
Sensitive and Specific Analyses of Colorectal Cancer Recurrence through Multiplex superRCA Mutation Detection in Blood Plasma

 , , ,
, , ,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Tissue and Plasma Samples
2.2. DNA Preparation
2.3. superRCA Assay Principle
2.4. Pre-Amplification
2.5. superRCA Reactions
2.6. Flow Cytometry
2.7. Ethical Approval
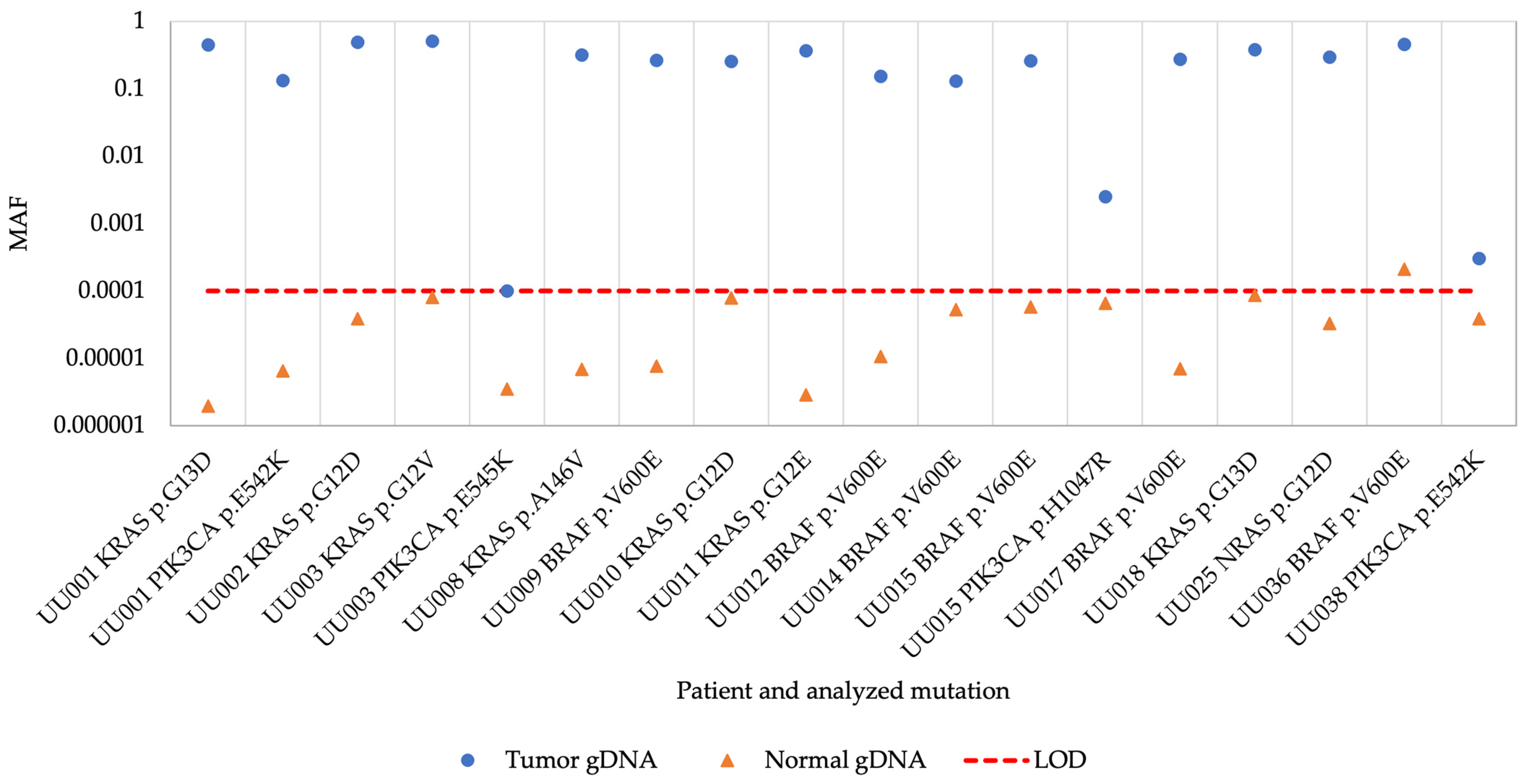
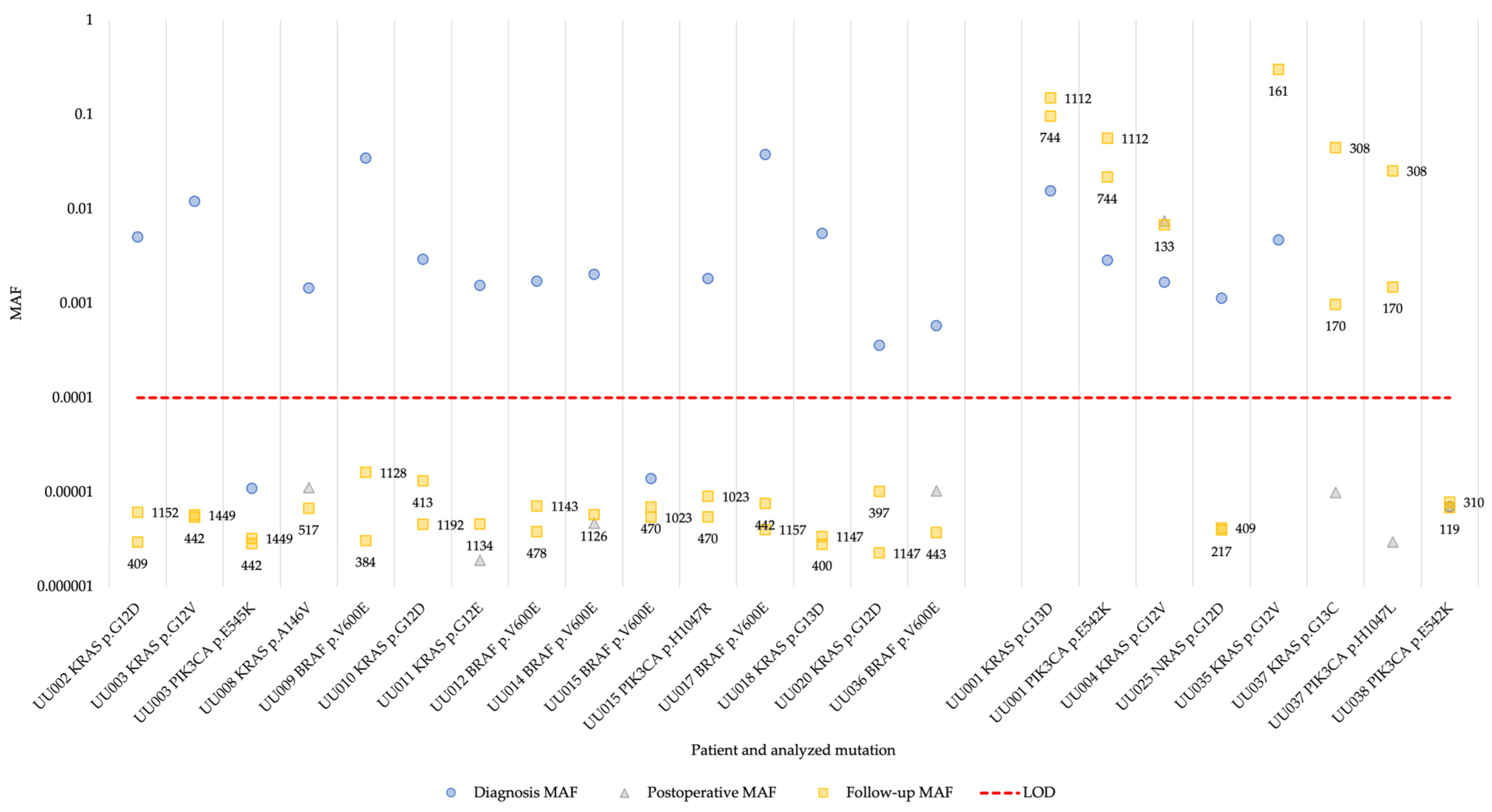
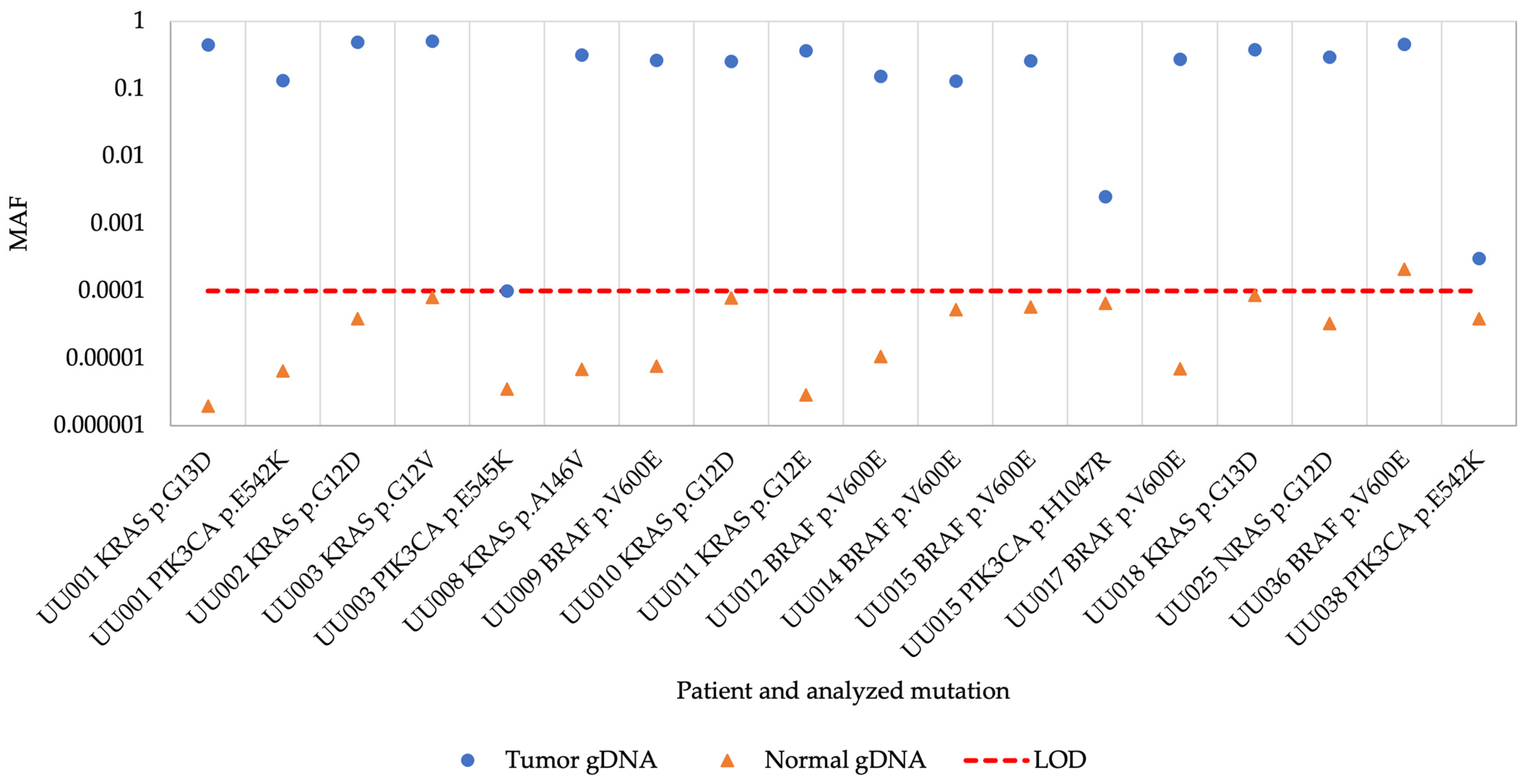
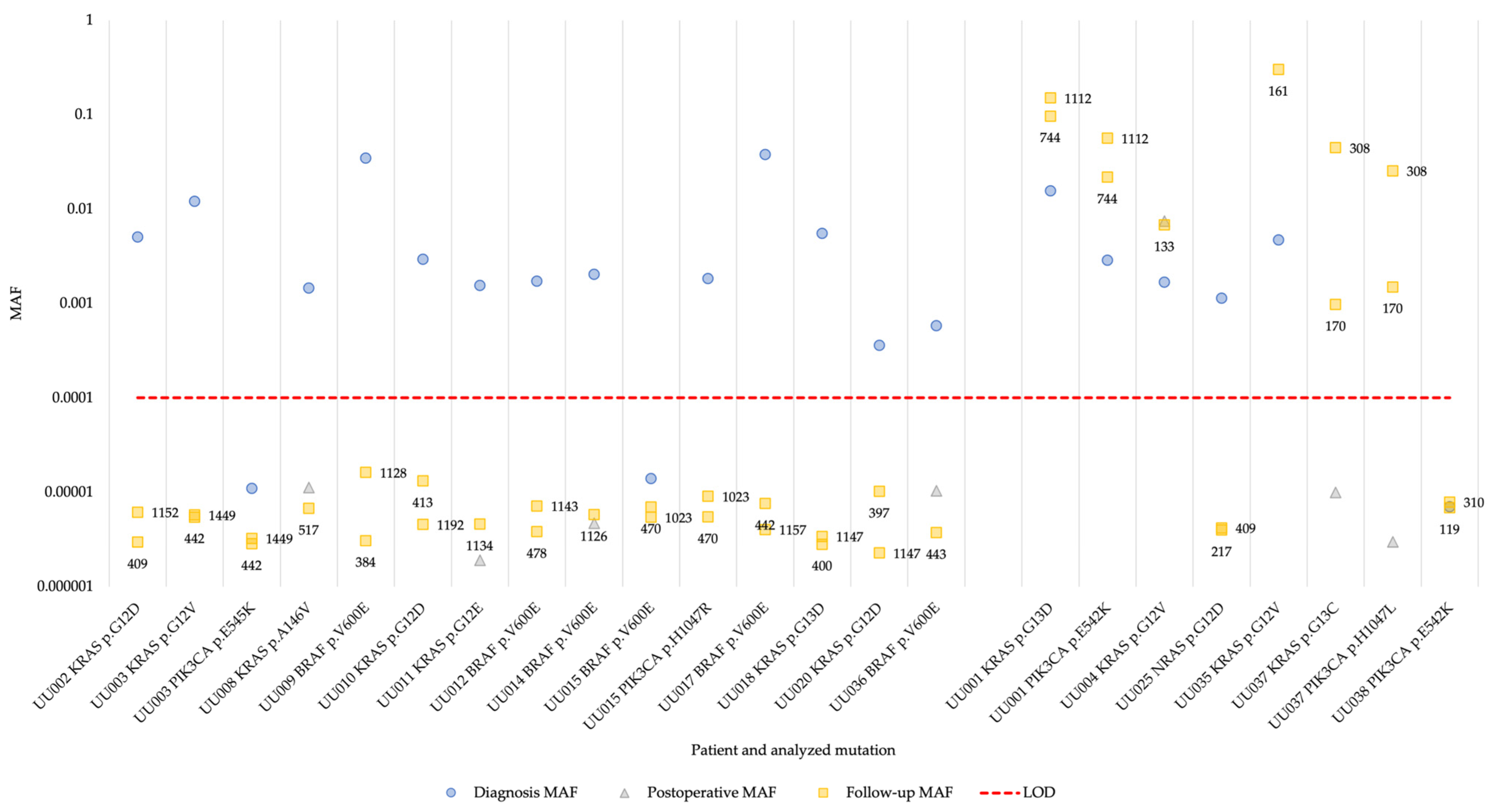
3. Results
3.1. Patient Cohort and cfDNA Purification
3.2. superRCA Analysis of Samples with Identified Hotspot Mutations
3.3. Multiplex superRCA Analysis of Wild-Type Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Osterman, E.; Glimelius, B. Recurrence Risk After Up-to-Date Colon Cancer Staging, Surgery, and Pathology: Analysis of the Entire Swedish Population. Dis. Colon. Rectum 2018, 61, 1016–1025. [Google Scholar] [CrossRef]
- Argilés, G.; Tabernero, J.; Labianca, R.; Hochhauser, D.; Salazar, R.; Iveson, T.; Laurent-Puig, P.; Quirke, P.; Yoshino, T.; Taieb, J.; et al. Localised colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef]
- McKiernan, J.M.; Buttyan, R.; Bander, N.H.; de la Taille, A.; Stifelman, M.D.; Emanuel, E.R.; Bagiella, E.; Rubin, M.A.; Katz, A.E.; Olsson, C.A.; et al. The detection of renal carcinoma cells in the peripheral blood with an enhanced reverse transcriptase-polymerase chain reaction assay for MN/CA9. Cancer 1999, 86, 492–497. [Google Scholar] [CrossRef]
- Loberg, R.D.; Fridman, Y.; Pienta, B.A.; Keller, E.T.; McCauley, L.K.; Taichman, R.S.; Pienta, K.J. Detection and isolation of circulating tumor cells in urologic cancers: A review. Neoplasia 2004, 6, 302–309. [Google Scholar] [CrossRef]
- Zygulska, A.L.; Pierzchalski, P. Novel Diagnostic Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 852. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, L.; Song, J.; Wang, G.; Li, P.; Li, W.; Luo, P.; Sun, X.; Wu, J.; Liu, Y.; et al. Liquid biopsy at the frontier of detection, prognosis and progression monitoring in colorectal cancer. Mol. Cancer 2022, 21, 86. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Yamaguchi, K.; Zembutsu, H. Clinical utility of circulating tumor DNA for colorectal cancer. Cancer Sci. 2019, 110, 1148–1155. [Google Scholar] [CrossRef]
- Taniguchi, H.; Nakamura, Y.; Kotani, D.; Yukami, H.; Mishima, S.; Sawada, K.; Shirasu, H.; Ebi, H.; Yamanaka, T.; Aleshin, A.; et al. CIRCULATE-Japan: Circulating tumor DNA-guided adaptive platform trials to refine adjuvant therapy for colorectal cancer. Cancer Sci. 2021, 112, 2915–2920. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Cohen, J.; Li, L.; Hong, W.; Christie, M.; Wong, H.L.; Kosmider, S.; Wong, R.; Thomson, B.; et al. Circulating tumor DNA dynamics and recurrence risk in patients undergoing curative intent resection of colorectal cancer liver metastases: A prospective cohort study. PLoS Med. 2021, 18, e1003620. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Boysen, A.K.; Pallisgaard, N.; Andersen, C.S.A.; Spindler, K.G. Circulating tumor DNA as a marker of minimal residual disease following local treatment of metastases from colorectal cancer. Acta Oncol. 2020, 59, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Zviran, A.; Schulman, R.C.; Shah, M.; Hill, S.T.K.; Deochand, S.; Khamnei, C.C.; Maloney, D.; Patel, K.; Liao, W.; Widman, A.J.; et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med. 2020, 26, 1114–1124. [Google Scholar] [CrossRef]
- Malla, M.; Loree, J.M.; Kasi, P.M.; Parikh, A.R. Using Circulating Tumor DNA in Colorectal Cancer: Current and Evolving Practices. J. Clin. Oncol. 2022, 40, 2846–2857. [Google Scholar] [CrossRef]
- Chen, L.; Eriksson, A.; Weström, S.; Pandzic, T.; Lehmann, S.; Cavelier, L.; Landegren, U. Ultra-sensitive monitoring of leukemia patients using superRCA mutation detection assays. Nat. Commun. 2022, 13, 4033. [Google Scholar] [CrossRef]
- Glimelius, B.; Melin, B.; Enblad, G.; Alafuzoff, I.; Beskow, A.; Ahlström, H.; Bill-Axelson, A.; Birgisson, H.; Björ, O.; Edqvist, P.H.; et al. U-CAN: A prospective longitudinal collection of biomaterials and clinical information from adult cancer patients in Sweden. Acta Oncol. 2018, 57, 187–194. [Google Scholar] [CrossRef]
- Dasari, A.; Morris, V.K.; Allegra, C.J.; Atreya, C.; Benson, A.B., 3rd; Boland, P.; Chung, K.; Copur, M.S.; Corcoran, R.B.; Deming, D.A.; et al. ctDNA applications and integration in colorectal cancer: An NCI Colon and Rectal-Anal Task Forces whitepaper. Nat. Rev. Clin. Oncol. 2020, 17, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Schøler, L.V.; Reinert, T.; Ørntoft, M.W.; Kassentoft, C.G.; Árnadóttir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445. [Google Scholar] [CrossRef] [PubMed]
- Regionala Cancercentrum i Samverkan. Nationellt Vårdprogram Tjock-och Ändtarmscancer. Available online: https://kunskapsbanken.cancercentrum.se/globalassets/cancerdiagnoser/tjock--och-andtarm-anal/vardprogram/nationellt-vardprogram-tjock-andtarmscancer.pdf (accessed on 9 June 2023).
- Wille-Jørgensen, P.; Syk, I.; Smedh, K.; Laurberg, S.; Nielsen, D.T.; Petersen, S.H.; Renehan, A.G.; Horváth-Puhó, E.; Påhlman, L.; Sørensen, H.T. Effect of More vs Less Frequent Follow-up Testing on Overall and Colorectal Cancer-Specific Mortality in Patients With Stage II or III Colorectal Cancer: The COLOFOL Randomized Clinical Trial. JAMA 2018, 319, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Van Seventer, E.E.; Siravegna, G.; Hartwig, A.V.; Jaimovich, A.; He, Y.; Kanter, K.; Fish, M.G.; Fosbenner, K.D.; Miao, B.; et al. Minimal Residual Disease Detection using a Plasma-only Circulating Tumor DNA Assay in Patients with Colorectal Cancer. Clin. Cancer Res. 2021, 27, 5586–5594. [Google Scholar] [CrossRef] [PubMed]
- Biagi, J.J.; Raphael, M.J.; Mackillop, W.J.; Kong, W.; King, W.D.; Booth, C.M. Association between time to initiation of adjuvant chemotherapy and survival in colorectal cancer: A systematic review and meta-analysis. JAMA 2011, 305, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, S.B.; Heo, S.; Lim, Y.; Lee, W.; Cho, S.H.; Ahn, J.; Kang, J.K.; Kim, S.Y.; Kim, H.P.; Bang, D.; et al. Personalised circulating tumour DNA assay with large-scale mutation coverage for sensitive minimal residual disease detection in colorectal cancer. Br. J. Cancer 2023, 129, 374–381. [Google Scholar] [CrossRef]
- Food and Drug Administration. Summary of Safety and Effectiveness Data (SSED) for Guardant360® CDx. Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf20/P200010S010B.pdf (accessed on 25 October 2023).
- Food and Drug Administration. Summary of Safety and Effectiveness Data (SSED) for FoundationOne® Liquid CDx. Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190032S005B.pdf (accessed on 25 October 2023).
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Henriksen, T.V.; Tarazona, N.; Frydendahl, A.; Reinert, T.; Gimeno-Valiente, F.; Carbonell-Asins, J.A.; Sharma, S.; Renner, D.; Hafez, D.; Roda, D.; et al. Circulating Tumor DNA in Stage III Colorectal Cancer, beyond Minimal Residual Disease Detection, toward Assessment of Adjuvant Therapy Efficacy and Clinical Behavior of Recurrences. Clin. Cancer Res. 2022, 28, 507–517. [Google Scholar] [CrossRef]
- Pascual, J.; Attard, G.; Bidard, F.C.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.D.; Montagut, C.; et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Patient ID | Stage | Recurrence | Hotspot Mutations | gDNA Availability | Plasma cfDNA Amount (ng) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Tumor | Normal | Diagnosis | Postoperative | Follow-Up | |||||
| UU001 | III | Yes | KRAS p.G13D + PIK3CA p.E542K | Yes | Yes | 35.7 | N/A | 27.0 | 18.0 |
| UU002 | III | No | KRAS p.G12D | Yes | Yes | 32.7 | N/A | 16.2 | 12.3 |
| UU003 | III | No | KRAS p.G12V + PIK3CA p.Q545K | Yes | Yes | 12.8 | N/A | 8.4 | 12.6 |
| UU004 | IV | Yes | KRAS p.G12V | No | No | 15.3 | 8.18 | 9.83 | N/A |
| UU005 | III | Yes | Wild-type | Yes | Yes | 24.9 | 17.4 | 41.7 | N/A |
| UU006 | III | Yes | Wild-type | No | No | 20.2 | N/A | 24.0 | 43.7 |
| UU007 | III | Yes | Wild-type | Yes | Yes | 15.9 | N/A | 12.3 | Leaky Tube |
| UU008 | II | No | KRAS p.A146V | Yes | Yes | 16.5 | 9.83 | 18.3 | N/A |
| UU009 | III | No | BRAF p.V600E | Yes | Yes | 19.3 | N/A | 10.5 | 17.7 |
| UU010 | III | No | KRAS p.G12D | Yes | Yes | 29.0 | N/A | 13.2 | 30.6 |
| UU011 | III | No | KRAS p.G12E | Yes | Yes | 29.0 | 16.8 | 10.5 | N/A |
| UU012 | III | No | BRAF p.V600E | Yes | Yes | 38.7 | N/A | 11.6 | 21.3 |
| UU013 | II | No | Wild-type | Yes | Yes | 16.5 | N/A | 21.3 | 28.2 |
| UU014 | III | No | BRAF p.V600E | Yes | Yes | 21.3 | 11.5 | 21.6 | N/A |
| UU015 | II | No | BRAF p.V600E + PIK3CA p.H1047R | Yes | Yes | 18.5 | N/A | 27.0 | 12.0 |
| UU016 | I | No | Wild-type | Yes | Yes | 15.5 | N/A | 30.9 | 20.4 |
| UU017 | II | No | BRAF p.V600E | Yes | Yes | 25.3 | N/A | 50.1 | 37.8 |
| UU018 | I | No | KRAS p.G13D | Yes | Yes | 19.6 | N/A | 13.8 | 23.4 |
| UU019 | II | No | Wild-type | Yes | Yes | 11.1 | 12.6 | 7.13 | N/A |
| UU020 | I | No | KRAS p.G12D | No | No | 32.1 | N/A | 20.4 | 20.7 |
| UU025 | IV | Yes | NRAS p.G12D | Yes | Yes | 35.7 | N/A | 32.1 | 26.7 |
| UU035 | III | Yes | KRAS p.G12V | No | No | 13.5 | N/A | N/A | 222.9 |
| UU036 | III | No | BRAF p.V600E | Yes | Yes | 16.5 | 27.0 | 15.9 | N/A |
| UU037 | III | Yes | KRAS p.G13C + PIK3CA p.H1047L | No | No | N/A | 24.0 | 18.0 | 23.4 |
| UU038 | III | Yes | PIK3CA p.E542K | Yes | Yes | 22.8 | N/A | 27.0 | 30.9 |
| Time from Surgery (Days) | |||||
|---|---|---|---|---|---|
| Patient ID | Diagnosis | Postoperative Plasma Sample | Follow-Up Plasma Samples | Recurrent Disease | |
| UU001 | −26 | N/A | +744 | +1112 | +286 |
| UU002 | −5 | N/A | +409 | +1152 | No |
| UU003 | −22 | N/A | +442 | +1449 | No |
| UU004 * | N/A | +42 | +133 | N/A | +130 |
| UU005 | −30 | +43 | +420 | N/A | +414 |
| UU006 | −110 | N/A | +113 | +868 | +382 |
| UU007 | −1 | N/A | +437 | +1214 | +1187 |
| UU008 | −30 | +60 | +517 | N/A | No |
| UU009 | −37 | N/A | +384 | +1128 | No |
| UU010 | −14 | N/A | +413 | +1192 | No |
| UU011 | −41 | +34 | +1134 | N/A | No |
| UU012 | −40 | N/A | +478 | +1143 | No |
| UU013 | −57 | N/A | +405 | +1127 | No |
| UU014 | −51 | +29 | +1126 | N/A | No |
| UU015 | −37 | N/A | +470 | +1023 | No |
| UU016 | −13 | N/A | +389 | +1115 | No |
| UU017 | −48 | N/A | +442 | +1157 | No |
| UU018 | −42 | N/A | +400 | +1147 | No |
| UU019 | −47 | +87 | +414 | N/A | No |
| UU020 | −68 | N/A | +397 | +1147 | No |
| UU025 | −68 | N/A | +217 | +409 | +422 |
| UU035 | −128 | N/A | +161 | N/A | +157 |
| UU036 | −103 | +93 | +443 | N/A | No |
| UU037 | N/A | +20 | +170 | +308 | +101 |
| UU038 | −104 | N/A | +119 | +310 | +289 |
| Characteristic | Cohort (%) |
|---|---|
| Sex | |
| Female | 10 (40%) |
| Male | 15 (60%) |
| Age at Diagnosis | |
| <75 | 15 (60%) |
| ≥75 | 10 (40%) |
| TNM stage | |
| I | 3 (12%) |
| II | 5 (20%) |
| III | 15 (60%) |
| IV | 2 (8%) |
| Primary Location | |
| Colon | 18 (72%) |
| Rectum | 7 (28%) |
| Malignancy Grade | |
| High Grade | 6 (24%) |
| Low Grade | 18 (72%) |
| NOS | 1 (4%) |
| Histology Subtype | |
| Adenocarcinoma | 20 (80%) |
| Mucinous Adenocarcinoma | 4 (16%) |
| NOS | 1 (4%) |
| Recurrent Disease | |
| Yes | 9 (36%) |
| No | 16 (64%) |
| Surgery | 24 (96%) |
| Neoadjuvant and Adjuvant Treatments | |
| Neoadjuvant Treatment | 5 (20%) |
| Chemotherapy | 1 (4%) |
| Radiotherapy | 1 (4%) |
| Chemoradiotherapy | 3 (12%) |
| Adjuvant Treatment | 11 (44%) |
| Chemotherapy | 11 (44%) |
| Radiotherapy | 0 |
| Chemoradiotherapy | 0 |
| Both Neoadjuvant and Adjuvant Treatment | 2 (8%) |
| No Neoadjuvant or Adjuvant Treatment | 11 (44%) |
| Palliative Treatment | 6 (24%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandberg, E.; Nunes, L.; Edqvist, P.-H.; Mathot, L.; Chen, L.; Edgren, T.; Al Nassralla, S.; Glimelius, B.; Landegren, U.; Sjöblom, T. Sensitive and Specific Analyses of Colorectal Cancer Recurrence through Multiplex superRCA Mutation Detection in Blood Plasma. Cancers 2024, 16, 549. https://doi.org/10.3390/cancers16030549
Sandberg E, Nunes L, Edqvist P-H, Mathot L, Chen L, Edgren T, Al Nassralla S, Glimelius B, Landegren U, Sjöblom T. Sensitive and Specific Analyses of Colorectal Cancer Recurrence through Multiplex superRCA Mutation Detection in Blood Plasma. Cancers. 2024; 16(3):549. https://doi.org/10.3390/cancers16030549
Chicago/Turabian StyleSandberg, Emma, Luís Nunes, Per-Henrik Edqvist, Lucy Mathot, Lei Chen, Tomas Edgren, Shahed Al Nassralla, Bengt Glimelius, Ulf Landegren, and Tobias Sjöblom. 2024. "Sensitive and Specific Analyses of Colorectal Cancer Recurrence through Multiplex superRCA Mutation Detection in Blood Plasma" Cancers 16, no. 3: 549. https://doi.org/10.3390/cancers16030549
APA StyleSandberg, E., Nunes, L., Edqvist, P.-H., Mathot, L., Chen, L., Edgren, T., Al Nassralla, S., Glimelius, B., Landegren, U., & Sjöblom, T. (2024). Sensitive and Specific Analyses of Colorectal Cancer Recurrence through Multiplex superRCA Mutation Detection in Blood Plasma. Cancers, 16(3), 549. https://doi.org/10.3390/cancers16030549