



Comparative Study of the Immune Microenvironment in Heterotopic Tumor Models
 , , ,
, , ,  , and
, and
Abstract
Simple Summary
Abstract
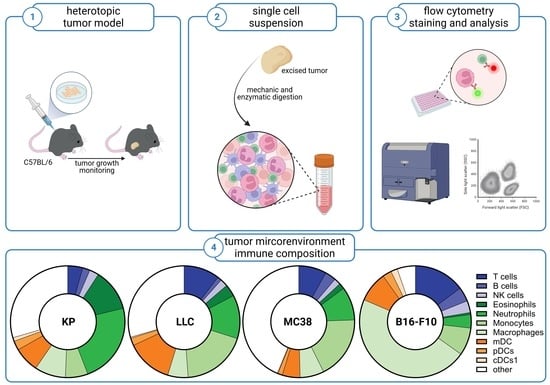
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Animal Studies and Tumor Models
2.3. Preparation of Single-Cell Suspensions
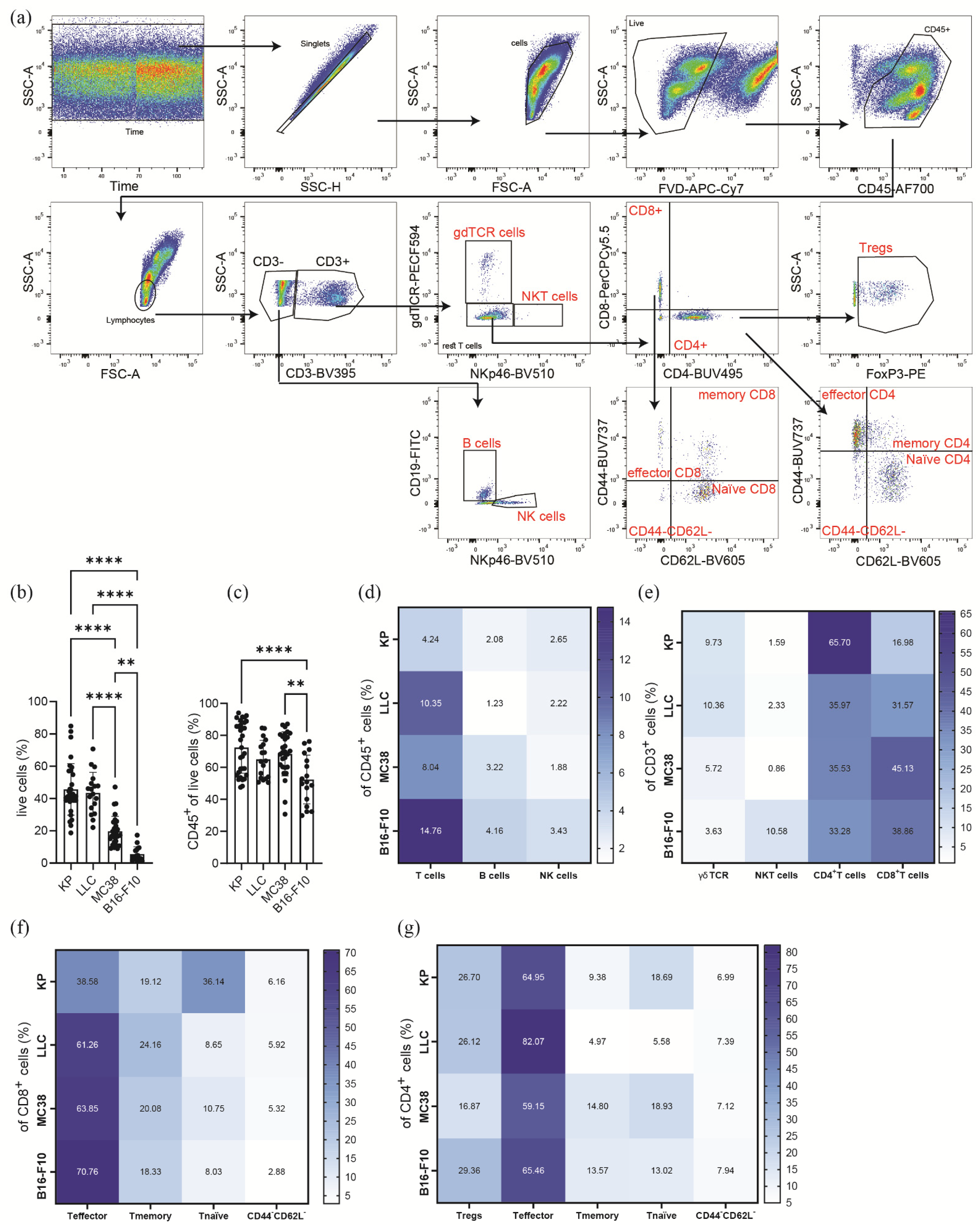
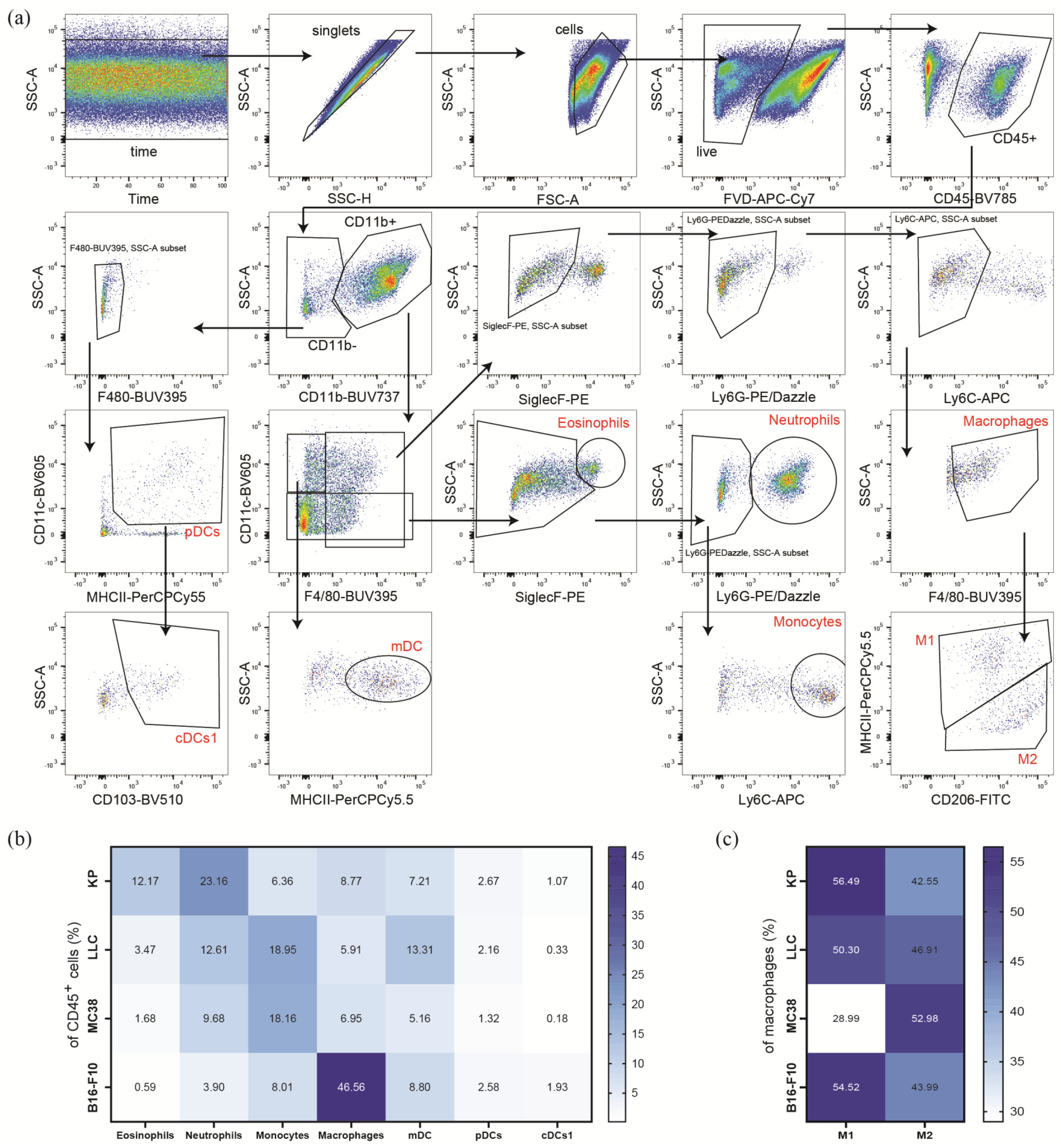
2.4. Flow Cytometric Phenotyping of Immune Cell Populations
2.5. Statistical Analysis
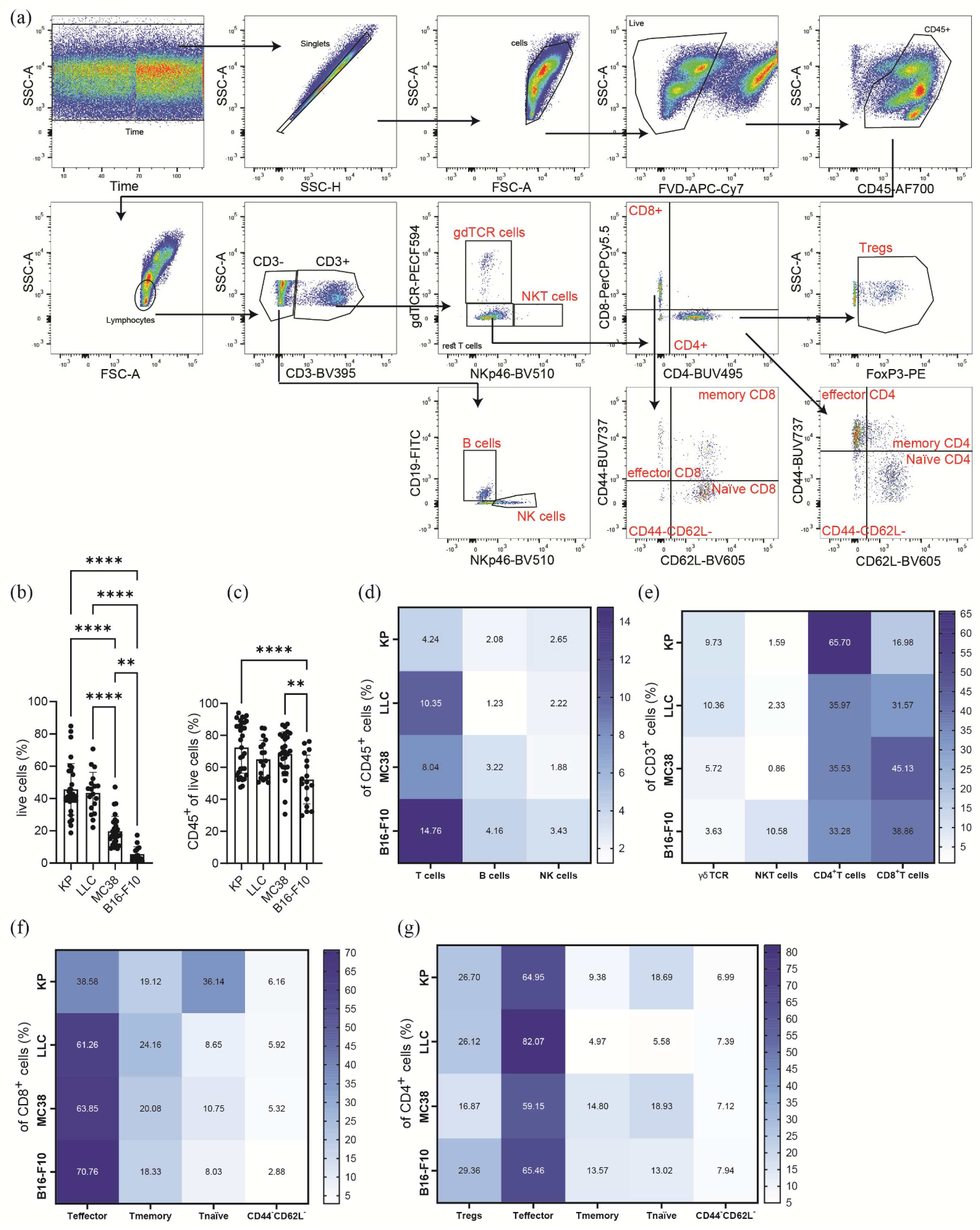
3. Results
3.1. Subcutaneous Tumors of Lung Cancer, Colon Cancer, and Melanoma Differ in Their Viability and Immune Cell Composition
3.2. Subcutaneous Tumors of Lung Cancer, Colon Cancer, and Melanoma Show Significantly Different Lymphoid Immune Cell Compositions of the TME
3.3. Subcutaneous Tumors of the Heterotopic Models Exhibit Distinct Compositions of Myeloid Immune Cells within Their TME
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virchow, R. As Based upon Physiological and Pathological Histology. Nutr. Rev. 1989, 47, 23–25. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem Cell Divisions, Somatic Mutations, Cancer Etiology, and Cancer Prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J.P. Targeting Epigenetic Readers in Cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune Surveillance of Tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, K.; Hana, D.; Chou, J.T.-T.; Singh, C.; Mackiewicz, A.; Kaczmarek, M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front. Immunol. 2021, 12, 1764. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.-H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The Evolving Tumor Microenvironment: From Cancer Initiation to Metastatic Outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Grisaru-Tal, S.; Itan, M.; Klion, A.D.; Munitz, A. A New Dawn for Eosinophils in the Tumour Microenvironment. Nat. Rev. Cancer 2020, 20, 594–607. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Xue, R.; Zhu, Z.; Farrukh, H.; Song, W.; Li, T.; Zheng, L.; Pan, C. Increasing Cure Rates of Solid Tumors by Immune Checkpoint Inhibitors. Exp. Hematol. Oncol. 2023, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Onoi, K.; Chihara, Y.; Uchino, J.; Shimamoto, T.; Morimoto, Y.; Iwasaku, M.; Kaneko, Y.; Yamada, T.; Takayama, K. Immune Checkpoint Inhibitors for Lung Cancer Treatment: A Review. J. Clin. Med. 2020, 9, 1362. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Hermel, D.J.; Sigal, D. The Emerging Role of Checkpoint Inhibition in Microsatellite Stable Colorectal Cancer. J. Pers. Med. 2019, 9, 5. [Google Scholar] [CrossRef]
- Tang, T.; Huang, X.; Zhang, G.; Hong, Z.; Bai, X.; Liang, T. Advantages of Targeting the Tumor Immune Microenvironment over Blocking Immune Checkpoint in Cancer Immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 72. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The Clinical Role of the TME in Solid Cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Zabransky, D.J.; Yarchoan, M.; Jaffee, E.M. Strategies for Heating Up Cold Tumors to Boost Immunotherapies. Annu. Rev. Cancer Biol. 2023, 7, 149–170. [Google Scholar] [CrossRef]
- Busch, S.E.; Hanke, M.L.; Kargl, J.; Metz, H.E.; MacPherson, D.; Houghton, A.M. Lung Cancer Subtypes Generate Unique Immune Responses. J. Immunol. 2016, 197, 4493–4503. [Google Scholar] [CrossRef]
- Kienzl, M.; Hasenoehrl, C.; Valadez-Cosmes, P.; Maitz, K.; Sarsembayeva, A.; Sturm, E.; Heinemann, A.; Kargl, J.; Schicho, R. IL-33 Reduces Tumor Growth in Models of Colorectal Cancer with the Help of Eosinophils. OncoImmunology 2020, 9, 1776059. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The Ever-Increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Busch, S.E.; Yang, G.H.Y.; Kim, K.-H.; Hanke, M.L.; Metz, H.E.; Hubbard, J.J.; Lee, S.M.; Madtes, D.K.; McIntosh, M.W.; et al. Neutrophils Dominate the Immune Cell Composition in Non-Small Cell Lung Cancer. Nat. Commun. 2017, 8, 14381. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Zhu, X.; Zhang, H.; Yang, G.H.Y.; Friesen, T.J.; Shipley, M.; Maeda, D.Y.; Zebala, J.A.; McKay-Fleisch, J.; Meredith, G.; et al. Neutrophil Content Predicts Lymphocyte Depletion and Anti-PD1 Treatment Failure in NSCLC. JCI Insight 2019, 4, e130850. [Google Scholar] [CrossRef]
- Valadez-Cosmes, P.; Maitz, K.; Kindler, O.; Mujkanovic, N.C.; Lueger, A.; Raftopoulou, S.; Kienzl, M.; Mihalic, Z.N.; Santiso, A.; Sarsembayeva, A.; et al. Myeloperoxidase Promotes a Tumorigenic Microenvironment in Non-Small Cell Lung Cancer. bioRxiv 2023. [Google Scholar] [CrossRef]
- Lechner, M.G.; Karimi, S.S.; Barry-Holson, K.; Angell, T.E.; Murphy, K.A.; Church, C.H.; Ohlfest, J.R.; Hu, P.; Epstein, A.L. Immunogenicity of Murine Solid Tumor Models as a Defining Feature of In Vivo Behavior and Response to Immunotherapy. J. Immunother. 2013, 36, 477–489. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Q.; Zhang, M.; Yu, Y.; Liu, X.; Cao, X. Fas Signal Promotes Lung Cancer Growth by Recruiting Myeloid-Derived Suppressor Cells via Cancer Cell-Derived PGE21. J. Immunol. 2009, 182, 3801–3808. [Google Scholar] [CrossRef]
- Chen, T.; Hu, R.; Wan, Y.; Sun, F.; Wang, Z.; Yue, J.; Chen, J.; Han, G.; Wei, G.; Dong, Z. Comprehensive Mutanome Analysis of Lewis Lung Cancer Reveals Immunogenic Neoantigens for Therapeutic Vaccines. Biochem. Biophys. Res. Commun. 2020, 525, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Agalioti, T.; Giannou, A.D.; Krontira, A.C.; Kanellakis, N.I.; Kati, D.; Vreka, M.; Pepe, M.; Spella, Μ.; Lilis, I.; Zazara, D.E.; et al. Mutant KRAS Promotes Malignant Pleural Effusion Formation. Nat. Commun. 2017, 8, 15205. [Google Scholar] [CrossRef]
- Li, H.Y.; McSharry, M.; Bullock, B.; Nguyen, T.T.; Kwak, J.; Poczobutt, J.M.; Sippel, T.R.; Heasley, L.E.; Weiser-Evans, M.C.; Clambey, E.T.; et al. The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol. Res. 2017, 5, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Y.; Shen, Y.; Qian, C.; Oupicky, D.; Sun, M. Targeting Pulmonary Tumor Microenvironment with CXCR4-Inhibiting Nanocomplex to Enhance Anti–PD-L1 Immunotherapy. Sci. Adv. 2020, 6, eaaz9240. [Google Scholar] [CrossRef]
- Jin, Y.; An, X.; Mao, B.; Sun, R.; Kumari, R.; Chen, X.; Shan, Y.; Zang, M.; Xu, L.; Muntel, J.; et al. Different Syngeneic Tumors Show Distinctive Intrinsic Tumor-Immunity and Mechanisms of Actions (MOA) of Anti-PD-1 Treatment. Sci. Rep. 2022, 12, 3278. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Myers, J.S.; Wang, F.; Wang, K.; Lucas, J.; Rosfjord, E.; Lucas, J.; Hooper, A.T.; Yang, S.; Lemon, L.A.; et al. Comparison of the Molecular and Cellular Phenotypes of Common Mouse Syngeneic Models with Human Tumors. BMC Genom. 2020, 21, 2. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Chen, L.; Souto, F.O.; Canasto-Chibuque, C.; Bongers, G.; Deshpande, M.; Harpaz, N.; Ko, H.M.; Kelley, K.; Furtado, G.C.; et al. Epithelial-Derived IL-33 Promotes Intestinal Tumorigenesis in Apc Min/+ Mice. Sci. Rep. 2017, 7, 5520. [Google Scholar] [CrossRef]
- Hasenoehrl, C.; Feuersinger, D.; Sturm, E.M.; Barnthaler, T.; Heitzer, E.; Graf, R.; Grill, M.; Pichler, M.; Beck, S.; Butcher, L.; et al. G Protein-Coupled Receptor GPR55 Promotes Colorectal Cancer and Has Opposing Effects to Cannabinoid Receptor 1. Int. J. Cancer 2018, 142, 121–132. [Google Scholar] [CrossRef]
- Lee, J.G.; Lee, Y.; Lee, A.; Park, C.H.; Han, D.S.; Eun, C.S. Role of the Global Gut Microbial Community in the Development of Colitis-Associated Cancer in a Murine Model. Biomed. Pharmacother. 2021, 135, 111206. [Google Scholar] [CrossRef]
- Jou, E.; Rodriguez-Rodriguez, N.; Ferreira, A.-C.F.; Jolin, H.E.; Clark, P.A.; Sawmynaden, K.; Ko, M.; Murphy, J.E.; Mannion, J.; Ward, C.; et al. An Innate IL-25–ILC2–MDSC Axis Creates a Cancer-Permissive Microenvironment for Apc Mutation–Driven Intestinal Tumorigenesis. Sci. Immunol. 2022, 7, eabn0175. [Google Scholar] [CrossRef]
- Guo, X.; Lei, R.; Zhou, Q.; Zhang, G.; Hu, B.; Liang, Y. Tumor Microenvironment Characterization in Colorectal Cancer to Identify Prognostic and Immunotherapy Genes Signature. BMC Cancer 2023, 23, 773. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yan, H.; Qiu, M.; Qu, X.; Wang, J.; Xu, S.; Zheng, Y.; Ge, M.; Yan, L.; Liang, L. Comprehensive Characterization of Tumor Microenvironment in Colorectal Cancer via Molecular Analysis. eLife 2023, 12, e86032. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; Vries, E.d.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495. [Google Scholar] [CrossRef] [PubMed]
- Lucarini, V.; Ziccheddu, G.; Macchia, I.; La Sorsa, V.; Peschiaroli, F.; Buccione, C.; Sistigu, A.; Sanchez, M.; Andreone, S.; D’Urso, M.T.; et al. IL-33 Restricts Tumor Growth and Inhibits Pulmonary Metastasis in Melanoma-Bearing Mice through Eosinophils. Oncoimmunology 2017, 6, e1317420. [Google Scholar] [CrossRef]
- Huang, L.; Chen, H.; Xu, Y.; Chen, J.; Liu, Z.; Xu, Q. Correlation of Tumor-infiltrating Immune Cells of Melanoma with Overall Survival by Immunogenomic Analysis. Cancer Med. 2020, 9, 8444–8456. [Google Scholar] [CrossRef]
- Courtney, A.N.; Tian, G.; Metelitsa, L.S. Natural Killer T Cells and Other Innate-like T Lymphocytes as Emerging Platforms for Allogeneic Cancer Cell Therapy. Blood 2023, 141, 869–876. [Google Scholar] [CrossRef]
- Ireson, C.R.; Alavijeh, M.S.; Palmer, A.M.; Fowler, E.R.; Jones, H.J. The Role of Mouse Tumour Models in the Discovery and Development of Anticancer Drugs. Br. J. Cancer 2019, 121, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, F.; Liu, R.; Shi, L.; Zhao, G.; Yan, Z. Gene Expression and Immune Infiltration in Melanoma Patients with Different Mutation Burden. BMC Cancer 2021, 21, 379. [Google Scholar] [CrossRef]
- Sarsembayeva, A.; Kienzl, M.; Gruden, E.; Ristic, D.; Maitz, K.; Valadez-Cosmes, P.; Santiso, A.; Hasenoehrl, C.; Brcic, L.; Lindenmann, J.; et al. Cannabinoid Receptor 2 Plays a Pro-Tumorigenic Role in Non-Small Cell Lung Cancer by Limiting Anti-Tumor Activity of CD8+ T and NK Cells. Front. Immunol. 2023, 13, 997115. [Google Scholar] [CrossRef]
- Kienzl, M.; Hasenoehrl, C.; Maitz, K.; Sarsembayeva, A.; Taschler, U.; Valadez-Cosmes, P.; Kindler, O.; Ristic, D.; Raftopoulou, S.; Santiso, A.; et al. Monoacylglycerol Lipase Deficiency in the Tumor Microenvironment Slows Tumor Growth in Non-Small Cell Lung Cancer. OncoImmunology 2021, 10, 1965319. [Google Scholar] [CrossRef]
- Carretero, R.; Sektioglu, I.M.; Garbi, N.; Salgado, O.C.; Beckhove, P.; Hämmerling, G.J. Eosinophils Orchestrate Cancer Rejection by Normalizing Tumor Vessels and Enhancing Infiltration of CD8+ T Cells. Nat. Immunol. 2015, 16, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lee, L.-F.; Fisher, T.S.; Jessen, B.; Elliott, M.; Evering, W.; Logronio, K.; Tu, G.H.; Tsaparikos, K.; Li, X.; et al. Combination of 4-1BB Agonist and PD-1 Antagonist Promotes Antitumor Effector/Memory CD8 T Cells in a Poorly Immunogenic Tumor Model. Cancer Immunol. Res. 2015, 3, 149–160. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kienzl, M.; Maitz, K.; Sarsembayeva, A.; Valadez-Cosmes, P.; Gruden, E.; Ristic, D.; Herceg, K.; Kargl, J.; Schicho, R. Comparative Study of the Immune Microenvironment in Heterotopic Tumor Models. Cancers 2024, 16, 295. https://doi.org/10.3390/cancers16020295
Kienzl M, Maitz K, Sarsembayeva A, Valadez-Cosmes P, Gruden E, Ristic D, Herceg K, Kargl J, Schicho R. Comparative Study of the Immune Microenvironment in Heterotopic Tumor Models. Cancers. 2024; 16(2):295. https://doi.org/10.3390/cancers16020295
Chicago/Turabian StyleKienzl, Melanie, Kathrin Maitz, Arailym Sarsembayeva, Paulina Valadez-Cosmes, Eva Gruden, Dusica Ristic, Karolina Herceg, Julia Kargl, and Rudolf Schicho. 2024. "Comparative Study of the Immune Microenvironment in Heterotopic Tumor Models" Cancers 16, no. 2: 295. https://doi.org/10.3390/cancers16020295
APA StyleKienzl, M., Maitz, K., Sarsembayeva, A., Valadez-Cosmes, P., Gruden, E., Ristic, D., Herceg, K., Kargl, J., & Schicho, R. (2024). Comparative Study of the Immune Microenvironment in Heterotopic Tumor Models. Cancers, 16(2), 295. https://doi.org/10.3390/cancers16020295