
Is Immunotherapy Beneficial in Patients with Oncogene-Addicted Non-Small Cell Lung Cancers? A Narrative Review


Abstract
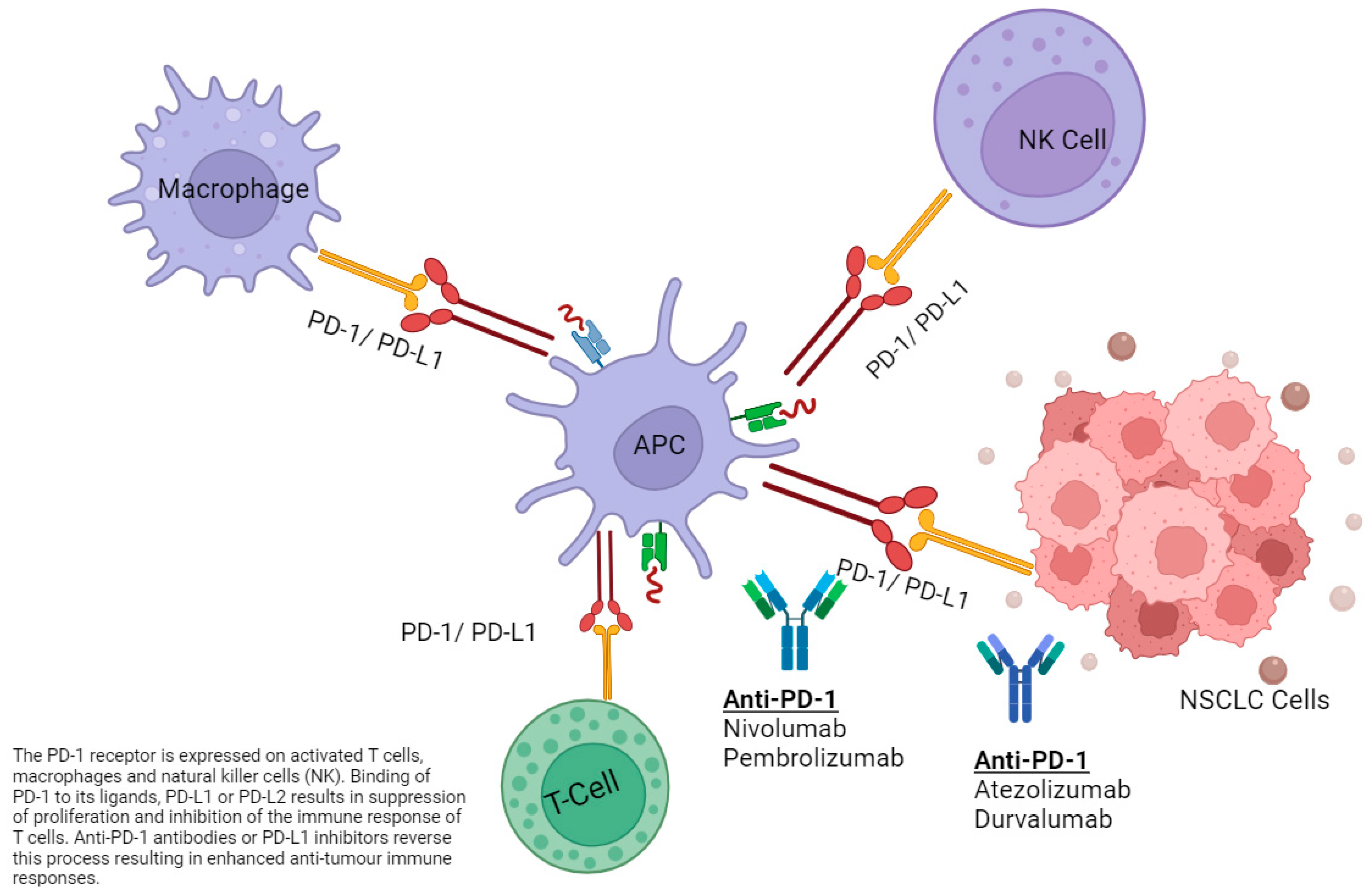
Simple Summary
Abstract
1. Introduction
2. Retrospective Data
3. KRAS
3.1. Efficacy
3.2. Safety

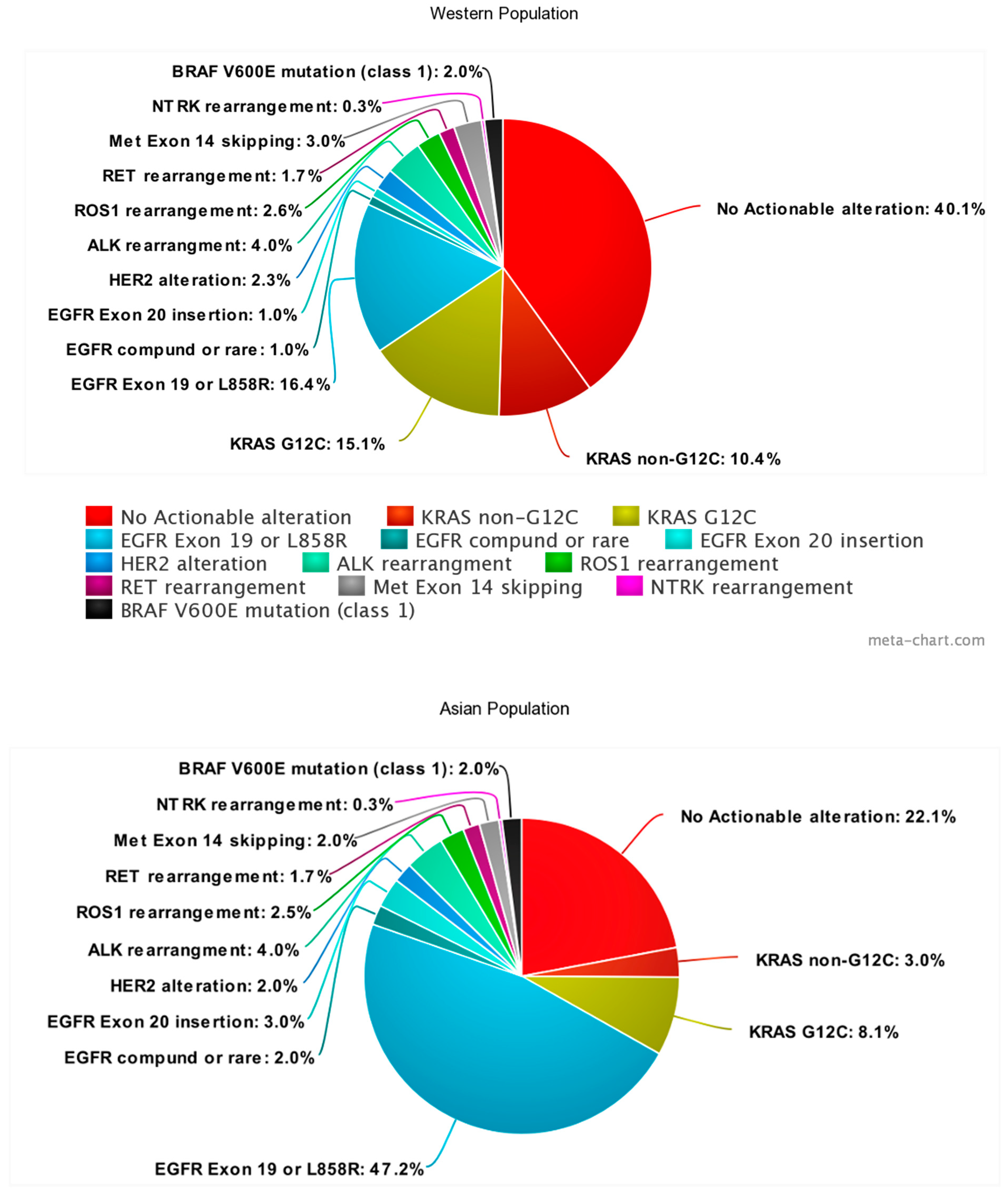
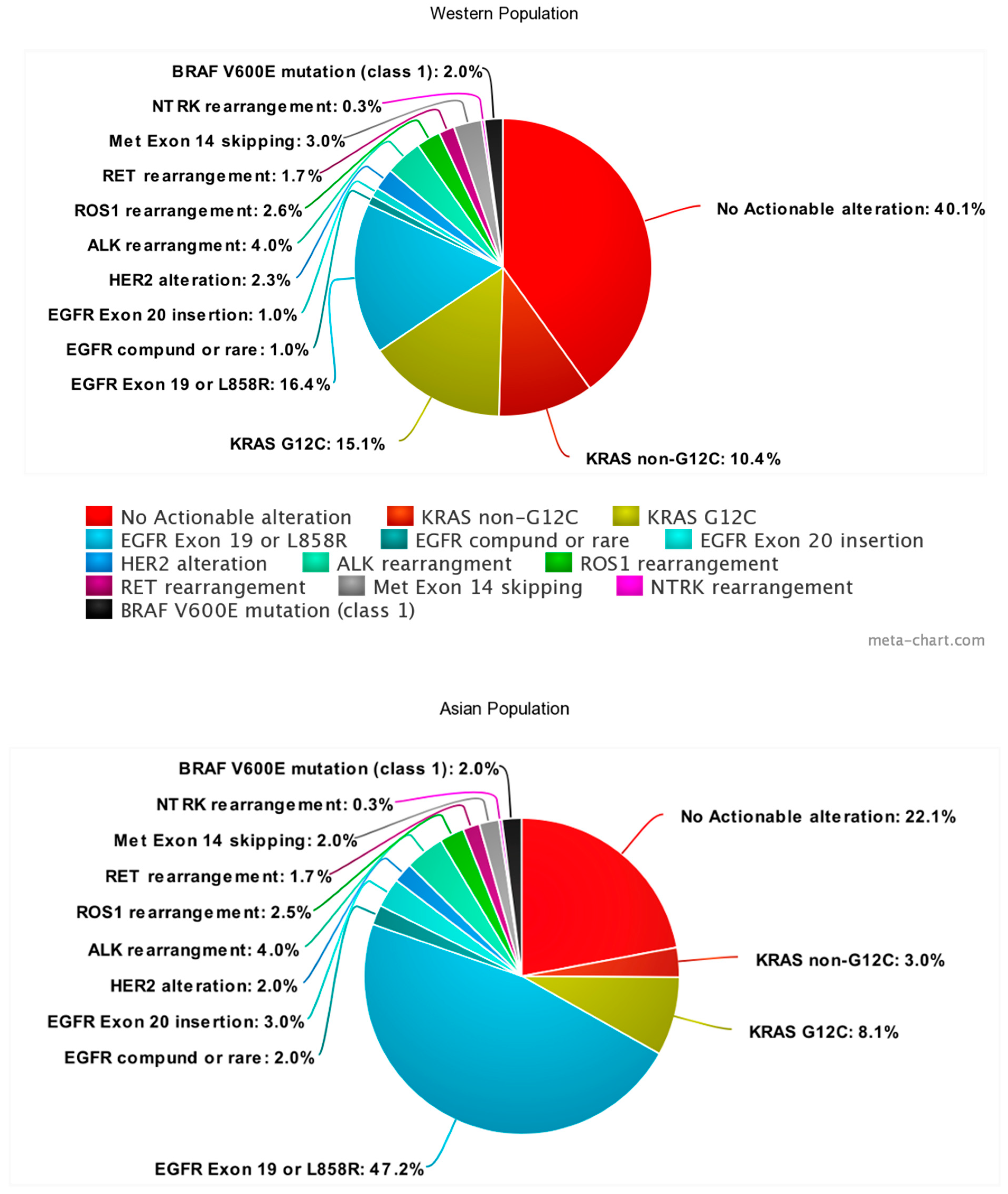{kind=link}
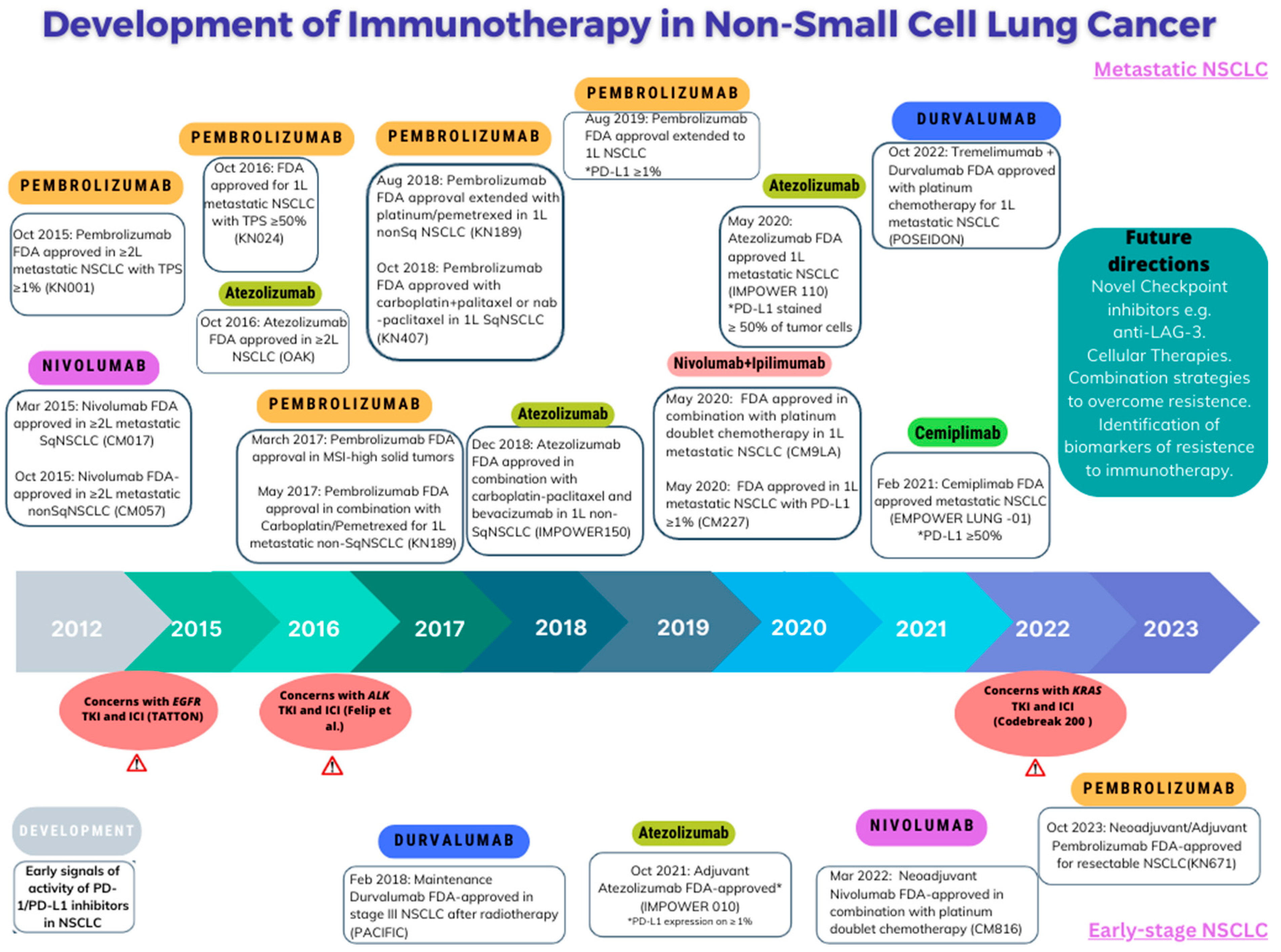{kind=link}
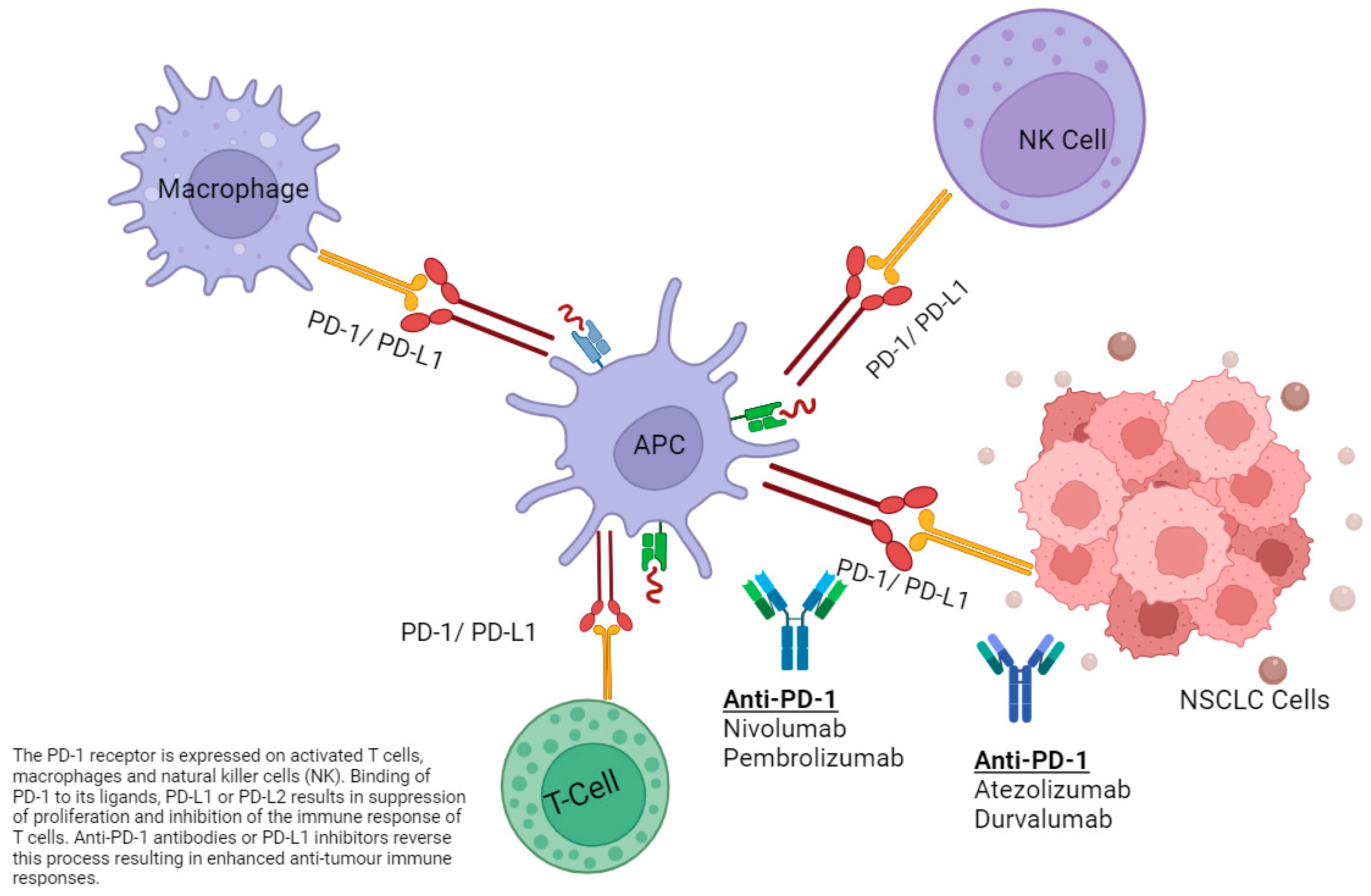{kind=link}
| Drug | Trial | Sample Size | PFS (Months) | OS (Months) | ORR% |
|---|---|---|---|---|---|
| KRAS | |||||
| Chemo ICI | Nakajima EC et al. [27] 2022 FDA pooled analysis | 1430 KRASm 39% (n = 557) KRASwt 61% (n = 873) (KRAS G12C n = 58) | Not reported | KRASm 22.4 (18.2-NR) vs. KRASwt 18.7 (16.0–25.2) KRAS G12C 20.8 (11.3-NR) [n = 58] | KRASm 46% vs. KRASwt 47% KRAS G12C 51% |
| ICI monotherapy | Nakajima EC et al. [27] 2022 FDA pooled analysis | 1430 KRASm 39% (n = 557) KRASwt 61% (n = 873) [KRAS G12C (n = 45)] | Not reported | KRASm 16.2 (11.1-NR) (n = 135) vs. KRASwt 14.9 (12.2–6.6) (n = 322) KRAS G12C 11.8 (8.2-NR) (n = 45) | 37% vs. 33% KRAS G12C 33% |
| Pembrolizumab | Herbst RS et al. [23] 2019 Keynote 042 1st line Phase 3 PDL-1 > 50% | Any KRAS mutation 30 vs. KRAS G12C 12 vs. No KRAS mutation 127 | Any KRAS mutation 12 (HR = 0.51, 0.29–0.87;95% 95% CI 8-NR) vs. KRAS G12C 15 (HR 0.27, 0.1–0.71; 95% CI 10-NR) vs. No KRAS mutation 6 (4–7) [HR 1.00 (0.75–1.34)] | Any KRAS mutation 29 m (HR = 0.42, 0.22–0.81; 95% CI 23-NR) vs. KRAS G12C NR (HR 0.28, 0.09–0.86; 95% CI 23-NR) vs. No KRAS mutation 15 (12–24) [HR 0.86 (0.63–1.18)] | Any KRAS mutation 56.7% (n = 30) (95% CI 37.4–74.5) vs. KRAS G12C 66.7% (n = 12) (95% CI 34.9–90.1) vs. No KRAS mutation 29.1 (n = 127) (21.4–37.9) |
| Pembrolizumab + Sotorasib Or Atezolizumab + sotorasib | Li BT et al. [37] 2022 Codebreak 100 Phase 2 1st line | 58 Atezolizumab + Sotorasib lead in N = 10 Atezolizumab + Sotorasib concurrent N = 10 Pembrolizumab + Sotorasib lead in N = 19 Pembrolizumab + Sotorasib concurrent N = 19 | Not reported | All patients 15.7 m (95% CI: 9.8, 17.8) Atezolizumab + Sotorasib lead in 8.1 (95% CI 2.5-NR) Atezolizumab + Sotorasib concurrent 11.5 (95% CI 5.0-NR) Pembrolizumab + Sotorasib lead in NR (95% CI 10.1-NR) Pembrolizumab + Sotorasib concurrent 14.1 (95% CI 6.2–17.8) | 29% (n = 17/58) |
| Pembrolizumab + Adagrasib | Jänne PA et al. [38] 2022 KRYSTAL-1 (phase 1b) + KRYSTAL-7 (phase 2) 1st line | 75 | Not reported | Not reported | 49% (n = 26/53) |
| EGFR | |||||
| Pembrolizumab (PDL1 > 50%) | Lisberg et al. [39] 2018 Phase 2 1st line | 10 | 119 days | NR | 0% |
| ABCP vs. ACP vs. BCP (A Atezolizumab B bevacizumab C Carboplatin P Paclitaxel) | Reck et al. [40] 2019 Impower150 1st line Phase 3 | 122 ABCP (34) vs. ACP (45) vs. BCP (43) | ABCP 10.2 vs. BCP 6.9 (HR 0.61) Sensitizing mutations ABCP vs. BCP (HR 0.41) | ABCP not estimable (NE) vs. 18.7 BCP Sensitizing mutations ABCP NE vs. 17.5 BCP (HR 0.31) | ABCP (70.6%) Vs. ACP (35.6%) Vs. BCP (41.9%) |
| Durvalumab + Gefitinib | Gibbons D et al. [41] 2016 Phase 1 1st line | 19 9 ARM A (concurrent) 10 ARM B (sequential) | Not reported | Not reported | ARM A 77.8% (n = 7) Arm B 80.0% (n = 8) |
| Nivolumab +Erlotinib | Gettinger S et al. [42] 2018 Phase 1 ≥1st line (prior chemotherapy excluded) | 21 | 5.1 (95% CI: 2.3–12.1) | 18.7 (95% CI: 7.3–NR) | 15% (n = 3 of 20) |
| Sintilimab + IBI305 + chemotherapy or Sintilimab + chemotherapy vs. chemotherapy alone. | Lu S et al. et al. [43] 2023 Orient-31 trial Phase 3 2nd line | 476 | Sintilimab, (anti-PD1) + chemotherapy 5.5 vs. Chemotherapy alone 4.3 Sintilimab + IBI305 + chemotherapy 7.2 vs. 4.3 Sintilimab + chemotherapy versus 19.2 Chemotherapy alone | Sintilimab + IBI305 + chemotherapy 21.1 vs. Sintilimab plus chemotherapy 20.5 vs. Chemotherapy alone 19.2 | 35% (n = 55/158) sintilimab + chemotherapy vs. 29% (n = 47/160) chemotherapy alone |
| Durvalumab + Osimertinib | Oxnard g et al. [44] 2022 TATTON trial Phase 1b 2nd line | 23 | Not reported | Not reported | 43% (approximate) |
| Nivolumab | Rizvi et al. [45] 2014 Checkmate 063 Phase 2 ≥2nd line | 117 | 1.9 (95% CI 1.8–3.2) | 8.2 (95% CI 6.1–10.9) | 14.5% (95% CI 8.7–22.2) |
| Durvalumab + Osimertinib vs. Osimertinib alone | Yang J et al. [46] 2019 CAURAL trial Phase 3 ≥2nd line * Terminated early due to the TATTON trial safety concerns | 14 | Combination NR vs. 19.3 Osimertinib | NR vs. NR * early termination | Combination 80% (n = 12 of 15) ([95% CI: 52–96]) vs. Osimertinib 64% (n = 9 of 14) ([95% CI: 35–87]). |
| Nivolumab | Mok T et al. [47] 2022 Checkmate 722 ≥2nd line Phase 3 | 294 | Nivolumab + chemotherapy 5.6 vs. chemotherapy 5.4 (HR0.75, p = 0.0528) | 19.4 vs. 15.9 (HR 0.82, p ≥ 0.05) | 31% vs. 27% * Exact number at risk unknown |
| Pembrolizumab | Chih-Hsin Yang J et al. [48] 2023 KEYNOTE 789 ≥2nd line Phase 3 | 492 | Pembrolizumab + chemotherapy 5.6 vs. chemotherapy 5.5 (HR 0.80; p = 0.0122) | 15.9 vs. 14.7 m (HR 0.84; p = 0.0362) | 29.0% vs. 27.1% |
| EGFR + ALK + combined | |||||
| Durvalumab * EGFR and ALK groups combined for analysis | Naidoo et al. [35] 2022 PACIFIC subgroup analysis Phase 3 -Stage III | 35 24 (durvalumab) vs. 11 (placebo) (Median follow up 42 months) | 11.2 vs. 10.9 (95% CI 7.3–20.7 vs. 1.9-NR, [HR 0.91] | 46.8 (95% CI 29.9-NR) vs. 43.0 (95% CI 14.9, NE) | 26% (95% CI, 10.2, 48.4) vs. 18.2% (95% CI 2.3, 51.8) |
| Durvalumab * (EGFR +/ALK+ combined) | Garassino MC et al. [49] 2018 ATLANTIC Phase 2 ≥3rd line | 444 111 in cohort 1 (EGFR+/ALK+) | PDL-1 ≤ 25% 1.9 (1.8–1.9) PDL-1 ≥ 25% 1.9 (1.8–3.6) | PDL-1 ≤ 25% 9.9 (4.2–13.0) PDL-1 ≥ 25% 13.3 (8.1-NR) | 14% [12.2% (n = 9 of 74) of PDL1 + ≥25% group 95% CI 5.7–21.8)] |
| ALK | |||||
| Nivolumab + Crizotinib | Spigel D et al. [50] 2018 CheckMate 370 Phase 1/2 1st line | 13 | Not reported | Not reported | 38% (n = 5 of 13) |
| Pembrolizumab + Crizotinib | Patel SP et al. [51] 2020 Phase 1b 1st line | Not reported | Not reported | Not reported | Not reported |
| Atezolizumab + Alectinib | Kim DW et al. [52] 2022 Phase 1b 1st line | 21 | NR (95% CI: 13–NR) | NR (95% CI: 33 –NR) | 86% (n = 18) |
| Avelumab + Lorlatinib | Shaw AT et al. [53] Javelin Lung 01 2018 Phase 1b ≥2nd line | 28 | Not reported | Not reported | 46.4% |
| Nivolumab + Ceritinib | Felip E et al. [2] 2017 Phase 1 1st or ≥2nd line | 36 | Not reported | Not reported |
63% (pretreated) 83% (TKI Naïve) |
| Durvalumab - | Naidoo et al. [35] 2022 PACIFIC subgroup analysis Phase 3 Stage III | 4 | 7.8 [95% CI, 3.9-NR] | Not reported | Not reported |
4. EGFR
4.1. Efficacy
| Drug | Trial | Sample Size | TRAE | IRAE |
|---|---|---|---|---|
| KRAS | ||||
| Pembrolizumab + Sotorasib or Atezolizumab + Sotorasib | Li BT et al. [37] 2022 Codebreak 100 Phase 2 1st line | 58 Atezolizumab + Sotorasib lead In N = 10 Atezolizumab + Sotorasib concurrent N = 10 Pembrolizumab + Sotorasib lead in N = 19 Pembrolizumab + Sotorasib concurrent N = 19 | All grade 88% (n = 51) G3/4 59% (n = 34) | AE of special interest Hepatotoxicity G3/4 43% (n = 25) |
| Pembrolizumab + Adagrasib | Jänne PA et al. [38] 2022 KRYSTAL-1 (phase 1b) + KRYSTAL-7 (phase 2) 1st line | 75 | All grade 83% G3/4.44% G3 Elevated lipase 11% G3 increased ALT/AST 8%/9% | Not reported |
| EGFR | ||||
| Pembrolizumab | Lisberg et al. [39] 2018 Phase 2 1st line PDL1 > 50% | 10 | 46% | 46% |
| ABCP vs. ACP vs. BCP (A, Atezolizumab B, bevacizumab C, Carboplatin P, Paclitaxel) | Reck et al. [40] 2019 Impower150 Phase 3 1st line | 122 ABCP (34) vs. ACP (45) vs. BCP (43) | G3/4 ABCP 64% (n = 21 of 33) vs. ACP 68% (n = 30 of 44) vs. BCP 64% (n = 28 of 44) + 1 G5 toxicity | ABCP 55% (n = 18) G3/4 9% (n = 3) vs. ACP 52% (n = 23), G3/4 9% (n = 4) vs. BCP 23% (n = 10), G3/4 2% (n = 1) *AE of special interest including irAE |
| Durvalumab + Gefitinib | Gibbons D et al. [41] 2016 Phase 1 1st line | 19 9 ARM A (Concurrent) 10 ARM B (Sequential) | All grade AE 100% | Not reported |
| Nivolumab + Erlotinib | Gettinger S et al. [42] 2018 Phase 1 ≥1st line (prior chemotherapy excluded) | 21 | All grade 100% (n = 21) rash (n = 10, 48%) fatigue (n = 6, 29%) paronychia (n = 6, 29%), skin fissures (n = 5, 24%) No G4 or 5 toxicities | All grade N = 18 (86%) 24% (n = 5) ≥G3 toxicities Diarrhea (n = 2), ALT+/− AST increase (n = 2) Weight loss (n = 1) |
| Sintilimab + IBI305 + chemotherapy or Sintilimab + chemotherapy vs. chemotherapy alone | Lu S et al. [43] 2023 Orient-31 trial Phase 3 2nd line | 476 | ≥3 56% (n = 88/158) sintilimab + IBI305 + chemotherapy group vs. 41% (n = 64/156) in the sintilimab + chemotherapy group vs. 49% (n = 79/160) in the chemotherapy alone group | (Investigator assessed, all grade) 26% (n = 41/156) sintilimab + chemotherapy vs. 16% ( n = 25/160) chemotherapy alone G5 1% (pneumonitis) vs. 1% (unknown) |
| Durvalumab + Osimertinib | Oxnard G et al. [44] 2022 TATTON trial Phase 1b 2nd line | 23 | All grade 100%. 39% discontinued due to TRAEs | Pneumonitis 22% [n = 2 at 3 mg/kg [n = 1 grade 2 n = 1 grade 3] n = 3 at 10 mg/kg [n = 1 grade 1, n = 1 grade 2 n = 1 grade 4] * Overall figure Not available |
| Nivolumab | Rizvi et al. [45] 2014 Checkmate 063 Phase 2 ≥2nd line | 117 | 74% (n = 87) G3/4 17% (n = 20) Fatigue 33% (n = 38) Asthenia 12% (n = 14) | G5 3% Rash 11%(n = 13) Pneumonitis 3% (n = 4) Diarrhea 3% (n = 3) |
| Durvalumab + Osimertinib vs. Osimertinib * Terminated early due to the TATTON trial safety concern | Yang J et al. [46] 2019 CAURAL trial Phase 3 ≥2nd line | 12 | 8% (n = 1) G3 rash (n = 8 [67%]) diarrhea (n = 6 [50%]) decreased appetite (n = 6 [50%]) | Possible irAE 58% (n = 7) -all G1/2 |
| Nivolumab | Mok T et al. [47] 2022 Checkmate 722 Phase 3 ≥2nd line | 294 | No new safety signals identified * Specific results awaited | * Specific results awaited |
| Pembrolizumab | Chih-Hsin Yang J et al. [48] 2023 KEYNOTE 789 Phase 3 ≥2nd line | 492 | ≥3 G3 43.7% Pembrolizumab + chemotherapy vs. 38.6% chemotherapy G5 AEs 0.4% vs. 0.8%. | ≥3 G3 IRAEs and infusion reactions occurred. 4.5% vs. 2.0% G5 0.4% vs. 0% |
| EGFR + ALK combined | ||||
| Durvalumab 3rd line or greater (EGFR+/ALK+) | Garassino MC et al. [49] 2018 ATLANTIC Phase 2 ≥3rd line | 444 111 in cohort 1 (EGFR+/ALK+) | All grade 48% G3 4% (n = 4) G4 2% (n = 2) | AE of special interest 25% (n = 28) irAE 12% (n = 13) G3/4 2% (n = 2) Pneumonitis 2% (n = 2) * 1 G5 pneumonitis * 2 days after starting erlotinib 65days post durvalumab |
| Durvalumab | Naidoo et al. [35] 2022 PACIFIC subgroup analysis Phase 3 1st line -Stage III | 35 (EGFR+ ALK combined) 24 (durvalumab) vs. 11 (placebo) | AEs leading to dose delays 71% vs. 18% | Radiation pneumonitis 10% vs. 4% All low grade. |
| ALK | ||||
| Nivolumab + Crizotinib | Spigel D et al. [50] 2018 CheckMate 370 Phase 1/2 1st line | 13 | 62% (n = 8/13) - at least 1 ≥ G3 toxicity, 8% (n = 1) G4 pneumonitis. * Trial was halted for safety | ≥G3 hepatotoxicity 38% (n = 5) 2 deaths considered -potential G5 toxicities in these patients. |
| Nivolumab + Ceritinib | Felip E et al. [2] 2017 Phase 1 1st or ≥2nd line | 36 | AEs occurring in ≥ 40% of patients: diarrhea (64%) rash (61%) ALT increase (56%) AST increase (44%) vomiting (42%) ≥G3 with ≥10% frequency ALT increase (22%) GGT increase (17%) amylase increase (11%) lipase increase (11%) | Not reported |
| Pembrolizumab + Crizotinib | Patel SP et al. [51] 2020 Phase 1b 1st line | 9 | 1 G5 pneumonia (determined to be due to disease progression) | dose limiting toxicities of ≥G3 44% (n = 4/9) * prior to maximum tolerated dose being identified
|
| Atezolizumab + Alectinib | Kim DW et al. [52] 2022 Phase 1b 1st line | 21 | All grade 95% G3 57% Most common G3 -rash (19%) 0 G4/5 events | All grade 86% G3 43% (n = 9) G3 Rash 19% (n = 4) G3 Dyspnoea 10% (n = 2) G3 ALT increase 10% (n = 2) |
| Avelumab + Lorlatinib | Javelin Lung 01 [53] 2018 Phase 1b ≥2nd line | 28 | 96.4% (n = 27) ≥G3 53.6% (n = 15) | Serious AE 39.3% (n = 11) Pneumonitis 7.1% (n = 2) AST increase, cerebral haemorrhage, confusional state, delirium and others - all 3.6% (n = 1) |
4.2. Safety
5. ALK
Safety
6. BRAF
6.1. Efficacy
6.2. Uncommon Oncogenic Alterations in NSCLCs—ROS1, RET, NTRK 1/2/3, HER-2, and MET
6.3. Effect of the Immune Microenvironment and Smoking Status on ICI Response
6.4. Future Directions
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hendriks, L.E.; Kerr, K.M.; Menis, J.; Mok, T.S.; Nestle, U.; Passaro, A.; Peters, S.; Planchard, D.; Smit, E.F.; Solomon, B.J.; et al. Oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Felip, E.; De Braud, F.G.; Maur, M.; Loong, H.H.F.; Shaw, A.T.; Vansteenkiste, J.F.; John, T.; Liu, G.; Lolkema, M.P.; Scott, J.W.; et al. Ceritinib plus nivolumab (NIVO) in patients (pts) with anaplastic lymphoma kinase positive (ALK+) advanced non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2017, 35 (Suppl. 15), 2502. [Google Scholar] [CrossRef]
- Mok, T.; Camidge, D.R.; Gadgeel, S.M.; Rosell, R.; Dziadziuszko, R.; Kim, D.W.; Perol, M.; Ou, S.-H.I.; Ahn, J.S.; Shat, A.T.; et al. Updated overall survival and final progression-free survival data for patients with treatment-naive advanced ALK-positive non-small-cell lung cancer in the ALEX study. Ann. Oncol. 2020, 31, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR -Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, M.; Herbst, R.S.; John, T.; Kato, T.; Majem, M.; Grohé, C.; Wang, J.; Goldman, J.W.; Lu, S.; Su, W.C.; et al. Overall Survival with Osimertinib in Resected EGFR -Mutated NSCLC. N. Engl. J. Med. 2023, 389, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Ahn, J.S.; Barlesi, F.; Dziadziuszko, R.; Nishio, M.; Shaw, A.T.; Bordogna, W.; Meyenberg, C.; Wu, Y.L. ALINA: A phase III study of alectinib versus chemotherapy as adjuvant therapy in patients with stage IB–IIIA anaplastic lymphoma kinase-positive (ALK+) non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2019, 37 (Suppl. 15), TPS8569. [Google Scholar] [CrossRef]
- Novello, S.; Kowalski, D.M.; Luft, A.; Gümüş, M.; Vicente, D.; Mazières, J.; Rodriguez-Cid, J.; Tafreshi, A.; Cheng, Y.; Lee, K.H.; et al. Pembrolizumab Plus Chemotherapy in Squamous Non-Small-Cell Lung Cancer: 5-Year Update of the Phase III KEYNOTE-407 Study. J. Clin. Oncol. 2023, 41, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Garassino, M.C.; Gadgeel, S.; Speranza, G.; Felip, E.; Esteban, E.; Dómine, M.; Hochmair, M.J.; Powell, S.F.; Bischoff, H.G.; Peled, N.; et al. Pembrolizumab Plus Pemetrexed and Platinum in Nonsquamous Non-Small-Cell Lung Cancer: 5-Year Outcomes from the Phase 3 KEYNOTE-189 Study. J. Clin. Oncol. 2023, 41, 1992–1998. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Lee, J.S.; Ciuleanu, T.E.; Bernabe Caro, R.; Nishio, M.; Urban, L.; Audigier-Valette, C.; Lupinacci, L.; Sangha, R.; Pluzanski, A.; et al. Five-Year Survival Outcomes With Nivolumab Plus Ipilimumab Versus Chemotherapy as First-Line Treatment for Metastatic Non-Small-Cell Lung Cancer in CheckMate 227. J. Clin. Oncol. 2023, 41, 1200–1212. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csöszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Five-Year Outcomes with Pembrolizumab Versus Chemotherapy for Metastatic Non–Small-Cell Lung Cancer with PD-L1 Tumor Proportion Score ‡ 50%. J. Clin. Oncol. 2021, 39, 2339–2349. [Google Scholar] [CrossRef]
- Hendriks, L.E.; Kerr, K.M.; Menis, J.; Mok, T.S.; Nestle, U.; Passaro, A.; Peters, S.; Planchard, D.; Smit, E.F.; Solomon, B.J.; et al. Non-oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 358–376. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Riudavets, M.; Auclin, E.; Mosteiro, M.; Dempsey, N.; Majem, M.; Lobefaro, R.; Lopez-Castro, R.; Bosch-Barrera, J.; Pilotto, S.; Escalera, E.; et al. Durvalumab consolidation in patients with unresectable stage III non-small cell lung cancer with driver genomic alterations. Eur. J. Cancer 2022, 167, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Cortiula, F.; De Ruysscher, D.; Steens, M.; Wijsman, R.; van der Wekken, A.; Alberti, M.; Hendricks, L.E.L. Adjuvant durvalumab after concurrent chemoradiotherapy for patients with unresectable stage III NSCLC harbouring uncommon genomic alterations. Eur. J. Cancer 2023, 184, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-year overall survival for patients with advanced non-small-cell lung cancer treated with pembrolizumab: Results from the phase i KEYNOTE-001 study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; von Pawel, J.; Park, K.; Rittmeyer, A.; Gandara, D.R.; Ponce Aix, S.; Han, J.Y.; Gadgeel, S.M.; Hida, T.; Cortinovis, D.L.; et al. Updated efficacy analysis including secondary population results for OAK: A randomized phase III study of atezolizumab versus docetaxel in patients with previously treated advanced non-small cell lung cancer. J. Thorac. Oncol. 2018, 13, 1156–1170. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vdal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef]
- Passiglia, F.; Cappuzzo, F.; Alabiso, O.; Bettini, A.C.; Bidoli, P.; Chiari, R.; Defferrari, C.; Delmonte, A.; Finocchiaro, G.; Francini, G.; et al. Efficacy of nivolumab in pre-treated non-small-cell lung cancer patients harbouring KRAS mutations. Br. J. Cancer 2019, 120, 57. [Google Scholar] [CrossRef]
- Sun, L.; Hsu, M.; Cohen, R.B.; Langer, C.J.; Mamtani, R.; Aggarwal, C. Association Between KRAS Variant Status and Outcomes With First-line Immune Checkpoint Inhibitor–Based Therapy in Patients With Advanced Non–Small-Cell Lung Cancer. JAMA Oncol. 2021, 7, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.; Lopes, G.; Kowalski, D.; Kasahara, K.; Wu, Y.-L.; De Castro, G.; Cho, B.; Turna, H.; Cristescu, R.; Aurora-Garg, D.; et al. LBA4 Association of KRAS mutational status with response to pembrolizumab monotherapy given as first-line therapy for PD-L1-positive advanced non-squamous NSCLC in Keynote-042. Ann. Oncol. 2019, 30, xi63–xi64. [Google Scholar] [CrossRef]
- Amanam, I.; Mambetsariev, I.; Gupta, R.; Achuthan, S.; Wang, Y.; Pharaon, R.; Massarelli, E.; Koczywas, M.; Reckamp, K.; Salgia, R. Role of immunotherapy and co-mutations on KRAS-mutant non-small cell lung cancer survival. J. Thorac. Dis. 2020, 12, 5086. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Ricciuti, B.; Rizvi, H.; Hellmann, M.D.; Yu, H.A.; Ladanyi, M.; Kris, M.G.; Arcila, M.E.; Rudin, C.M.; Lito, P.; et al. Chemo-immunotherapy outcomes of KRAS-G12C mutant lung cancer compared to other molecular subtypes of KRAS-mutant lung cancer. J. Clin. Oncol. 2021, 39 (Suppl. 15), 9088. [Google Scholar] [CrossRef]
- Gadgeel, S.; Rodriguez-Abreu, D.; Felip, E.; Esteban, E.; Speranza, G.; Reck, M.; Hui, R.; Boyer, M.; Garon, E.B.; Horinouchi, H.; et al. KRAS mutational status efficacy in KEYNOTE-189: Pembrolizumab (pembro) plus chemotherapy (chemo) vs placebo plus chemo as first-line therapy for metastatic non-squamous, NSCLC. Ann. Oncol. 2019, 30, xi64–xi65. [Google Scholar] [CrossRef]
- Nakajima, E.C.; Ren, Y.; Vallejo, J.J.; Akinboro, O.; Mishra-Kalyani, P.S.; Larkins, E.A.; Dresner, N.L.; Tang, S.; Pazdur, R.; Beaver, J.A.; et al. Outcomes of first-line immune checkpoint inhibitors with or without chemotherapy according to KRAS mutational status and PD-L1 expression in patients with advanced NSCLC: FDA pooled analysis. J. Clin. Oncol. 2022, 40 (Suppl. 16), 9001. [Google Scholar] [CrossRef]
- Lauko, A.; Kotecha, R.; Barnett, A.; Li, H.; Tatineni, V.; Ali, A.; Patil, P.; Mohammadi, A.M.; Chao, S.T.; Murphy, E.S.; et al. Impact of KRAS mutation status on the efficacy of immunotherapy in lung cancer brain metastases. Sci. Rep. 2021, 11, 18174. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Cancales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 861–878. [Google Scholar] [CrossRef]
- Negrao, M.V.; Araujo, H.A.; Lamberti, G.; Cooper, A.J.; Akhave, N.S.; Zhou, T.; Delasos, L.; Hicks, J.K.; Aldea, M.; Minuti, G.; et al. Co-mutations KRASG12C inhibitor efficacy in advanced, NSCLC. Cancer Discov. 2023, 13, 1556–1571. [Google Scholar] [CrossRef]
- Davis, A.P.; Cooper, W.A.; Boyer, M.; Lee, J.H.; Pavlakis, N.; Kao, S.C. Efficacy of immunotherapy in KRAS-mutant non-small-cell lung cancer with comutations. Immunotherapy 2021, 13, 941–952. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Arbour, K.; Lin, J.; Vajdi, A.; Tolstorukov, M.; Hong, L.; Zhange, J.; Tolstorukov, M.Y.; Li, Y.Y.; Spurr, L.F.; et al. P14.26 Diminished Efficacy of PD-(L)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma is Impacted by KRAS Mutation Status. J. Thorac. Oncol. 2021, 16, S341. [Google Scholar] [CrossRef]
- Dudnik, E.; Bshara, E.; Grubstein, A.; Fridel, L.; Shochat, T.; Roisman, L.C.; Ilouze, M.; Rozenblum, A.B.; Geva, S.; Zer, A.; et al. Rare targetable drivers (RTDs) in non-small cell lung cancer (NSCLC): Outcomes with immune check-point inhibitors (ICPi). Lung Cancer 2018, 124, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Antonia, S.J.; Wu, Y.L.; Cho, B.C.; Thiyagarajah, P.; Mann, H.; Newton, M.; Faivre-Finn, C. Durvalumab (durva) after chemoradiotherapy (CRT) in unresectable, stage III, EGFR mutation-positive (EGFRm) NSCLC: A post hoc subgroup analysis from PACIFIC. J. Clin. Oncol. 2022, 40 (Suppl. 16), 8541. [Google Scholar] [CrossRef]
- Chour, A.; Denis, J.; Lafitte, C.; Mascaux, C.; Zysman, M.; Lemaitre, A.; Swalduz, A.; Gounant, V.; Cortot, A.; Darrason, M.; et al. Sotorasib-induced liver and non-liver toxicity associated with sequential sotorasib following anti-PD(L)1 in KRASG12C mutant. Lung Cancer 2022, 33, S49–S50. [Google Scholar] [CrossRef]
- Li, B.T.; Falchook, G.S.; Durm, G.A.; Burns, T.; Skoulidis, F.; Ramalingam, S.S.; Spira, A.; Bestvina, C.M.; Foldberg, S.B.; Veluswamy, R.; et al. CodeBreaK 100/101: First Report of Safety/Efficacy of Sotorasib in Combination with Pembrolizumab or Atezolizumab in Advanced KRAS p.G12C NSCLC. In Proceedings of the 2022 World Conference on Lung Cancer, Vienna, Austria, 6–9 August 2022. [Google Scholar]
- Janne, P.A.; Smit, E.F.; De Marinis, F.; laskin, J.; Domine, M.; Gadgeel, S.; Garassino, M.C.; Lu, S.; Spira, A.S.; Kang, V.; et al. Preliminary Safety and Efficacy of Adagrasib with Pembrolizumab in Treatment-Naïve Patients with Advanced Non-Small Cell Lung Cancer (NSCLC) harboring a KRASG12C mutation | OncologyPRO. Available online: https://oncologypro.esmo.org/meeting-resources/esmo-immuno-oncology-congress/preliminary-safety-and-efficacy-of-adagrasib-with-pembrolizumab-in-treatment-naive-patients-with-advanced-non-small-cell-lung-cancer-nsclc-harbor# (accessed on 6 February 2023).
- Lisberg, A.; Cummings, A.; Goldman, J.W.; Bornazyan, K.; Reese, N.; Wang, T.; Coluzzi, P.; Ledezma, B.; Mendenhall, M.; Hunt, J.; et al. A Phase II Study of Pembrolizumab in EGFR-Mutant, PD-L1+, Tyrosine Kinase Inhibitor Naïve Patients With Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1138–1145. [Google Scholar] [CrossRef]
- Reck, M.; Mok, T.S.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sinilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Chow, L.Q.; Kim, D.W.; Kim, S.W.; Yeh, T.; Song, X.; Jiang, H.; Taylor, R.; Karakunnel, J.; Creelan, B. 57OEfficacy safety tolerability of MEDI4736 (durvalumab [D]); a human IgG1 anti-programmed cell death-ligand-1 (PD-L1) antibody combined with gefitinib (G): A phase I expansion in TKI-naïve patients (pts) with EGFR mutant, NSCLC. J. Thorac. Oncol. 2016, 11, S79. [Google Scholar] [CrossRef]
- Gettinger, S.; Hellmann, M.D.; Chow, L.Q.M.; Borghaei, H.; Antonia, S.; Brahmer, J.R.; Goldman, J.W.; Gerber, D.E.; Juergens, R.A.; Shepherd, F.A.; et al. Nivolumab Plus Erlotinib in Patients With EGFR-Mutant Advanced, NSCLC. J. Thorac. Oncol. 2018, 13, 1363–1372. [Google Scholar] [CrossRef]
- Lu, S.; Wu, L.; Jian, H.; Cheng, Y.; Wang, Q.; Fang, J.; Wang, Z.; Hu, Y.; Han, L.; Sun, M.; et al. Sintilimab plus chemotherapy for patients with EGFR-mutated non-squamous non-small-cell lung cancer with disease progression after EGFR tyrosine-kinase inhibitor therapy (ORIENT-31): Second interim analysis from a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2023, 11, 624–636. [Google Scholar] [PubMed]
- Oxnard, G.R.; Yang, J.C.H.; Yu, H.; Kim, S.W.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Chow, L.Q.M.; Borghaei, H.; Shen, Y.; Harbison, C.; Alaparthy, S.; Chen, A.; Gettinger, S. Safety and response with nivolumab (anti-PD-1; BMS-936558, ONO-4538) plus erlotinib in patients (pts) with epidermal growth factor receptor mutant (EGFR MT) advanced NSCLC. J. Clin. Oncol. 2014, 32 (Suppl. 15), 8022. [Google Scholar] [CrossRef]
- Chih-Hsin Yang, J.; Shepherd, F.A.; Kim, D.W.; Lee, G.W.; Lee, J.S.; Chang, G.C.; Leek, S.S.; Wei, Y.-F.; Lee, Y.G.; Laus, G.; et al. Osimertinib Plus Durvalumab versus Osimertinib Monotherapy in EGFR T790M-Positive NSCLC following Previous EGFR TKI Therapy: CAURAL Brief Report. J. Thorac. Oncol. 2019, 14, 933–939. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Nakagawa, K.; Park, K.; Ohe, Y.; Girard, N.; Kim, H.R.; Wu, Y.-L.; Gainor, J.; Lee, S.-H.; Chiu, C.-H.; et al. LBA8 Nivolumab (NIVO) + chemotherapy (chemo) vs chemo in patients (pts) with EGFR-mutated metastatic non-small cell lung cancer (mNSCLC) with disease progression after EGFR tyrosine kinase inhibitors (TKIs) in CheckMate 722. Ann. Oncol. 2022, 33, S1561–S1562. [Google Scholar] [CrossRef]
- Yang, J.C.H.; Lee, D.H.; Lee, J.S.; Fan, Y.; de Marinis, F.; Okamoto, I.; Inoue, T.; Rodriguez, J.C.; Zhang, L.; Yang, C.; et al. Pemetrexed and platinum with or without pembrolizumab for tyrosine kinase inhibitor (TKI)-resistant, EGFR-mutant, metastatic nonsquamous NSCLC: Phase 3 KEYNOTE-789 study. Am. Soc. Clin. Oncol. 2023, 41 (Suppl. 17), LBA9000. [Google Scholar] [CrossRef]
- Garassino, M.C.; Cho, B.C.; Kim, J.H.; Mazières, J.; Vansteenkiste, J.; Lena, H.; Corral, J.J.; Gray, J.; Powderly, J.; Chouaid, C.; et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): An open-label, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 521. [Google Scholar] [CrossRef]
- Spigel, D.R.; Reynolds, C.; Waterhouse, D.; Garon, E.B.; Chandler, J.; Babu, S.; Thurmes, P.; Spira, A.; Jotte, R.; Zhu, J.; et al. Phase 1/2 Study of the Safety and Tolerability of Nivolumab Plus Crizotinib for the First-Line Treatment of Anaplastic Lymphoma Kinase Translocation—Positive Advanced Non-Small Cell Lung Cancer (CheckMate 370). J. Thorac. Oncol. 2018, 13, 682–688. [Google Scholar] [CrossRef]
- Patel, S.P.; Pakkala, S.; Pennell, N.A.; Reckamp, K.L.; Lanzalone, S.; Polli, A.; Tarazi, J.; Robert-Vizcarrondo, F. Phase Ib Study of Crizotinib plus Pembrolizumab in Patients with Previously Untreated Advanced Non-Small Cell Lung Cancer with ALK Translocation. Oncologist 2020, 25, 562. [Google Scholar] [CrossRef]
- Kim, D.W.; Gadgeel, S.; Gettinger, S.N.; Riely, G.J.; Oxnard, G.R.; Mekhail, T.; Schmid, P.; Dowlati, A.; Heist, R.S.; Wozniak, A.J.; et al. Brief Report: Safety and Antitumor Activity of Alectinib Plus Atezolizumab From a Phase 1b Study in Advanced ALK-Positive NSCLC. JTO Clin. Res. Rep. 2022, 3, 100367. [Google Scholar] [CrossRef]
- Shaw, A.T.; Lee, S.H.; Ramalingham, S.S.; Bauer, T.M.; Boyer, M.J.; Carcereny Costa, E.; Felip, E.; Han, J.-Y.; Hida, T.; Hughes, B.; et al. Avelumab (anti-PD-L1) in combination with crizotinib or lorlatinib in patients with previously treated advanced NSCLC: Phase 1b results from JAVELIN Lung 101. J. Clin. Oncol. 2018, 36 (Supply. 15), 9008. [Google Scholar] [CrossRef]
- Spigel, D.R.; Faivre-Finn, C.; Gray, J.E.; Vicente, D.; Planchard, D.; Paz-Ares, L.; Vansteenkiste, J.F.; Garassino, M.C.; Hui, R.; Quantin, X.; et al. Five-Year Survival Outcomes From the PACIFIC Trial: Durvalumab After Chemoradiotherapy in Stage III Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 1301. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kin, H.-H.; Dubos Arvis, C.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Nogami, N.; Barlesi, F.; Socinski, M.A.; Reck, M.; Thomas, C.A.; Cappuzzo, F.; Mok, T.; Finley, G.; Aerts, J.G.; Orlandi, F.; et al. IMpower150 Final Exploratory Analyses for Atezolizumab Plus Bevacizumab and Chemotherapy in Key NSCLC Patient Subgroups With EGFR Mutations or Metastases in the Liver or Brain. J. Thorac. Oncol. 2022, 17, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Man, J.; Lord, S.; Links, M.; Gebski, V.; Mok, T.; Tang, J.C.-H. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non-Small Cell Lung Cancer-A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Felip, E.; Altorki, N.; Zhou, C.; Csőszi, T.; Vynnychenko, I.; Goloborodko, O.; Luft, A.; Akopov, A.; Martinez-Marti, A.; Kenmotsu, H.; et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB–IIIA non-small-cell lung cancer (IMpower010): A randomised, multicentre, open-label, phase 3 trial. Lancet 2021, 398, 1344–1357. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Arbour, K.C.; Rizvi, H.; Iqbal, A.N.; Gadgeel, S.M.; Girshman, J.; Kris, M.G.; Reily, G.J.; Yu, H.A.; Hellmann, M.D. Severe immune-related adverse events are common with sequential PD-(L)1 blockade and osimertinib. Ann. Oncol. 2019, 30, 839–844. [Google Scholar] [CrossRef]
- Cui, W.; Cotter, C.; Sreter, K.B.; Heelan, K.; Creamer, D.; Basu, T.N.; Handy, J.; Walsh, S.; Popat, S. Case of Fatal Immune-Related Skin Toxicity From Sequential Use of Osimertinib After Pembrolizumab: Lessons for Drug Sequencing in Never-Smoking Non–Small-Cell Lung Cancer. JCO Oncol. Pr. 2020, 16, 842–844. [Google Scholar] [CrossRef]
- Oshima, Y.; Tanimoto, T.; Yuji, K.; Tojo, A. EGFR-TKI-associated interstitial pneumonitis in nivolumab-treated patients with non-small cell lung cancer. JAMA Oncol. 2018, 4, 1112–1115. [Google Scholar] [CrossRef]
- Jahanzeb, M.; Lin, H.M.; Pan, X.; Yin, Y.; Baumann, P.; Langer, C.J. Immunotherapy Treatment Patterns and Outcomes Among ALK-Positive Patients With Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 49–57. [Google Scholar] [CrossRef]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer (NSCLC): A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585. [Google Scholar] [CrossRef]
- Sankar, K.; Nagrath, S.; Ramnath, N. Immunotherapy for ALK-Rearranged Non-Small Cell Lung Cancer: Challenges Inform Promising Approaches. Cancers 2021, 13, 1476. [Google Scholar] [CrossRef] [PubMed]
- Voena, C.; Menotti, M.; Mastini, C.; Di Giacomo, F.; Longo, D.L.; Castella, B.; Merlo, M.E.B.; Ambrogio, C.; Wang, Q.; Minero, V.G.; et al. Efficacy of a Cancer Vaccine against ALK-Rearranged Lung Tumors. Cancer Immunol. Res. 2015, 3, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189. [Google Scholar] [CrossRef]
- Mason, R.; Dearden, H.C.; Nguyen, B.; Soon, J.A.; Smith, J.L.; Randhawa, M.; Mant, A.; Warburton, L.; Lo, S.; Meniawy, T.; et al. Combined ipilimumab and nivolumab first-line and after BRAF-targeted therapy in advanced melanoma. Pigment. Cell Melanoma Res. 2020, 33, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gatzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Dong, J.; Li, B.; Lin, D.; Zhou, Q.; Huang, D. Advances in Targeted Therapy and Immunotherapy for Non-small Cell Lung Cancer Based on Accurate Molecular Typing. Front. Pharmacol. 2019, 10, 2019. [Google Scholar] [CrossRef] [PubMed]
- Guisier, F.; Dubos-Arvis, C.; Viñas, F.; Doubre, H.; Ricordel, C.; Ropert, S.; Janicot, H.; Bernardi, M.; Fournel, P.; Lamy, R.; et al. Efficacy and Safety of Anti-PD-1 Immunotherapy in Patients With Advanced NSCLC With BRAF, HER2, or MET Mutations or RET Translocation: GFPC 01-2018. J. Thorac. Oncol. 2020, 15, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Dudnik, E.; Peled, N.; Nechushtan, H.; Wollner, M.; Onn, A.; Agbarya, A.; Moskovitz, M.; Keren, S.; Popovits-Hadari, N.; Urban, D.; et al. BRAF Mutant Lung Cancer: Programmed Death Ligand 1 Expression, Tumor Mutational Burden, Microsatellite Instability Status, and Response to Immune Check-Point Inhibitors. J. Thorac. Oncol. 2018, 13, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Qian, J.; Chen, Z.; Zhang, B.; Chen, P.; Zhang, L.; Li, J.; Zhang, H.; Wang, C. Short-term response to immune-chemotherapy and immune features of a ceritinib-resistant patient with ROS1-rearranged lung adenocarcinoma. J. Immunother. Cancer 2021, 9, e001967. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, N.J.; Schneider, J.L.; Patil, T.; Zhu, V.W.; Goldman, D.A.; Yang, S.R.; Li, J.; Zhang, H.; Wang, C. Response to Immune Checkpoint Inhibition as Monotherapy or in Combination With Chemotherapy in Metastatic ROS1-Rearranged Lung Cancers. JTO Clin. Res. Rep. 2021, 2, 100187. [Google Scholar] [PubMed]
- Liu, Z.; Zhao, K.; Wei, S.; Liu, C.; Zhou, J.; Gou, Q.; Wu, X.; Yang, Z.; Yang, Y.; Peng, Y.; et al. ROS1-fusion protein induces PD-L1 expression via MEK-ERK activation in non-small cell lung cancer. Oncoimmunology 2020, 9, 1758003. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Kikuchi, R.; Iwai, Y.; Ito, M.; Tsukamoto, H.; Yamazaki, K.; Nakamura, H.; Aoshiba, K. Immune Checkpoint Inhibitors in ROS1-Rearranged Non–Small Cell Lung Cancer: A Report of Two Cases. J. Thorac. Oncol. 2019, 14, e165–e167. [Google Scholar]
- Offin, M.; Guo, R.; Wu, S.L.; Sabari, J.; Land, J.D.; Ni, A.; Montecalvo, J.; Halpenny, D.F.; Buie, L.W.; Pak, T.; et al. Immunophenotype and Response to Immunotherapy of RET-Rearranged Lung Cancers. JCO Precis. Oncol. 2019, 3, PO.18.00386. [Google Scholar] [CrossRef]
- Lu, C.; Dong, X.R.; Zhao, J.; Zhang, X.C.; Chen, H.J.; Zhou, Q.; Tu, H.-Y.; Ai, X.-H.; Chen, X.-F.; An, G.-L.; et al. Association of genetic and immuno-characteristics with clinical outcomes in patients with RET-rearranged non-small cell lung cancer: A retrospective multicenter study. J. Hematol. Oncol. 2020, 13, 37. [Google Scholar] [CrossRef]
- Ekman, S. How selecting best therapy for metastatic NTRK fusion-positive non-small cell lung cancer? Transl. Lung Cancer Res. 2020, 9, 2535. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; Van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Feverman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Shen, R.; Buonocore, D.; Olah, Z.T.; Ni, A.; Ginsberg, M.S.; Ulaner, G.A.; Offin, M.; Feldman, D.; Hembrough, T.; et al. Ado-Trastuzumab Emtansine for Patients With HER2-Mutant Lung Cancers: Results From a Phase II Basket Trial. J. Clin. Oncol. 2018, 36, 2532–2537. [Google Scholar] [CrossRef] [PubMed]
- Elamin, Y.Y.; Robichaux, J.P.; Carter, B.W.; Altan, M.; Gibbons, D.L.; Fossella, F.V.; Lam, V.K.; Patel, A.B.; Negrao, M.V.; Le, X.; et al. Poziotinib for Patients With HER2 Exon 20 Mutant Non–Small-Cell Lung Cancer: Results From a Phase II Trial. J. Clin. Oncol. 2022, 40, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Smit, E.F.; Goto, Y.; Nakagawa, K.; Udagawa, H.; Mazières, J.; Nagasaka, M.; Bazhenova, L.; Saltos, A.N.; Felip, E.; et al. Trastuzumab Deruxtecan in HER2 -Mutant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2022, 386, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Riudavets, M.; Sullivan, I.; Abdayem, P.; Planchard, D. Targeting HER2 in non-small-cell lung cancer (NSCLC): A glimpse of hope? An updated review on therapeutic strategies in NSCLC harbouring HER2 alterations. ESMO Open 2021, 6, 100260. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Kawazoe, A.; Yañez, P.; Li, N.; Lonardi, S.; Kolesnik, O.; Barajas, O.; Bai, Y.; Shen, L.; Tang, Y.; et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature 2021, 600, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018, 167, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): A single-arm, multicentre, phase 1b-2 trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Sabari, J.K.; Leonardi, G.C.; Shu, C.A.; Umeton, R.; Montecalvo, J.; Ni, A.; Chen, R.; Dienstag, J.; Mrad, C.; Bergagnini, I.; et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann. Oncol. 2018, 29, 2085–2091. [Google Scholar] [CrossRef]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.H.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villacruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor Activity of Crizotinib in Lung Cancers Harboring a MET Exon 14 Alteration. Nat. Med. 2020, 26, 47. [Google Scholar] [CrossRef]
- Mayenga, M.; Assié, J.B.; Monnet, I.; Massiani, M.A.; Tabeze, L.; Friard, S.; Fraboulet, S.; Metivier, A.-C.; Chouaid, C.; Zemoura, L.; et al. Durable responses to immunotherapy of non-small cell lung cancers harboring MET exon-14-skipping mutation: A series of 6 cases. Lung Cancer 2020, 150, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Geng, H.; Liu, Y.; Liu, L.; Chen, Y.; Wu, F.; Liu, Z.; Ling, S.; Wang, Y.; Zhou, L. Hot and cold tumors: Immunological features and the therapeutic strategies. MedComm 2023, 4, e343. [Google Scholar] [CrossRef] [PubMed]
- Soo, R.A.; Lim, S.M.; Syn, N.L.; Teng, R.; Soong, R.; Mok, T.S.K.; Cho, B.C. Immune checkpoint inhibitors in epidermal growth factor receptor mutant non-small cell lung cancer: Current controversies and future directions. Lung Cancer 2018, 115, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Azuma, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Tanizaki, J.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; et al. Induction of PD-L1 Expression by the EML4-ALK Oncoprotein and Downstream Signaling Pathways in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 4014–4021. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Chen, N.; Fang, W.; Zhan, J.; Liu, Q.; Kang, S.; He, X.; Liu, L.; Huang, J.; Chen, Y.; et al. Upregulation of PD-L1 by EML4-ALK fusion protein mediates the immune escape in ALK positive NSCLC: Implication for optional anti-PD-1/PD-L1 immune therapy for ALK-TKIs sensitive and resistant NSCLC patients. Oncoimmunology 2016, 5, e1094598. [Google Scholar] [CrossRef] [PubMed]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Ng, T.L.; Liu, Y.; Dimou, A.; Patil, T.; Aisner, D.L.; Dong, Z.; Jiang, T.; Su, S.; Wu, C.; Ren, S.; et al. Predictive value of oncogenic driver subtype, programmed death-1 ligand (PD-L1) score, and smoking status on the efficacy of PD-1/PD-L1 inhibitors in patients with oncogene-driven non–small cell lung cancer. Cancer 2019, 125, 1038–1049. [Google Scholar] [CrossRef]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Tarhini, A.A.; Cohen, G.I.; Truong, T.G.; Moon, H.H.; Davar, D.; O’Rourke, M.; Stephenson, J.J.; et al. Combination Dabrafenib and Trametinib Versus Combination Nivolumab and Ipilimumab for Patients With Advanced BRAF-Mutant Melanoma: The DREAMseq Trial-ECOG-ACRIN EA6134. J. Clin. Oncol. 2022, 41, 186–197. [Google Scholar] [CrossRef]
- Kwan, A.K.; Piazza, G.A.; Keeton, A.B.; Leite, C.A. The path to the clinic: A comprehensive review on direct KRASG12C inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 27. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McMahon, D.J.; McLaughlin, R.; Naidoo, J. Is Immunotherapy Beneficial in Patients with Oncogene-Addicted Non-Small Cell Lung Cancers? A Narrative Review. Cancers 2024, 16, 527. https://doi.org/10.3390/cancers16030527
McMahon DJ, McLaughlin R, Naidoo J. Is Immunotherapy Beneficial in Patients with Oncogene-Addicted Non-Small Cell Lung Cancers? A Narrative Review. Cancers. 2024; 16(3):527. https://doi.org/10.3390/cancers16030527
Chicago/Turabian StyleMcMahon, David John, Ronan McLaughlin, and Jarushka Naidoo. 2024. "Is Immunotherapy Beneficial in Patients with Oncogene-Addicted Non-Small Cell Lung Cancers? A Narrative Review" Cancers 16, no. 3: 527. https://doi.org/10.3390/cancers16030527
APA StyleMcMahon, D. J., McLaughlin, R., & Naidoo, J. (2024). Is Immunotherapy Beneficial in Patients with Oncogene-Addicted Non-Small Cell Lung Cancers? A Narrative Review. Cancers, 16(3), 527. https://doi.org/10.3390/cancers16030527