
KRAS, a New Target for Precision Medicine in Colorectal Cancer?

 , , , , , , , , and
, , , , , , , , and
Abstract
Simple Summary
Abstract
1. Introduction
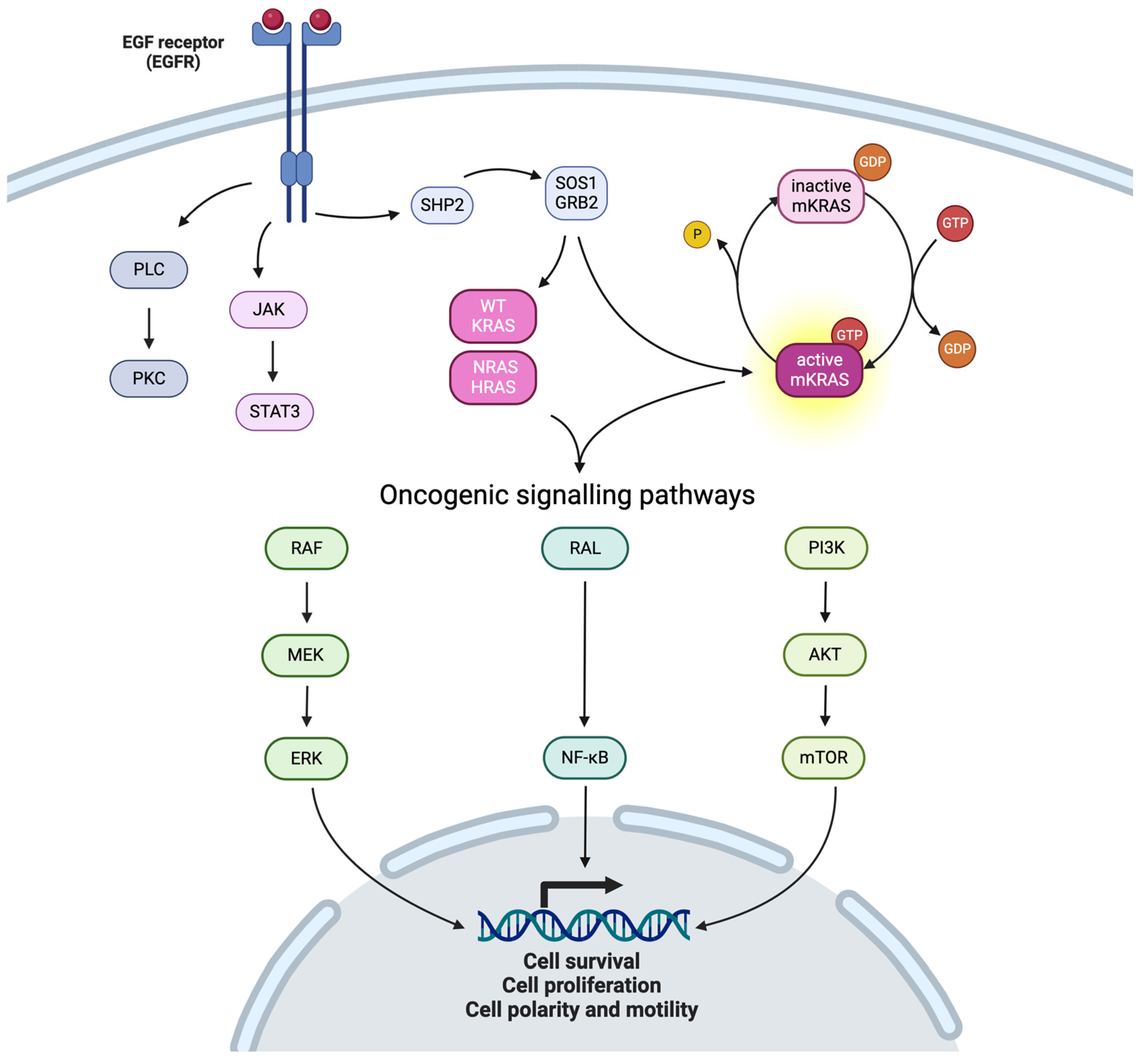
2. KRAS Pathway
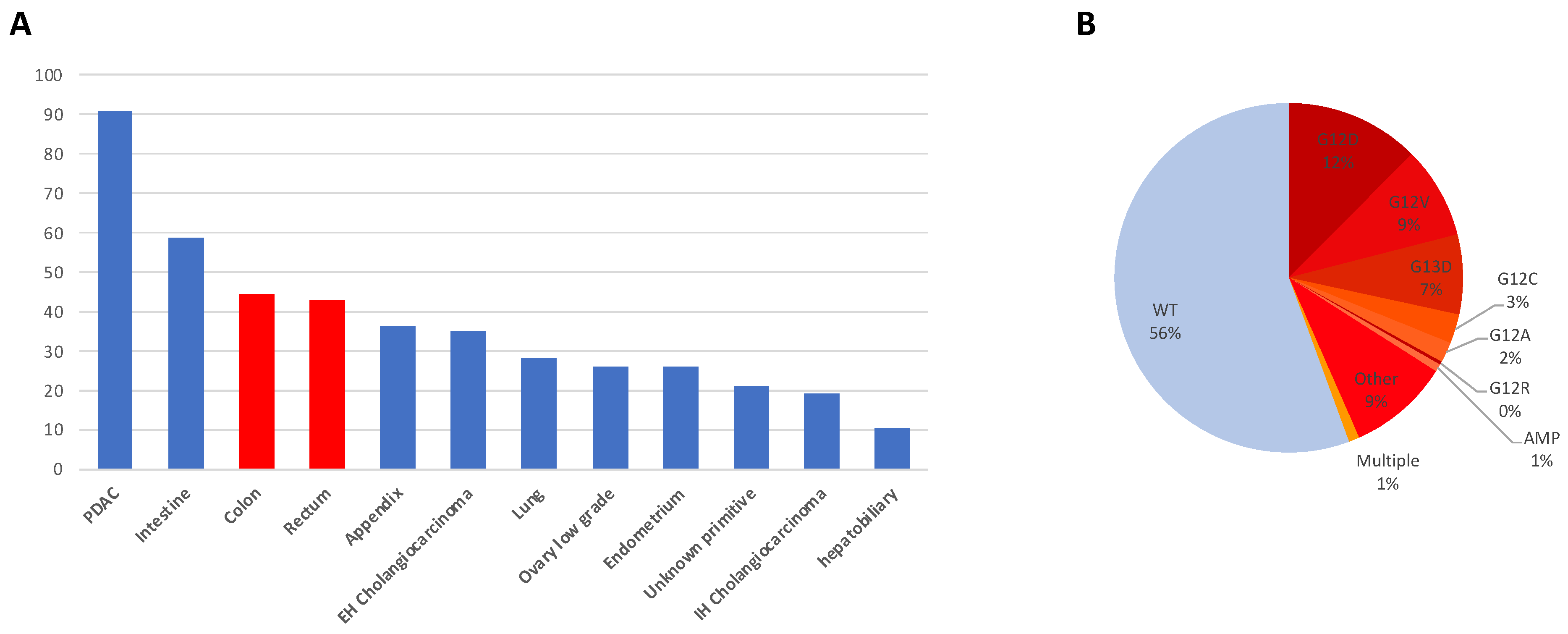
- Point mutations: Point mutations involve the substitution of a single nucleotide base within the KRAS gene, resulting in amino acid alterations in the KRAS protein. The most common point mutations in CRC affect codons 12, 13, and 61 of the KRAS gene. Codon 12 mutations, such as G12D and G12V, are particularly prevalent, accounting for approximately 50% of KRAS mutations in CRC (respectively 28% and 19% for G12D and G12V). G13D is another frequent point mutation (17%) [1]. Codon 61 mutations: mutations at codon 61 of the KRAS gene, such as Q61H and Q61L, are less common but still significant in CRC. These mutations account for approximately 5–10% of KRAS mutations in CRC and are associated with aggressive tumor phenotypes and resistance to targeted therapies;
- Insertions and deletions or amplifications: Insertions and deletions (indels) in the KRAS gene can lead to frameshift mutations, disrupting the reading frame and generating aberrant KRAS protein variants while less frequent compared to point mutations, as well as amplifications of KRAS;
- Rare mutations: In addition to the commonly observed mutations, CRC may harbor rare or uncommon KRAS mutations that involve other codons or unusual nucleotide changes. While individually rare, these mutations collectively contribute to the complexity of KRAS-driven tumorigenesis.
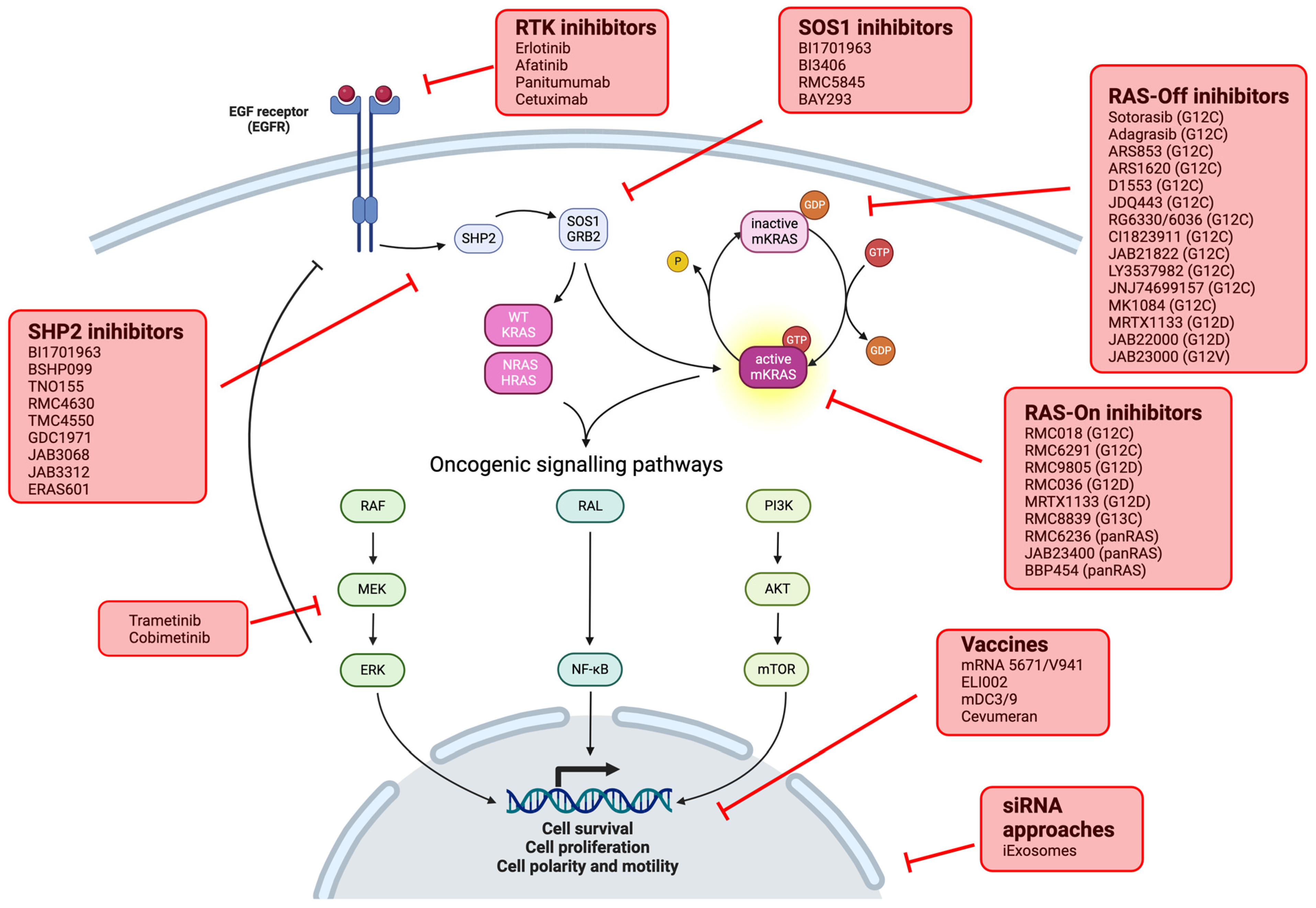
3. KRAS Inhibition: KRAS G12C
3.1. Alone

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adagrasib (n = 43) [24] | Sotorasib (n = 62) [22] | |
|---|---|---|
| Objective response rate | 23% (12–39%) | 9.7% (3.6–19.9%) |
| Disease control rate | 86% | 82.3% (70.5–90.8) |
| Median duration of response | 4.3 months (2.3–4.4) | 4.2 months (2.9–8.5) |
| Progression-free survival | 5.6 months (4.1–8.3) | 4.0 months (2.8–4.2) |
| Overall survival | 19.8 months (12.5–23.0) | 10.6 months (7.7–15.6) |
| Grade 3–4 toxicities | 34% | 10% |
| Dose reduction | 39% | 18% |
| Discontinuation for toxicity | 0% | 2% |
3.2. Combination with Other Drugs
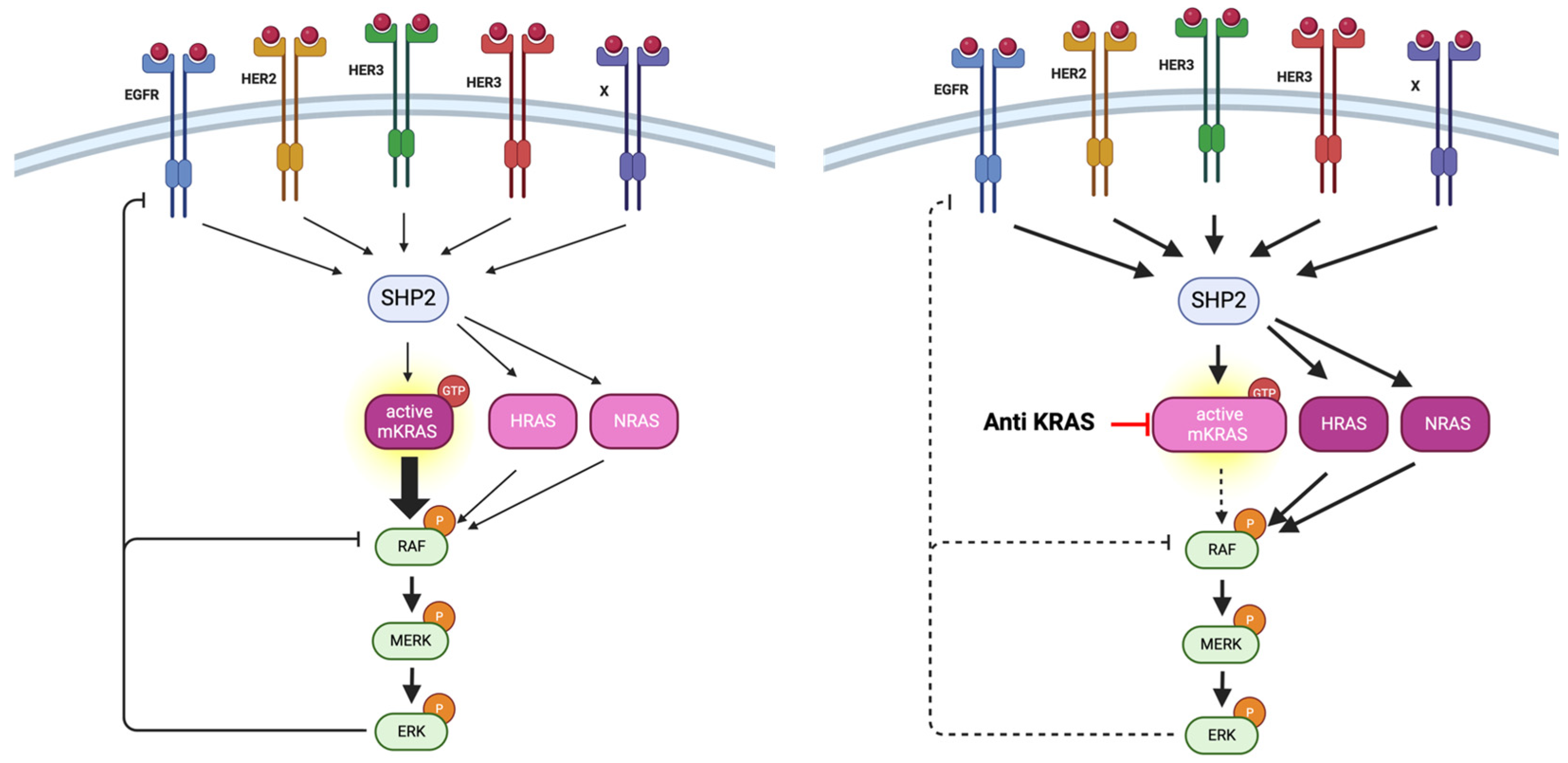
4. Other Strategies to Target KRAS
4.1. Other Codon Specific Inhibitor: MRTX1133 (KRASG12D)
4.1.1. Preclinical Data
4.1.2. Combination Perspectives
EGFR
Immune Checkpoint Inhibitors
4.2. Other Therapeutic Class
4.2.1. Pan-KRAS Inhibitors
4.2.2. Indirect Inhibition
4.2.3. Cancer Vaccines
4.2.4. CAR-T Cells
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hofmann, M.H.; Gerlach, D.; Misale, S.; Petronczki, M.; Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov. 2022, 12, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Arnedos, M.; Baras, A.S.; Baselga, J.; Zhang, H.; AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Väyrynen, V.; Wirta, E.-V.; Seppälä, T.; Sihvo, E.; Mecklin, J.-P.; Vasala, K.; Kellokumpu, I. Incidence and Management of Patients with Colorectal Cancer and Synchronous and Metachronous Colorectal Metastases: A Population-Based Study. BJS Open 2020, 4, 685–692. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus Bevacizumab versus FOLFIRI plus Bevacizumab as First-Line Treatment of Patients with Metastatic Colorectal Cancer: Updated Overall Survival and Molecular Subgroup Analyses of the Open-Label, Phase 3 TRIBE Study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Tournigand, C.; André, T.; Achille, E.; Lledo, G.; Flesh, M.; Mery-Mignard, D.; Quinaux, E.; Couteau, C.; Buyse, M.; Ganem, G.; et al. FOLFIRI Followed by FOLFOX6 or the Reverse Sequence in Advanced Colorectal Cancer: A Randomized GERCOR Study. J. Clin. Oncol. 2004, 22, 229–237. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Cancer Stat Facts: Colorectal Cancer SEER 18 2011–2017. 2022. Available online: https://seer.cancer.gov/statfacts/html/colorect.html (accessed on 7 October 2024).
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Yaeger, R.; Spira, A.I.; Pelster, M.S.; Sabari, J.K.; Hafez, N.; Barve, M.; Velastegui, K.; Yan, X.; Shetty, A.; et al. Adagrasib in Advanced Solid Tumors Harboring a KRASG12C Mutation. J. Clin. Oncol. 2023, 41, 4097–4106. [Google Scholar] [CrossRef]
- Ostrem, J.M.L.; Shokat, K.M. Direct Small-Molecule Inhibitors of KRAS: From Structural Insights to Mechanism-Based Design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Ratner, N.; Miller, S.J. A RASopathy Gene Commonly Mutated in Cancer: The Neurofibromatosis Type 1 Tumour Suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-Targeted Therapies: Is the Undruggable Drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Xue, J.Y.; Lito, P. Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell 2020, 183, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Vial, E.; Pouysségur, J. Regulation of Tumor Cell Motility by ERK Mitogen-Activated Protein Kinases. Ann. N. Y. Acad. Sci. 2004, 1030, 208–218. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of Oncogenic KRAS in the Diagnosis, Prognosis and Treatment of Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and Saturation Analysis of Cancer Genes across 21 Tumour Types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Wang, H.; Chi, L.; Yu, F.; Dai, H.; Gao, C.; Si, X.; Wang, Z.; Liu, L.; Zheng, J.; Shan, L.; et al. Annual Review of KRAS Inhibitors in 2022. Eur. J. Med. Chem. 2023, 249, 115124. [Google Scholar] [CrossRef]
- Fakih, M.G.; Kopetz, S.; Kuboki, Y.; Kim, T.W.; Munster, P.N.; Krauss, J.C.; Falchook, G.S.; Han, S.-W.; Heinemann, V.; Muro, K.; et al. Sotorasib for Previously Treated Colorectal Cancers with KRASG12C Mutation (CodeBreaK100): A Prespecified Analysis of a Single-Arm, Phase 2 Trial. Lancet Oncol. 2022, 23, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients with Advanced KRASG12C Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Weiss, J.; Pelster, M.S.; Spira, A.I.; Barve, M.; Ou, S.-H.I.; Leal, T.A.; Bekaii-Saab, T.S.; Paweletz, C.P.; Heavey, G.A.; et al. Adagrasib with or without Cetuximab in Colorectal Cancer with Mutated KRAS G12C. N. Engl. J. Med. 2023, 388, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Uboha, N.V.; Pelster, M.S.; Bekaii-Saab, T.S.; Barve, M.; Saltzman, J.; Sabari, J.K.; Peguero, J.A.; Paulson, A.S.; Jänne, P.A.; et al. Efficacy and Safety of Adagrasib plus Cetuximab in Patients with KRASG12C-Mutated Metastatic Colorectal Cancer. Cancer Discov. 2024, 14, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Cowzer, D.; Zameer, M.; Conroy, M.; Kolch, W.; Duffy, A.G. Targeting KRAS in Pancreatic Cancer. J. Pers. Med. 2022, 12, 1870. [Google Scholar] [CrossRef]
- Akhave, N.S.; Biter, A.B.; Hong, D.S. Mechanisms of Resistance to KRASG12C-Targeted Therapy. Cancer Discov. 2021, 11, 1345–1352. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Huang, H.-Y.; Lin, Z.; Ranieri, M.; Li, S.; Sahu, S.; Liu, Y.; Ban, Y.; Guidry, K.; Hu, H.; et al. Genome-Wide CRISPR Screens Identify Multiple Synthetic Lethal Targets That Enhance KRASG12C Inhibitor Efficacy. Cancer Res. 2023, 83, 4095–4111. [Google Scholar] [CrossRef]
- Ryan, M.B.; Coker, O.; Sorokin, A.; Fella, K.; Barnes, H.; Wong, E.; Kanikarla, P.; Gao, F.; Zhang, Y.; Zhou, L.; et al. KRASG12C-Independent Feedback Activation of Wild-Type RAS Constrains KRASG12C Inhibitor Efficacy. Cell Rep. 2022, 39, 110993. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal Cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef]
- Feng, J.; Hu, Z.; Xia, X.; Liu, X.; Lian, Z.; Wang, H.; Wang, L.; Wang, C.; Zhang, X.; Pang, X. Feedback Activation of EGFR/Wild-Type RAS Signaling Axis Limits KRASG12D Inhibitor Efficacy in KRASG12D-Mutated Colorectal Cancer. Oncogene 2023, 42, 1620–1633. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab Monotherapy and Cetuximab plus Irinotecan in Irinotecan-Refractory Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Schwartzberg, L.S.; Rivera, F.; Karthaus, M.; Fasola, G.; Canon, J.-L.; Hecht, J.R.; Yu, H.; Oliner, K.S.; Go, W.Y. PEAK: A Randomized, Multicenter Phase II Study of Panitumumab plus Modified Fluorouracil, Leucovorin, and Oxaliplatin (mFOLFOX6) or Bevacizumab plus mFOLFOX6 in Patients with Previously Untreated, Unresectable, Wild-Type KRAS Exon 2 Metastatic Colorectal Cancer. J. Clin. Oncol. 2014, 32, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Kuboki, Y.; Yaeger, R.; Fakih, M.; Strickler, J.H.; Masuishi, T.; Kim, E.J.-H.; Bestvina, C.M.; Langer, C.J.; Krauss, J.C.; Puri, S.; et al. 45MO Sotorasib in Combination with Panitumumab in Refractory KRAS G12C-Mutated Colorectal Cancer: Safety and Efficacy for Phase Ib Full Expansion Cohort. Ann. Oncol. 2022, 33, S1445–S1446. [Google Scholar] [CrossRef]
- Fakih, M.; Salvatore, L.; Esaki, T.; Modest, D.P.; Páez Lopez-Bravo, D.; Taieb, J.; Karamouzis, M.; Ruiz-Garcia, E.; Kim, T.W.; Kuboki, Y.; et al. Overall Survival (OS) of Phase 3 CodeBreaK 300 Study of Sotorasib plus Panitumumab (Soto+pani) versus Investigator’s Choice of Therapy for KRAS G12C-Mutated Metastatic Colorectal Cancer (mCRC). JCO 2024, 42, LBA3510. [Google Scholar] [CrossRef]
- Fakih, M.G.; Salvatore, L.; Esaki, T.; Modest, D.P.; Lopez-Bravo, D.P.; Taieb, J.; Karamouzis, M.V.; Ruiz-Garcia, E.; Kim, T.-W.; Kuboki, Y.; et al. Sotorasib plus Panitumumab in Refractory Colorectal Cancer with Mutated KRAS G12C. N. Engl. J. Med. 2023, 389, 2125–2139. [Google Scholar] [CrossRef]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-Tumor Efficacy of a Potent and Selective Non-Covalent KRASG12D Inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef]
- The KRASG12D Inhibitor MRTX1133 Elucidates KRAS-Mediated Oncogenesis. Nat. Med. 2022, 28, 2017–2018. [CrossRef]
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.-K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023, 13, 298–311. [Google Scholar] [CrossRef]
- Gulay, K.C.M.; Zhang, X.; Pantazopoulou, V.; Patel, J.; Esparza, E.; Pran Babu, D.S.; Ogawa, S.; Weitz, J.; Ng, I.; Mose, E.S.; et al. Dual Inhibition of KRASG12D and Pan-ERBB Is Synergistic in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2023, 83, 3001–3012. [Google Scholar] [CrossRef]
- Dougan, S.K. The Pancreatic Cancer Microenvironment. Cancer J. 2017, 23, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Mainardi, S. With a Little Help from My T Friends: T Cells Increase Efficacy of KRAS (G12D) Inhibitors. Cell Rep. Med. 2023, 4, 100950. [Google Scholar] [CrossRef] [PubMed]
- Watterson, A.; Coelho, M.A. Cancer Immune Evasion through KRAS and PD-L1 and Potential Therapeutic Interventions. Cell Commun. Signal. 2023, 21, 45. [Google Scholar] [CrossRef]
- La Vecchia, S.; Sebastián, C. Metabolic Pathways Regulating Colorectal Cancer Initiation and Progression. Semin. Cell Dev. Biol. 2020, 98, 63–70. [Google Scholar] [CrossRef]
- Qi, S.-M.; Dong, J.; Xu, Z.-Y.; Cheng, X.-D.; Zhang, W.-D.; Qin, J.-J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef]
- Farnaby, W.; Koegl, M.; McConnell, D.B.; Ciulli, A. Transforming Targeted Cancer Therapy with PROTACs: A Forward-Looking Perspective. Curr. Opin. Pharmacol. 2021, 57, 175–183. [Google Scholar] [CrossRef]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an Undruggable Pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef]
- Kim, D.; Herdeis, L.; Rudolph, D.; Zhao, Y.; Böttcher, J.; Vides, A.; Ayala-Santos, C.I.; Pourfarjam, Y.; Cuevas-Navarro, A.; Xue, J.Y.; et al. Pan-KRAS Inhibitor Disables Oncogenic Signalling and Tumour Growth. Nature 2023, 619, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Koltun, E.; Cregg, J.; Rice, M.A.; Whalen, D.M.; Freilich, R.; Jiang, J.; Hansen, R.; Bermingham, A.; Knox, J.E.; Dinglasan, J.; et al. Abstract 1260: First-in-Class, Orally Bioavailable KRASG12V(ON) Tri-Complex Inhibitors, as Single Agents and in Combinations, Drive Profound Anti-Tumor Activity in Preclinical Models of KRASG12V Mutant Cancers. Cancer Res. 2021, 81, 1260. [Google Scholar] [CrossRef]
- Valencia-Sama, I.; Ladumor, Y.; Kee, L.; Adderley, T.; Christopher, G.; Robinson, C.M.; Kano, Y.; Ohh, M.; Irwin, M.S. NRAS Status Determines Sensitivity to SHP2 Inhibitor Combination Therapies Targeting the RAS-MAPK Pathway in Neuroblastoma. Cancer Res. 2020, 80, 3413–3423. [Google Scholar] [CrossRef] [PubMed]
- Sheffels, E.; Kortum, R.L. The Role of Wild-Type RAS in Oncogenic RAS Transformation. Genes 2021, 12, 662. [Google Scholar] [CrossRef]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS Nucleotide Cycling Underlies the SHP2 Phosphatase Dependence of Mutant BRAF-, NF1- and RAS-Driven Cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef]
- Zhao, Y.; Xue, J.Y.; Lito, P. Suppressing Nucleotide Exchange to Inhibit KRAS-Mutant Tumors. Cancer Discov. 2021, 11, 17–19. [Google Scholar] [CrossRef]
- Chen, Y.-N.P.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric Inhibition of SHP2 Phosphatase Inhibits Cancers Driven by Receptor Tyrosine Kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- Garcia Fortanet, J.; Chen, C.H.-T.; Chen, Y.-N.P.; Chen, Z.; Deng, Z.; Firestone, B.; Fekkes, P.; Fodor, M.; Fortin, P.D.; Fridrich, C.; et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J. Med. Chem. 2016, 59, 7773–7782. [Google Scholar] [CrossRef]
- Ou, S.I.; Koczywas, M.; Ulahannan, S.; Janne, P.; Pacheco, J.; Burris, H.; McCoach, C.; Wang, J.S.; Gordon, M.; Haura, E.; et al. A12 The SHP2 Inhibitor RMC-4630 in Patients with KRAS-Mutant Non-Small Cell Lung Cancer: Preliminary Evaluation of a First-in-Man Phase 1 Clinical Trial. J. Thorac. Oncol. 2020, 15, S15–S16. [Google Scholar] [CrossRef]
- Brana, I.; Shapiro, G.; Johnson, M.L.; Yu, H.A.; Robbrecht, D.; Tan, D.S.-W.; Siu, L.L.; Minami, H.; Steeghs, N.; Hengelage, T.; et al. Initial Results from a Dose Finding Study of TNO155, a SHP2 Inhibitor, in Adults with Advanced Solid Tumors. J. Clin. Oncol. 2021, 39, 3005. [Google Scholar] [CrossRef]
- Johnson, M.L.; Gort, E.; Pant, S.; Lolkema, M.P.; Sebastian, M.; Scheffler, M.; Hwang, J.; Dünzinger, U.; Riemann, K.; Kitzing, T.; et al. 524P A Phase I, Open-Label, Dose-Escalation Trial of BI 1701963 in Patients (Pts) with KRAS Mutated Solid Tumours: A Snapshot Analysis. Ann. Oncol. 2021, 32, S591–S592. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Simnica, D.; Kobold, S. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med. 2022, 387, 573. [Google Scholar] [CrossRef]
| Criteria | Adagrasib + Cetuximab (n = 28) [24] | Sotorasib + Panitumumab (n = 53) [34,36] |
|---|---|---|
| Objective response rate % (CI95%) | 46% (28–66) | 26.4% (15.3–40.3) |
| Median duration of response Months (CI95%) | 7.6 (5.0–NR) | 4.4 (2.8–6.3) |
| Median progression-free survival Months (CI95%) | 6.9 (5.4–8.1) | 5.6 (4.2–7.6) |
| Median overall survival Months (CI5%) | 13.4 (9.5–20.1) | NR (9.6–NR) (960 mg) and 11.9 (7.5–NR) (240 mg) |
| NCT Number | Drug | Target | Main Sponsor | Study Status |
|---|---|---|---|---|
| Specific inhibitor alone | ||||
| NCT05485974 | HBI-2438 | KRASG12C | HUYABIO International, LLC. (San Diego, CA, USA) | RECRUITING |
| NCT05462717 | RMC-6291 | KRASG12C | Revolution Medicines, Inc. (Redwood City, CA, USA) | ACTIVE_NOT_RECRUITING |
| NCT06244771 | FMC-376 | KRASG12C | Frontier Medicines Corporation (San Fransisco, CA, USA) | RECRUITING |
| NCT04121286 | JAB-3312 | KRASG12C | Jacobio Pharmaceuticals Co., Ltd. (Beijing, China) | RECRUITING |
| NCT06385925 | TSN1611 | KRASG12D | Tyligand Bioscience (Shanghai, China) | RECRUITING |
| NCT05737706 | MRTX1133 | KRASG12D | Mirati Therapeutics Inc. (San Diego, CA, USA) | RECRUITING |
| NCT06364696 | ASP4396 | KRASG12D | Astellas Pharma Inc.(Tokyo, Japan) | RECRUITING |
| NCT06403735 | QLC1101 | KRASG12D | Qilu Pharmaceutical Co., Ltd. (Madrid, Spain) | RECRUITING |
| NCT06040541 | RMC-9805|RMC-6236 | KRASG12D/KRASG12X | Revolution Medicines, Inc. (Redwood City, CA, USA) | RECRUITING |
| Specific inhibitor in combinaiton | ||||
| NCT06412198 | Cetuximab|Cemiplimab|Adagrasib | KRASG12C | M.D. Anderson Cancer Center (Houston, TX, USA) | RECRUITING |
| NCT05194995 | JAB-21822|Cetuximab | KRASG12C | Jacobio Pharmaceuticals Co., Ltd. (Beijing, China) | ACTIVE_NOT_RECRUITING |
| NCT04330664 | MRTX849|TNO155 | KRASG12C + SHP2 | Mirati Therapeutics Inc. (San Diego, CA, USA) | ACTIVE_NOT_RECRUITING |
| NCT05288205 | JAB-21822|JAB-3312 | KRASG12C + SHP2 | Jacobio Pharmaceuticals Co., Ltd. (Beijing, China) | RECRUITING |
| NCT06039384 | INCB099280|adagrasib | KRASG12C | Incyte Corporation (Geneva, Switzerland) | ACTIVE_NOT_RECRUITING |
| NCT05198934 | Sotorasib|Standard treatment | KRASG12C | Amgen (Thousand Oaks, CA, USA) | ACTIVE_NOT_RECRUITING |
| NCT04956640 | LY3537982|standard treamtent | KRASG12C | Eli Lilly and Company (Indianapolis, IN, USA) | RECRUITING |
| NCT05578092 | MRTX0902|MRTX849 | KRASG12C + SOS1 | Mirati Therapeutics Inc. (San Diego, CA, USA) | RECRUITING |
| NCT04699188 | JDQ443|TNO155 | KRASG12C +SHP2 | Novartis Pharmaceuticals (Reuil Malmaison, France) | RECRUITING |
| NCT04449874 | GDC-6036|Standard treamtent | KRASG12C | Genentech, Inc. (San Franscisco, CA, USA) | RECRUITING |
| NCT05358249 | JDQ443|trametinib|Ribociclib | KRASG12C | Novartis Pharmaceuticals (Reuil Malmaison, France) | ACTIVE_NOT_RECRUITING |
| NCT06026410 | KO-2806|Cabozantinib|Adagrasib | KRASG12C, farnesyl transferase | Kura Oncology, Inc. (San Diego, CA, USA) | RECRUITING |
| NCT06252649 | Sotorasib|standard treatment | KRASG12C | Amgen (Thousand Oaks, CA, USA) | RECRUITING |
| NCT06586515 | LY3962673|standard treatment | KRASG12D | Eli Lilly and Company (Indianapolis, IN, USA) | NOT_YET_RECRUITING |
| NCT04793958 | MRTX849|standard treatment | KRASG12D | Mirati Therapeutics Inc. (San Diego, CA, USA) | ACTIVE_NOT_RECRUITING |
| NCT03785249 | MRTX849|standard treatment | KRASG12D | Mirati Therapeutics Inc. (San Diego, CA, USA) | RECRUITING |
| NCT05722327 | MRTX849|standard treatment | KRASG12D | M.D. Anderson Cancer Center (Houston, TX, USA) | RECRUITING |
| NCT06599502 | AZD0022|Cetuximab | KRASG12D | AstraZeneca (London, UK) | NOT_YET_RECRUITING |
| panKRAS | ||||
| NCT06078800 | YL-17231 | KRAS | Shanghai YingLi Pharmaceutical Co., Ltd. (Shanghai, China) | RECRUITING |
| NCT06607185 | LY4066434|standard treatment | KRAS | Eli Lilly and Company (Indianapolis, IN, USA) | NOT_YET_RECRUITING |
| NCT06447662 | PF-07934040|standard treatment | KRAS | Pfizer (New York, NY, USA) | RECRUITING |
| NCT06585488 | BGB-53038|Tislelizumab|Cetuximab | KRAS | BeiGene (Beiging, China) | NOT_YET_RECRUITING |
| NCT06445062 | RMC-6236|Standard treatment | KRAS | Revolution Medicines, Inc. (Redwood City, CA, USA) | RECRUITING |
| NCT05379985 | RMC-6236 | KRAS | Revolution Medicines, Inc. (Redwood City, CA, USA) | RECRUITING |
| RAF/MEK | ||||
| NCT05786924 | BDTX-4933 | RAF | Black Diamond Therapeutics, Inc. (Cambridge, MA, USA) | RECRUITING |
| NCT06194877 | BGB-3245|Panitumumab | RAF | MapKure, LLC (Stamford, CT, USA) | RECRUITING |
| NCT05200442 | VS-6766 | RAF/MEK | University of Chicago (Chicago, IL, USA) | RECRUITING |
| NCT06270082 | IK-595 | MEK/RAF | Ikena Oncology (Boston, MA, USA) | RECRUITING |
| NCT05163028 | HBI-2376 | SHP2 | HUYABIO International, LLC. San Diego, CA, USA) | RECRUITING |
| Cancer vaccines | ||||
| NCT04117087 | KRAS peptide vaccine|Nivolumab|Ipilimumab | KRAS vaccine | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (Adelaide, Australia) | RECRUITING |
| NCT04853017 | ELI-002 2P | KRAS vaccine | Elicio Therapeutics (Boston, MA, USA) | ACTIVE_NOT_RECRUITING |
| NCT06411691 | KRAS Vaccine|Balstilimab|Botensilimab | KRAS vaccine | Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (Adelaide, Australia) | NOT_YET_RECRUITING |
| NCT05726864 | ELI-002 7P | KRAS vaccine | Elicio Therapeutics (Boston, MA, USA) | RECRUITING |
| CART | ||||
| NCT06105021 | AFNT-211 | CART | Affini-T Therapeutics, Inc. (Boston, MA, USA) | RECRUITING |
| NCT06253520 | KRAS TCR-Transduced PBL | CART | National Cancer Institute (NCI) (Bethesda, MD, USA) | RECRUITING |
| NCT06487377 | IX001 TCR-T cells | CART | Shanghai Pudong Hospital (Shanghai, China) | RECRUITING |
| NCT06218914 | NT-112 | CART | Neogene Therapeutics, Inc. (Santa Monica, CA, USA) | RECRUITING |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boilève, A.; Smolenschi, C.; Lambert, A.; Boige, V.; Delaye, M.; Camilleri, G.M.; Tarabay, A.; Valéry, M.; Fuerea, A.; Pudlarz, T.; et al. KRAS, a New Target for Precision Medicine in Colorectal Cancer? Cancers 2024, 16, 3455. https://doi.org/10.3390/cancers16203455
Boilève A, Smolenschi C, Lambert A, Boige V, Delaye M, Camilleri GM, Tarabay A, Valéry M, Fuerea A, Pudlarz T, et al. KRAS, a New Target for Precision Medicine in Colorectal Cancer? Cancers. 2024; 16(20):3455. https://doi.org/10.3390/cancers16203455
Chicago/Turabian StyleBoilève, Alice, Cristina Smolenschi, Aurélien Lambert, Valérie Boige, Matthieu Delaye, Géraldine M. Camilleri, Anthony Tarabay, Marine Valéry, Alina Fuerea, Thomas Pudlarz, and et al. 2024. "KRAS, a New Target for Precision Medicine in Colorectal Cancer?" Cancers 16, no. 20: 3455. https://doi.org/10.3390/cancers16203455
APA StyleBoilève, A., Smolenschi, C., Lambert, A., Boige, V., Delaye, M., Camilleri, G. M., Tarabay, A., Valéry, M., Fuerea, A., Pudlarz, T., Mathieu, J. R. R., Jaulin, F., Hollebecque, A., & Ducreux, M. (2024). KRAS, a New Target for Precision Medicine in Colorectal Cancer? Cancers, 16(20), 3455. https://doi.org/10.3390/cancers16203455