CYP26A1 Links WNT and Retinoic Acid Signaling: A Target to Differentiate ALDH+ Stem Cells in APC-Mutant CRC

 , ,
, ,
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Inducing wt-APC Expression
2.2. Cell Proliferation
2.3. WNT/β-Catenin Activity
2.4. NanoString Profiling
2.5. Western Blotting and Densitometry
2.6. Flow Cytometry and Fluorescence Activated Cell Sorting
2.7. Determining Synergistic, Additive, or Antagonistic Anti-Proliferative Effects of CYP26A1 and WNT Signaling Inhibitors
2.8. Patient Survival Studies
2.9. Statistical Analysis
3. Results
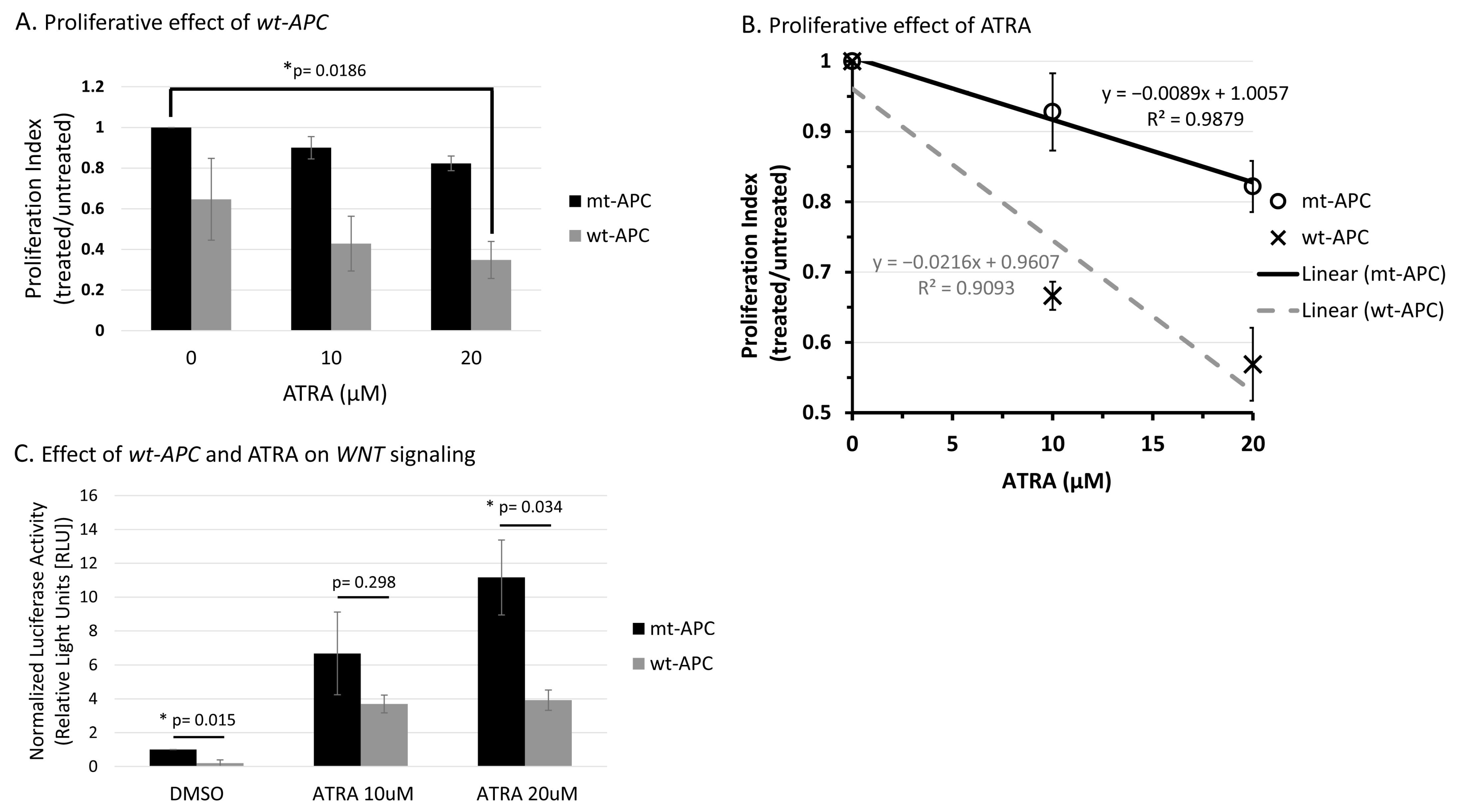
3.1. Inducing wt-APC Decreases WNT Signaling and Reduces Expression of WNT Target Genes
3.2. ATRA Promotes WNT/β-Catenin Activity, wt-APC Attenuates ATRA’s Effect
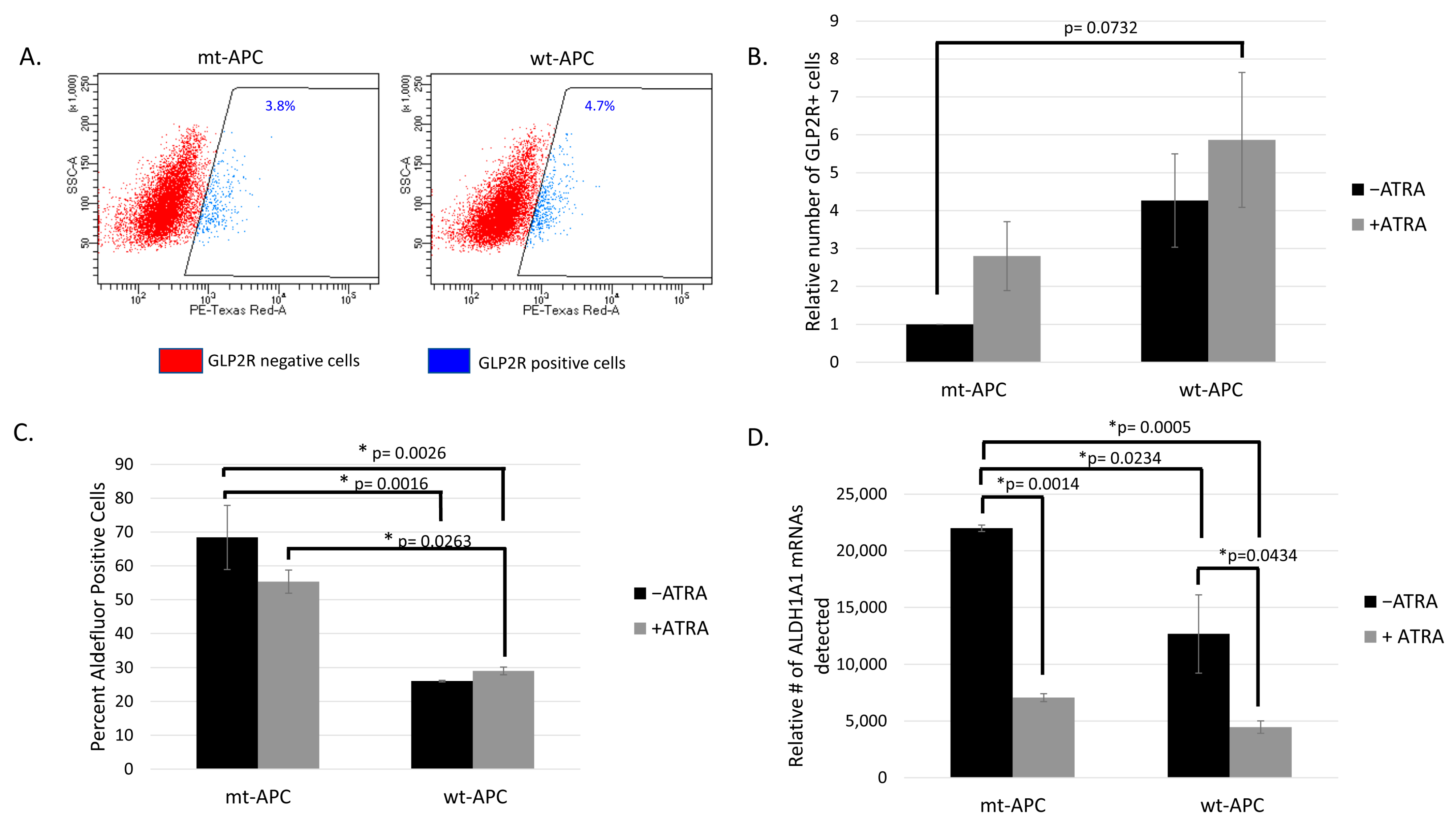
3.3. Inducing wt-APC Decreases ALDH+ Stem Cells and Increases NEC Differentiation
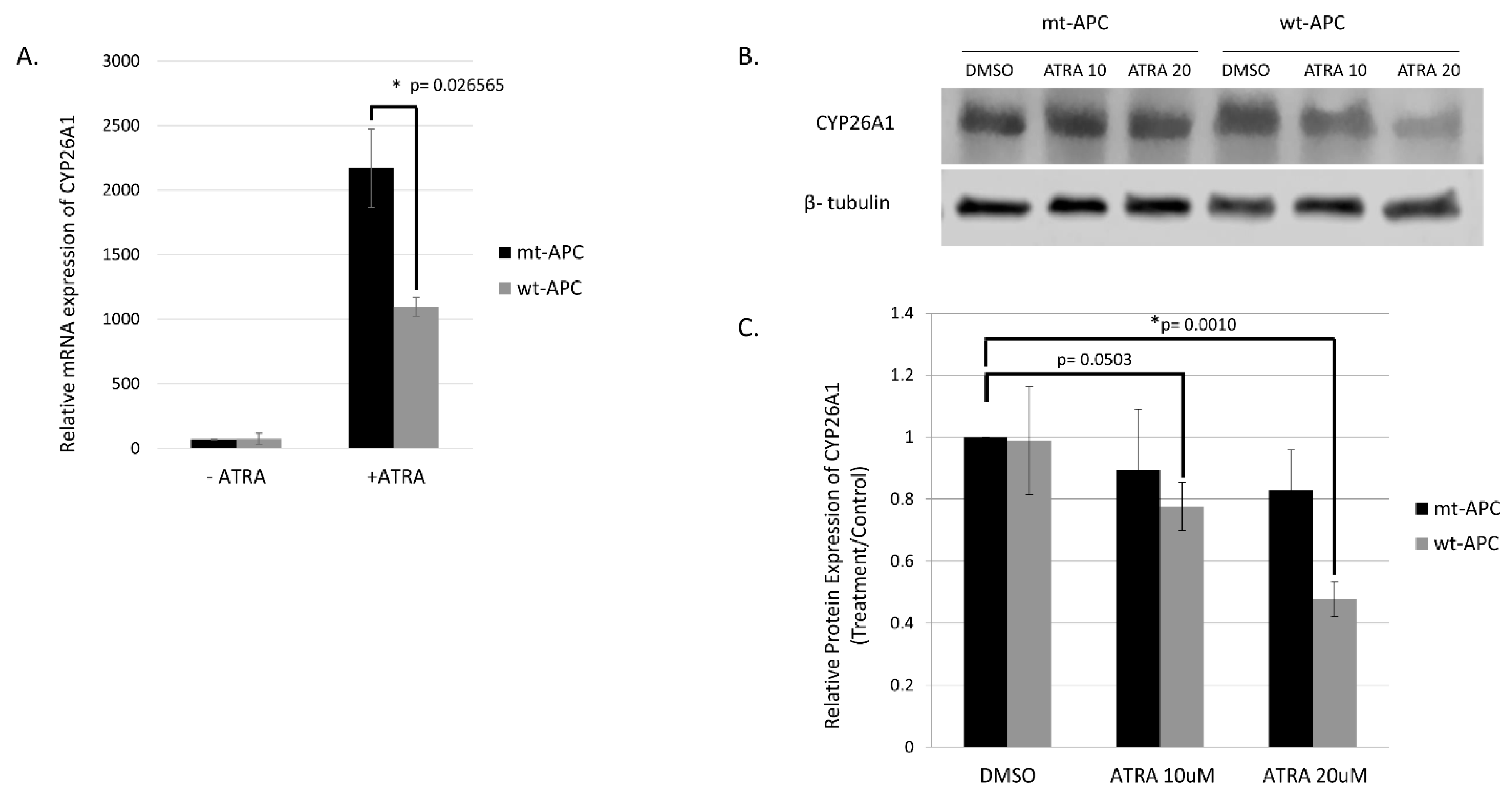
3.4. Expression Profiling Identified CYP26A1 as a Link between WNT and RA Signaling
3.5. CYP26A1 Inhibitor Agents Sensitize CRC Cells to the Anti-Proliferative Effect of Drugs That Downregulate WNT Signaling
3.6. Analysis of Human CRC Cases Indicates CYP26A1 Predicts Survival of Patients with Wild-type APC Tumors
4. Discussion
4.1. Main Findings of Our Study
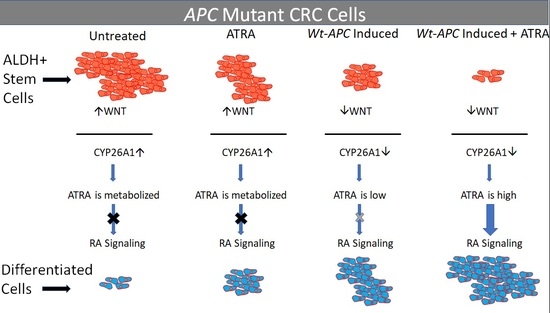
4.2. Studying the Effect of wt-APC on ATRA Response Indicates That WNT Signaling, via Its Target Gene CYP26A1, Regulates RA Signaling in the Differentiation of ALDH+ SCs
4.3. Studying the Effect of APC Mutation on NEC Differentiation Indicates That Decreased RA Signaling Contributes to the Overpopulation of ALDH+ SCs That Drives the Development of CRC
4.4. Clinical Significance of Our Results That Show CYP26A1 Inhibitors, Which Block RA Metabolism, Sensitize CRC Cells to the Anti-Proliferative Effect of Drugs That Downregulate WNT Signaling
4.5. Study of Human CRC Cases Demonstrates That CYP26A1 Predicts Patient Survival According to APC Genotype
4.6. Relations and Roles of CYP26A1 in the Context of Decreased RA Signaling and Increased ALDH+ SCs in APC-Mutant CRCs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Boman, B.M.; Huang, E. Human colon cancer stem cells: A new paradigm in gastrointestinal oncology. J. Clin. Oncol. 2008, 26, 2828–2838. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde Dehydrogenase 1 Is a Marker for Normal and Malignant Human Colonic Stem Cells (SC) and Tracks SC Overpopulation during Colon Tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Radford, I.R.; Lobachevsky, P.N. An enteroendocrine cell-based model for a quiescent intestinal stem cell niche. Cell Prolif. 2006, 39, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Ma, I.; Allan, A.L. The Role of Human Aldehyde Dehydrogenase in Normal and Cancer Stem Cells. Stem Cell Rev. Rep. 2010, 7, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-P450 Aldehyde Oxidizing Enzymes: The Aldehyde Dehydrogenase Superfamily. Expert Opin. Drug Metab. Toxicol. 2008, 4, 697–720. [Google Scholar] [CrossRef]
- Allahverdiyev, A.M.; Bagirova, M.; Nehir, O.; Yaman, S.; Sefik, E.; Cakir, R.; Canim, S.; Elcicek, S.; Yesilkir, S. Aldehyde Dehydrogenase: Cancer and Stem Cells. In Dehydrogenases; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Das, B.C.; Thapa, P.; Karki, R.; Das, S.; Mahapatra, S.; Liu, T.-C.; Torregroza, I.; Wallace, D.P.; Kambhampati, S.; Van Veldhuizen, P.; et al. Retinoic Acid Signaling Pathways in Development and Diseases. Bioorg. Med. Chem. 2014, 22, 673–683. [Google Scholar] [CrossRef]
- Zhang, T.; Ahn, K.; Emerick, B.; Modarai, S.R.; Opdenaker, L.M.; Palazzo, J.; Schleiniger, G.; Fields, J.Z.; Boman, B.M. APC mutations in human colon lead to decreased neuroendocrine maturation of ALDH+ stem cells that alters GLP-2 and SST feedback signaling: Clue to a link between WNT and retinoic acid signalling in colon cancer development. PLoS ONE 2020, 15, e0239601. [Google Scholar] [CrossRef]
- Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Apoptosis and APC in Colorectal Tumorigenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 7950–7954. [Google Scholar] [CrossRef]
- Groden, J.; Joslyn, G.; Samowitz, W.; Jones, D.; Bhattacharyya, N.; Spirio, L.; Thliveris, A.; Robertson, M.; Egan, S.; Meuth, M.; et al. Response of colon cancer cell lines to the introduction of APC, a colon-specific tumor suppressor gene. Cancer Res. 1995, 55, 1531–1539. [Google Scholar]
- Zhang, T.; Otevrel, T.; Gao, Z.; Gao, Z.; Ehrlich, S.M.; Fields, J.Z.; Boman, B.M. Evidence That APC Regulates Survivin Expression: A Possible Mechanism Contributing to the Stem Cell Origin of Colon Cancer. Cancer Res. 2001, 61, 8664–8667. [Google Scholar]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2008, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. APC restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Modarai, S.R.; Gupta, A.; Opdenaker, L.M.; Kowash, R.; Masters, G.; Viswanathan, V.; Zhang, T.; Fields, J.Z.; Boman, B.M. The anti-cancer effect of retinoic acid signaling in CRC occurs via decreased growth of ALDH+ colon cancer stem cells and increased differentiation of stem cells. Oncotarget 2018, 9, 34658–34669. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.W.; Palle, K. Aldehyde dehydrogenases in cancer stem cells: Potential as therapeutic targets. Ann. Transl. Med. 2016, 4, 518. [Google Scholar] [CrossRef] [PubMed]
- Hunsu, V.O.; Facey, C.O.B.; Fields, J.Z.; Boman, B.M. Retinoids as Chemo-Preventive and Molecular-Targeted Anti-Cancer Therapies. Int. J. Mol. Sci. 2021, 22, 7731. [Google Scholar] [CrossRef]
- Facey, C.O.B.; Boman, B.M. Retinoids in Treatment of Colorectal Cancer. In Colorectal Cancer; IntechOpen: Rijeka, Croatia, 2020; Available online: https://www.intechopen.com/online-first/retinoids-in-treatment-of-colorectal-cancer (accessed on 14 July 2021).
- Modarai, S.R.; Opdenaker, L.M.; Viswanathan, V.; Fields, J.Z.; Boman, B.M. Somatostatin Signaling via SSTR1 Contributes to the Quiescence of Colon Cancer Stem Cells. BMC Cancer 2016, 16, 941. [Google Scholar] [CrossRef] [PubMed]
- Boman, B.M.; Fields, J.Z. An APC:WNT Counter-Current-Like Mechanism Regulates Cell Division Along the Human Colonic Crypt Axis: A Mechanism That Explains How APC Mutations Induce Proliferative Abnormalities That Drive Colon Cancer Development. Front. Oncol. 2013, 3, 244. [Google Scholar] [CrossRef]
- Available online: https://Nanostring.Com/Products/Ncounter-Assays-Panels/Oncology/Ncounter-Pancancer-Pathways-Panel/ (accessed on 17 August 2023).
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein–Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Liu, Q.; Yin, X.; Languino, L.R.; Altieri, D.C. Evaluation of drug combination effect using a Bliss independence dose-response surface model. Stat. Biopharm. Res. 2018, 10, 112–122. [Google Scholar] [CrossRef]
- Available online: http://Web.Stanford.Edu/Group/Nusselab/Cgi-Bin/Wnt/ (accessed on 17 August 2023).
- Boon, E.M.J.; van der Neut, R.; van de Wetering, M.; Clevers, H.; Pals, S.T. Wnt Signaling Regulates Expression of the Receptor Tyrosine Kinase Met in Colorectal Cancer. Cancer Res. 2002, 62, 5126–5128. [Google Scholar]
- Mann, B.; Gelos, M.; Siedow, A.; Hanski, M.L.; Gratchev, A.; Ilyas, M.; Bodmer, W.F.; Moyer, M.P.; Riecken, E.O.; Buhr, H.J.; et al. Target Genes of β-Catenin–T Cell-Factor/Lymphoid-Enhancer-Factor Signaling in Human Colorectal Carcinomas. Proc. Natl. Acad. Sci. USA 1999, 96, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Wielenga, V.J.M.; Smits, R.; Korinek, V.; Smit, L.; Kielman, M.; Fodde, R.; Clevers, H.; Pals, S.T. Expression of CD44 in APC and Tcf Mutant Mice Implies Regulation by the WNT Pathway. Am. J. Pathol. 1999, 154, 515–523. [Google Scholar] [CrossRef] [PubMed]
- He, T.-C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c- MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Rennoll, S.; Yochum, G. Regulation of MYC gene expression by aberrant Wnt/β-catenin signaling in colorectal cancer. World J. Biol. Chem. 2015, 6, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://David.Ncifcrf.Gov/ (accessed on 27 August 2022).
- Shelton, D.N.; Sandoval, I.T.; Eisinger, A.; Chidester, S.; Ratnayake, A.; Ireland, C.M.; Jones, D.A. Up-Regulation of CYP26A1 in Adenomatous Polyposis Coli-Deficient Vertebrates via a WNT-Dependent Mechanism: Implications for Intestinal Cell Differentiation and Colon Tumor Development. Cancer Res. 2006, 66, 7571–7577. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat, B.; Mehta, K.; Lopez-Berestein, G. Regulation of a highly specific retinoic acid-4-hydroxylase (CYP26A1) enzyme and all-trans-retinoic acid metabolism in human intestinal, liver, endothelial, and acute promyelocytic leukemia cells. Leuk. Lymphoma 2005, 46, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar] [CrossRef]
- Rhinn, M.; Dollé, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef]
- Nadauld, L.D.; Sandoval, I.T.; Chidester, S.; Yost, H.J.; Jones, D.A. Adenomatous polyposis coli control of retinoic acid biosynthesis is critical for zebrafish intestinal development and differentiation. J. Biol. Chem. 2004, 279, 51581–51589. [Google Scholar] [CrossRef]
- Nadauld, L.D.; Chidester, S.; Shelton, D.N.; Rai, K.; Broadbent, T.; Sandoval, I.T.; Peterson, P.W.; Manos, E.J.; Ireland, C.M.; Yost, H.J.; et al. Dual roles for adenomatous polyposis coli in regulating retinoic acid biosynthesis and Wnt during ocular development. Proc. Natl. Acad. Sci. USA 2006, 103, 13409–13414. [Google Scholar] [CrossRef]
- Nelson, C.; Buttrick, B.; Isoherranen, N. Therapeutic potential of the inhibition of the retinoic acid hydroxylases CYP26A1 and CYP26B1 by xenobiotics. Curr. Top. Med. Chem. 2013, 13, 1402–1428. [Google Scholar] [CrossRef] [PubMed]
- Penny, H.L.; Prestwood, T.R.; Bhattacharya, N.; Sun, F.; Kenkel, J.A.; Davidson, M.G.; Shen, L.; Zuniga, L.A.; Seeley, E.S.; Pai, R.; et al. Restoring retinoic acid attenuates intestinal inflammation and tumorigenesis in APCMin/+ mice. Cancer Immunol. Res. 2016, 4, 917–926. [Google Scholar] [CrossRef]
- Brown, G.T.; Cash, B.G.; Blihoghe, D.; Johansson, P.; Alnabulsi, A.; Murray, G.I. The expression and prognostic significance of retinoic acid metabolising enzymes in colorectal cancer. PLoS ONE 2014, 9, e90776. [Google Scholar] [CrossRef] [PubMed]
- Osanai, M.; Takasawa, A.; Takasawa, K.; Kyuno, D.; Ono, Y.; Magara, K. Retinoic acid metabolism in cancer: Potential feasibility of retinoic acid metabolism blocking therapy. Med. Mol. Morphol. 2023, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stevison, F.; Jing, J.; Tripathy, S.; Isoherranen, N. Role of Retinoic Acid-Metabolizing Cytochrome P450s, CYP26, in Inflammation and Cancer. Adv. Pharmacol. 2015, 74, 373–412. [Google Scholar] [CrossRef] [PubMed]
- Idres, N.; Benoît, G.; Flexor, M.A.; Lanotte, M.; Chabot, G.G. Granulocytic differentiation of human NB4 promyelocytic leukemia cells induced by all-trans retinoic acid metabolites. Cancer Res. 2001, 61, 700–705. [Google Scholar] [PubMed]
- Zhu, J.-W.; Shi, X.G.; Chu, H.Y.; Tong, J.H.; Wang, Z.; Naoe, T.; Waxman, S.M.; Chen, S.; Chen, Z. Effect of retinoic acid isomers on proliferation, differentiation and PML relocalization in the APL cell line NB4. Leukemia 1995, 9, 302–309. [Google Scholar]
- Fang, J.; Chen, S.-J.; Tong, J.-H.; Wang, Z.-G.; Chen, G.-Q. Treatment of acute promyelocytic leukemia with ATRA and As2O3: A model of molecular-target-based cancer therapy. Cancer Biol. Ther. 2002, 1, 614–620. [Google Scholar] [CrossRef]
- Yilmaz, M.; Kantarjian, H.; Ravandi, F. Acute promyelocytic leukemia current treatment algorithms. Blood Cancer J. 2021, 11, 123. [Google Scholar] [CrossRef]
- Stahl, M.; Tallman, M.S. Acute Promyelocytic Leukemia (APL): Remaining challenges towards a cure for ALL. Leuk. Lymphoma 2019, 60, 3107–3115. [Google Scholar] [CrossRef]
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2011, 226, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.; Usuba, W.; Kohama, I.; Ochiya, T. Drug resistance driven by cancer stem cells and their niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Merhej, G.; Saravanan, S.; Chen, H. Cancer resistance to immunotherapy: What is the role of cancer stem cells? Cancer Drug Resist. 2022, 5, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Ramachandran, S.; Gupta, N.; Kaushik, I.; Srivastava, S.K. Cancer cells stemness: A doorstep to targeted therapy. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165424. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.M.; Zhang, J.G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Sig. Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A.P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165339. [Google Scholar] [CrossRef]
- Billingsley, J.L.; Yevdokimova, V.; Ayoub, K.; Benoit, Y.D. Colorectal cancer is borrowing blueprints from intestinal ontogenesis. Cancers 2023, 15, 4928. [Google Scholar] [CrossRef]
- Muralikrishnan, V.; Hurley, T.D.; Nephew, K.P. Targeting aldehyde dehydrogenases to eliminate cancer stem cells in gynecologic malignancies. Cancers 2020, 12, 961. [Google Scholar] [CrossRef]
- Eckerdt, F.; Platanias, L.C. Emerging role of glioma stem cells in mechanisms of therapy resistance. Cancers 2023, 15, 3458. [Google Scholar] [CrossRef]
- Frąszczak, K.; Barczyński, B. The role of cancer stem cell markers in ovarian cancer. Cancers 2024, 16, 40. [Google Scholar] [CrossRef]
- Tu, S.-M.; Aydin, A.M.; Maraboyina, S.; Chen, Z.; Singh, S.; Gokden, N.; Langford, T. Stem cell origin of cancer: Clinical implications for cancer immunity and immunotherapy. Cancers 2023, 15, 5385. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, A.; Snook, A.E.; Waldman, S.A. Stem cells as therapeutic targets in colorectal cancer. Per. Med. 2021, 18, 171–183. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Facey, C.O.B.; Hunsu, V.O.; Zhang, C.; Osmond, B.; Opdenaker, L.M.; Boman, B.M. CYP26A1 Links WNT and Retinoic Acid Signaling: A Target to Differentiate ALDH+ Stem Cells in APC-Mutant CRC. Cancers 2024, 16, 264. https://doi.org/10.3390/cancers16020264
Facey COB, Hunsu VO, Zhang C, Osmond B, Opdenaker LM, Boman BM. CYP26A1 Links WNT and Retinoic Acid Signaling: A Target to Differentiate ALDH+ Stem Cells in APC-Mutant CRC. Cancers. 2024; 16(2):264. https://doi.org/10.3390/cancers16020264
Chicago/Turabian StyleFacey, Caroline O. B., Victoria O. Hunsu, Chi Zhang, Brian Osmond, Lynn M. Opdenaker, and Bruce M. Boman. 2024. "CYP26A1 Links WNT and Retinoic Acid Signaling: A Target to Differentiate ALDH+ Stem Cells in APC-Mutant CRC" Cancers 16, no. 2: 264. https://doi.org/10.3390/cancers16020264
APA StyleFacey, C. O. B., Hunsu, V. O., Zhang, C., Osmond, B., Opdenaker, L. M., & Boman, B. M. (2024). CYP26A1 Links WNT and Retinoic Acid Signaling: A Target to Differentiate ALDH+ Stem Cells in APC-Mutant CRC. Cancers, 16(2), 264. https://doi.org/10.3390/cancers16020264