
Combinational Pulsing of TAAs Enforces Dendritic Cell-Based Immunotherapy through T-Cell Proliferation and Interferon-γ Secretion in LLC1 Mouse Model

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Isolation and Generation of TAAs-Pulsed Dendritic Cells
2.3. Characterization
2.4. Apoptosis and Necrosis Assay
2.5. Splenic T-Cell Population Analysis
2.6. T-Cell Proliferation and IFN-γ ELISA Assay
2.7. Animal Management
2.8. Histology
2.9. Statistical Analysis
3. Results
3.1. Cytotoxicity Test for Combinational TAAs Pulsing
3.2. Anti-Cancer Effect of TAAs-Pulsed mDCs in LLC1 Mouse Model
3.3. Analysis of Mouse Splenic T-Cell Subpopulations
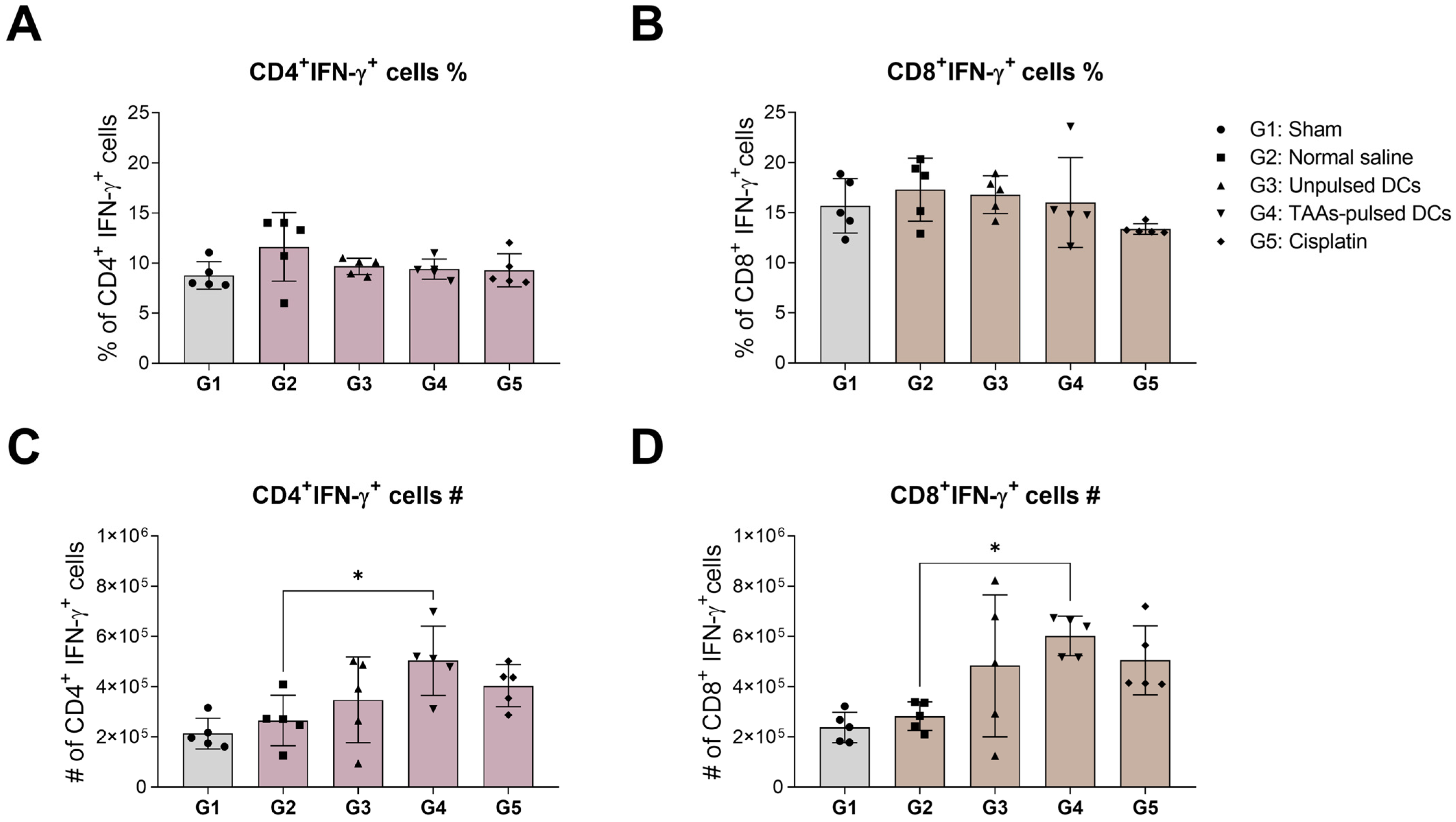
3.4. IFN-γ Expression in Mouse Splenic CD4+ and CD8+ T Cells
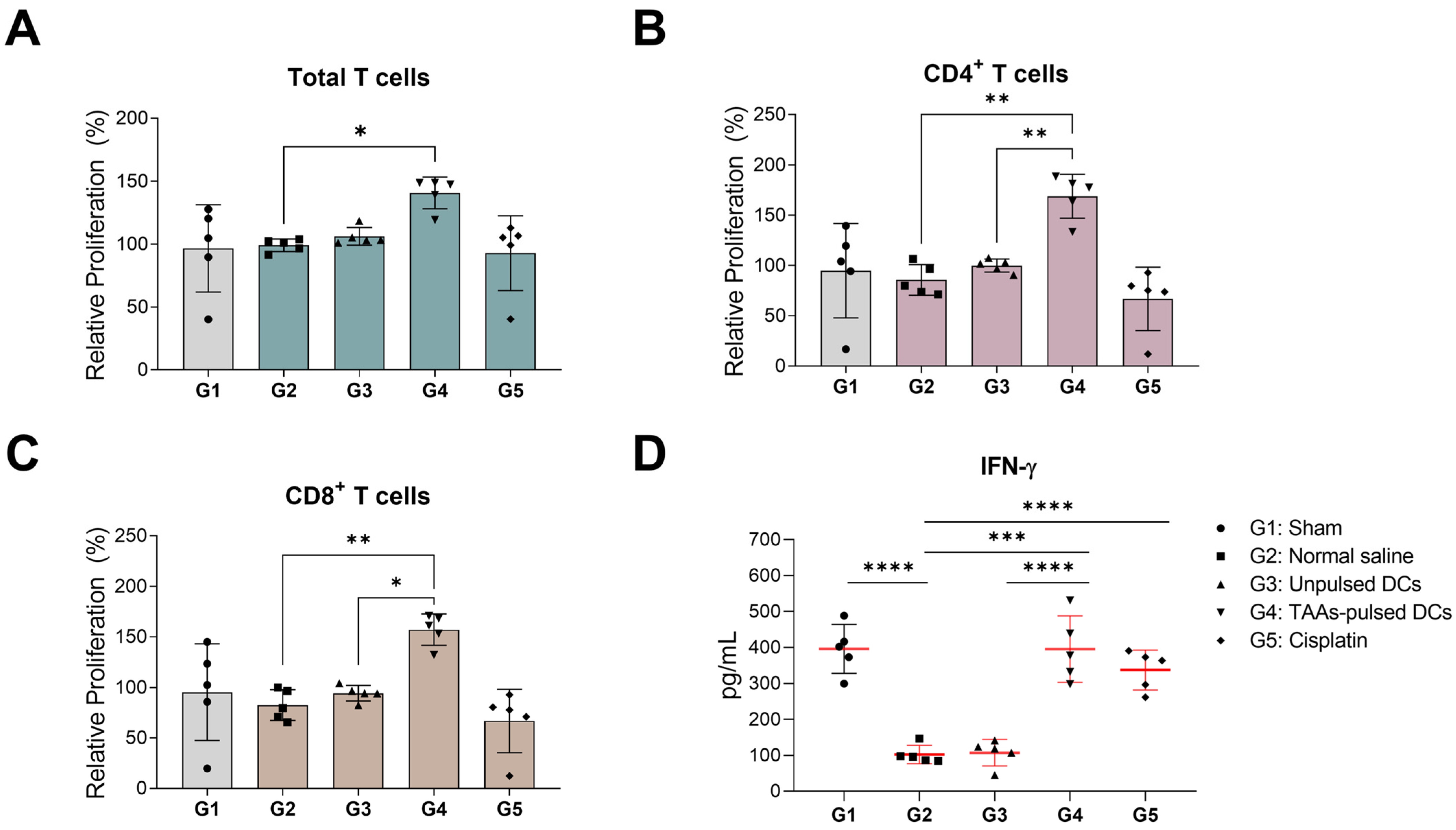
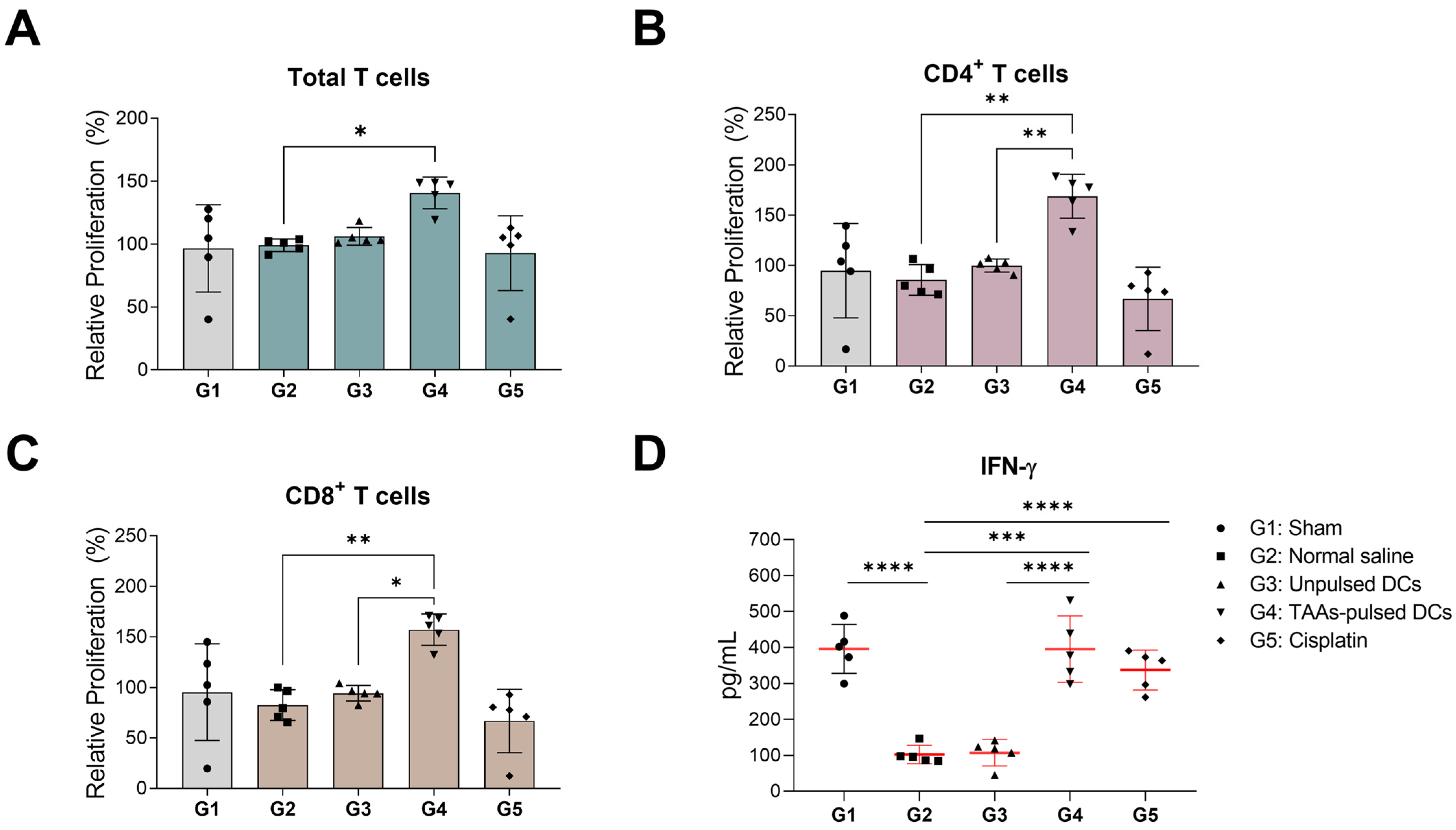
3.5. In Vitro Proliferation and IFN-γ Secretion of Mouse Splenocytes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ganti, A.K.; Klein, A.B.; Cotarla, I.; Seal, B.; Chou, E. Update of Incidence, Prevalence, Survival, and Initial Treatment in Patients With Non-Small Cell Lung Cancer in the US. JAMA Oncol. 2021, 7, 1824–1832. [Google Scholar] [CrossRef]
- Bouleftour, W.; Rowinski, E.; Louati, S.; Sotton, S.; Wozny, A.S.; Moreno-Acosta, P.; Mery, B.; Rodriguez-Lafrasse, C.; Magne, N. A Review of the Role of Hypoxia in Radioresistance in Cancer Therapy. Med. Sci. Monit. 2021, 27, e934116. [Google Scholar] [CrossRef]
- Pignon, J.P.; Tribodet, H.; Scagliotti, G.V.; Douillard, J.Y.; Shepherd, F.A.; Stephens, R.J.; Dunant, A.; Torri, V.; Rosell, R.; Seymour, L.; et al. Lung adjuvant cisplatin evaluation: A pooled analysis by the LACE Collaborative Group. J. Clin. Oncol. 2008, 26, 3552–3559. [Google Scholar] [CrossRef]
- Guo, Q.; Liu, L.; Chen, Z.; Fan, Y.; Zhou, Y.; Yuan, Z.; Zhang, W. Current treatments for non-small cell lung cancer. Front. Oncol. 2022, 12, 945102. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; de Vries, I.J.M.; Bol, K.F. Dendritic Cell Cancer Therapy: Vaccinating the Right Patient at the Right Time. Front. Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Cesaire, M.; Montanari, J.; Curcio, H.; Lerouge, D.; Gervais, R.; Demontrond, P.; Balosso, J.; Chevalier, F. Radioresistance of Non-Small Cell Lung Cancers and Therapeutic Perspectives. Cancers 2022, 14, 2829. [Google Scholar] [CrossRef]
- Palata, O.; Podzimkova Hradilova, N.; Mysikova, D.; Kutna, B.; Mrazkova, H.; Lischke, R.; Spisek, R.; Adkins, I. Detection of tumor antigens and tumor-antigen specific T cells in NSCLC patients: Correlation of the quality of T cell responses with NSCLC subtype. Immunol. Lett. 2020, 219, 46–53. [Google Scholar] [CrossRef]
- Saltos, A.; Khalil, F.; Smith, M.; Li, J.; Schell, M.; Antonia, S.J.; Gray, J.E. Clinical associations of mucin 1 in human lung cancer and precancerous lesions. Oncotarget 2018, 9, 35666–35675. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wu, C.; Xia, Y.; Zhong, Z.; Liu, X.; Xu, J.; Cui, F.; Chen, B.; Roe, O.D.; Li, A.; et al. WT1 promotes cell proliferation in non-small cell lung cancer cell lines through up-regulating cyclin D1 and p-pRb in vitro and in vivo. PLoS ONE 2013, 8, e68837. [Google Scholar] [CrossRef]
- Ma, T.; Gu, J.; Wen, H.; Xu, F.; Ge, D. BIRC5 Modulates PD-L1 Expression and Immune Infiltration in Lung Adenocarcinoma. J. Cancer 2022, 13, 3140–3150. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Heiser, A.; Boczkowski, D.; Majumdar, A.; Naoe, M.; Lebkowski, J.S.; Vieweg, J.; Gilboa, E. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat. Med. 2000, 6, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.; Ingels, J.; Van Lint, S.; Vandekerckhove, B.; Vermaelen, K. Dendritic Cell-Based Immunotherapy in Lung Cancer. Front. Immunol. 2020, 11, 620374. [Google Scholar] [CrossRef]
- Wagner, N.; Michiels, J.F.; Schedl, A.; Wagner, K.D. The Wilms’ tumour suppressor WT1 is involved in endothelial cell proliferation and migration: Expression in tumour vessels in vivo. Oncogene 2008, 27, 3662–3672. [Google Scholar] [CrossRef]
- Wagner, K.D.; Cherfils-Vicini, J.; Hosen, N.; Hohenstein, P.; Gilson, E.; Hastie, N.D.; Michiels, J.F.; Wagner, N. The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat. Commun. 2014, 5, 5852. [Google Scholar] [CrossRef]
- Lakshmanan, I.; Ponnusamy, M.P.; Macha, M.A.; Haridas, D.; Majhi, P.D.; Kaur, S.; Jain, M.; Batra, S.K.; Ganti, A.K. Mucins in lung cancer: Diagnostic, prognostic, and therapeutic implications. J. Thorac. Oncol. 2015, 10, 19–27. [Google Scholar] [CrossRef]
- Zhou, C.; Zhu, Y.; Lu, B.; Zhao, W.; Zhao, X. Survivin expression modulates the sensitivity of A549 lung cancer cells resistance to vincristine. Oncol. Lett. 2018, 16, 5466–5472. [Google Scholar] [CrossRef]
- Liu, N.; Guo, X.H.; Liu, J.P.; Cong, Y.S. Role of telomerase in the tumour microenvironment. Clin. Exp. Pharmacol. Physiol. 2020, 47, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Pineiro-Hermida, S.; Bosso, G.; Sanchez-Vazquez, R.; Martinez, P.; Blasco, M.A. Telomerase deficiency and dysfunctional telomeres in the lung tumor microenvironment impair tumor progression in NSCLC mouse models and patient-derived xenografts. Cell Death Differ. 2023, 30, 1585–1600. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, Y.; Zhang, S.; Kong, G.; Li, Z. Association between cellular immune response and spleen weight in mice with hepatocellular carcinoma. Oncol. Lett. 2021, 22, 625. [Google Scholar] [CrossRef] [PubMed]
- Abd Hamid, M.; Yao, X.; Waugh, C.; Rosendo-Machado, S.; Li, C.; Rostron, T.; Frankland, J.; Peng, Y.; Dong, T. Defective Interferon Gamma Production by Tumor-Specific CD8(+) T Cells Is Associated with 5′Methylcytosine-Guanine Hypermethylation of Interferon Gamma Promoter. Front. Immunol. 2020, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Mithoowani, H.; Febbraro, M. Non-Small-Cell Lung Cancer in 2022: A Review for General Practitioners in Oncology. Curr. Oncol. 2022, 29, 1828–1839. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Zhang, L.; Kandadi, H.; Yang, H.; Cham, J.; He, T.; Oh, D.Y.; Sheikh, N.A.; Fong, L. Long-term Sculpting of the B-cell Repertoire following Cancer Immunotherapy in Patients Treated with Sipuleucel-T. Cancer Immunol. Res. 2020, 8, 1496–1507. [Google Scholar] [CrossRef]
- Mullins, D.W.; Sheasley, S.L.; Ream, R.M.; Bullock, T.N.; Fu, Y.X.; Engelhard, V.H. Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J. Exp. Med. 2003, 198, 1023–1034. [Google Scholar] [CrossRef]
- Yoshitake, Y.; Fukuma, D.; Yuno, A.; Hirayama, M.; Nakayama, H.; Tanaka, T.; Nagata, M.; Takamune, Y.; Kawahara, K.; Nakagawa, Y.; et al. Phase II clinical trial of multiple peptide vaccination for advanced head and neck cancer patients revealed induction of immune responses and improved OS. Clin. Cancer Res. 2015, 21, 312–321. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-U.; Kim, S.-H.; Lee, S.-H.; Ji, M.-J.; Jin, J.-A.; So, H.-J.; Song, M.-L.; Lee, H.-K.; Kang, T.-W. Combinational Pulsing of TAAs Enforces Dendritic Cell-Based Immunotherapy through T-Cell Proliferation and Interferon-γ Secretion in LLC1 Mouse Model. Cancers 2024, 16, 409. https://doi.org/10.3390/cancers16020409
Lee J-U, Kim S-H, Lee S-H, Ji M-J, Jin J-A, So H-J, Song M-L, Lee H-K, Kang T-W. Combinational Pulsing of TAAs Enforces Dendritic Cell-Based Immunotherapy through T-Cell Proliferation and Interferon-γ Secretion in LLC1 Mouse Model. Cancers. 2024; 16(2):409. https://doi.org/10.3390/cancers16020409
Chicago/Turabian StyleLee, Jae-Ung, Sang-Heon Kim, Sung-Hoon Lee, Min-Jae Ji, Jeong-Ah Jin, Hyung-Joon So, Myoung-Lim Song, Hong-Ki Lee, and Tae-Wook Kang. 2024. "Combinational Pulsing of TAAs Enforces Dendritic Cell-Based Immunotherapy through T-Cell Proliferation and Interferon-γ Secretion in LLC1 Mouse Model" Cancers 16, no. 2: 409. https://doi.org/10.3390/cancers16020409
APA StyleLee, J.-U., Kim, S.-H., Lee, S.-H., Ji, M.-J., Jin, J.-A., So, H.-J., Song, M.-L., Lee, H.-K., & Kang, T.-W. (2024). Combinational Pulsing of TAAs Enforces Dendritic Cell-Based Immunotherapy through T-Cell Proliferation and Interferon-γ Secretion in LLC1 Mouse Model. Cancers, 16(2), 409. https://doi.org/10.3390/cancers16020409