, thrombosis |
| Phase 1 Study of EGFRvIII-Directed CAR T Cells Combined with PD-1 Inhibition in Patients With Newly Diagnosed: MGMT-Unmethylated Glioblastoma | NCT03726515 | Phase 1 | EGFRvIII | Completed | No results posted | No results posted |
| Phase I Study of Cellular ImmunoTx Using Memory Enriched T Cells Lentivirally Transduced to Express an IL13Rα2-Specific, Hinge-Optimized, 41BB-Costimulatory Chimeric Receptor and a Truncated CD19 for Pts With Rec/Ref Malignant Glioma | NCT02208362 | Phase 1 | IL13Rα2 | Active, not recruiting | -- | -- |
| A Phase I Clinical Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Antitumor Activity of SNC-109 CAR-T Cell Therapy in Subjects With Recurrent Glioblastoma | NCT05868083 | Phase 1 | HER2, IL13Rα2, EGFR, EGFRvIII | Recruiting | -- | -- |
| A Pilot Study of Chimeric Antigen Receptor (CAR) T Cells Targeting B7-H3 Antigen in Treating Patients with Recurrent and Refractory Glioblastoma | NCT04385173 | Phase 1 | B7-H3 | Recruiting | -- | -- |
| B7-H3-Targeted Chimeric Antigen Receptor (CAR) T Cells in Treating Patients With Recurrent or Refractory Glioblastoma | NCT04077866 | Phase 1 | B7-H3 | Recruiting | -- | -- |
| An Open, Single-Arm Phase 1 Study to Evaluate the Safety/Preliminary Effectiveness and Determine the Maximal Tolerated Dose of B7-H3-targeting CAR-T Cell Therapy in Treating Recurrent Glioblastomas | NCT05241392 | Phase 1 | B7-H3 | Recruiting | -- | -- |
| A Phase 1 Study to Evaluate IL13Rα2-Targeted Chimeric Antigen Receptor (CAR) T Cells Combined with Checkpoint Inhibition for Patients With Resectable Recurrent Glioblastoma | NCT04003649 | Phase 1 | IL13Rα2 | Recruiting | -- | -- |
| A Phase I Clinical Trial of NKG2D-based CAR T-cell Injection for Subjects With Relapsed/Refractory NKG2DL+ Solid Tumors | NCT05131763 | Phase 1 | NKG2D | Recruiting | -- | -- |
| A Phase 1 Study to Evaluate Chimeric Antigen Receptor (CAR) T Cells with a Chlorotoxin Tumor-Targeting Domain for Patients with MMP2+ Recurrent or Progressive Glioblastoma | NCT04214392 | Phase 1 | Chlorotoxin | Recruiting | -- | -- |
| A Phase 1 Study to Evaluate IL13Rα2-Targeted Chimeric Antigen Receptor (CAR) T Cells for Adult Patients with Leptomeningeal Glioblastoma, Ependymoma, or Medulloblastoma | NCT04661384 | Phase 1 | IL13Rα2 | Recruiting | -- | -- |
| Loc3CAR: Locoregional Delivery of B7-H3-specific Chimeric Antigen Receptor Autologous T Cells for Pediatric Patients With Primary CNS Tumors | NCT05835687 | Phase 1 | B7-H3 | Recruiting | -- | -- |
| INCIPIENT: Intraventricular CARv3-TEAM-E T Cells for Patients With GBM | NCT05660369 | Phase 1 | EGFRvIII | Recruiting | -- | -- |
| Phase I Study of Intraventricular Infusion of T Cells Expressing B7-H3 Specific Chimeric Antigen Receptors (CAR) in Subjects With Recurrent or Refractory Glioblastoma | NCT05366179 | Phase 1 | B7-H3 | Recruiting | -- | -- |
| Phase I Clinical Trial of Locoregionally (LR) Delivered Autologous B7-H3 Chimeric Antigen Receptor T-Cells (B7-H3CART) in Adults with Recurrent Glioblastoma Multiforme | NCT05474378 | Phase 1 | B7-H3 | Recruiting | -- | -- |
| Phase 1, Open-label Study Evaluating the Safety and Feasibility of CART-EGFR-IL13Ra2 Cells in Patients with EGFR-Amplified Recurrent | NCT05168423 | Phase 1 | IL13Rα2, EGFR | Recruiting | -- | -- |
| Phase I Study of Cellular Immunotherapy Using Memory-Enriched T Cells Lentivirally Transduced to Express a HER2-Specific, Hinge-Optimized, 41BB-Costimulatory Chimeric Receptor and a Truncated CD19 for Patients with Recurrent/Refractory Malignant Glioma | NCT03389230 | Phase 1 | HER2 | Active, not recruiting | -- | -- |
| Phase 1 Study of Autologous Tris-CAR-T Cell Locoregional Immunotherapy for Recurrent Glioblastoma | NCT05577091 | Phase 1 | IL7Rα | Not yet recruiting | -- | -- |
| Pilot Study of UWNKG2D CAR-T in Treating Patients with Recurrent Glioblastoma | NCT04717999 | Phase 1 | NKG2D | Not yet recruiting | -- | -- |
| An Open Clinical Study to Evaluate the Safety, Tolerance, and Initial Efficacy of Epidermal Growth Factor Receptor Variant III Chimeric Antigen Receptor T (EGFRvIII CAR-T) in the Treatment of Recurrent Glioblastoma | NCT05802693 | Phase 1 | EGFRvIII | Not yet recruiting | -- | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losurdo, A.; Di Muzio, A.; Cianciotti, B.C.; Dipasquale, A.; Persico, P.; Barigazzi, C.; Bono, B.; Feno, S.; Pessina, F.; Santoro, A.; et al. T Cell Features in Glioblastoma May Guide Therapeutic Strategies to Overcome Microenvironment Immunosuppression. Cancers 2024, 16, 603. https://doi.org/10.3390/cancers16030603
Losurdo A, Di Muzio A, Cianciotti BC, Dipasquale A, Persico P, Barigazzi C, Bono B, Feno S, Pessina F, Santoro A, et al. T Cell Features in Glioblastoma May Guide Therapeutic Strategies to Overcome Microenvironment Immunosuppression. Cancers. 2024; 16(3):603. https://doi.org/10.3390/cancers16030603
Chicago/Turabian StyleLosurdo, Agnese, Antonio Di Muzio, Beatrice Claudia Cianciotti, Angelo Dipasquale, Pasquale Persico, Chiara Barigazzi, Beatrice Bono, Simona Feno, Federico Pessina, Armando Santoro, and et al. 2024. "T Cell Features in Glioblastoma May Guide Therapeutic Strategies to Overcome Microenvironment Immunosuppression" Cancers 16, no. 3: 603. https://doi.org/10.3390/cancers16030603
APA StyleLosurdo, A., Di Muzio, A., Cianciotti, B. C., Dipasquale, A., Persico, P., Barigazzi, C., Bono, B., Feno, S., Pessina, F., Santoro, A., & Simonelli, M. (2024). T Cell Features in Glioblastoma May Guide Therapeutic Strategies to Overcome Microenvironment Immunosuppression. Cancers, 16(3), 603. https://doi.org/10.3390/cancers16030603