

Glutamine Metabolism and Prostate Cancer

 ,
,  , ,
, , 
Abstract
Simple Summary
Abstract

1. Glutamine Metabolism: An Overview
2. The Impact of Gln Metabolism on Tumor Immune Response
3. The Role of Gln Metabolism in Prostate Cancer Development and Progression
4. Targeting Glutamine Metabolism as a Strategy to Enhance Therapy Outcomes in Cancer
5. Clinical Trials Targeting Gln Metabolism in Prostate Cancer
6. The Interplay between Glutamine Metabolism and Androgen Signaling
7. The Impact of Gln Metabolism on Tumor Chemotherapy Resistance
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Sponagel, J.; Jones, J.K.; Frankfater, C.; Zhang, S.; Tung, O.; Cho, K.; Tinkum, K.L.; Gass, H.; Nunez, E.; Spitz, D.R.; et al. Sex differences in brain tumor glutamine metabolism reveal sex-specific vulnerabilities to treatment. Med 2022, 3, 792–811.e12. [Google Scholar] [CrossRef] [PubMed]
- Najumudeen, A.K.; Ceteci, F.; Fey, S.K.; Hamm, G.; Steven, R.T.; Hall, H.; Nikula, C.J.; Dexter, A.; Murta, T.; Race, A.M.; et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat. Genet. 2021, 53, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Encarnacion-Rosado, J.; Sohn, A.S.W.; Biancur, D.E.; Lin, E.Y.; Osorio-Vasquez, V.; Rodrick, T.; Gonzalez-Baerga, D.; Zhao, E.; Yokoyama, Y.; Simeone, D.M.; et al. Targeting pancreatic cancer metabolic dependencies through glutamine antagonism. Nat. Cancer 2023, 5, 85–89. [Google Scholar] [CrossRef]
- Kodama, M.; Toyokawa, G.; Sugahara, O.; Sugiyama, S.; Haratake, N.; Yamada, Y.; Wada, R.; Takamori, S.; Shimokawa, M.; Takenaka, T.; et al. Modulation of host glutamine anabolism enhances the sensitivity of small cell lung cancer to chemotherapy. Cell Rep. 2023, 42, 112899. [Google Scholar] [CrossRef]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Löck, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef]
- Nomoto, A.; Nishinami, S.; Shiraki, K. Solubility Parameters of Amino Acids on Liquid-Liquid Phase Separation and Aggregation of Proteins. Front. Cell Dev. Biol. 2021, 9, 691052. [Google Scholar] [CrossRef]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. Membrane transporters for the special amino acid glutamine: Structure/function relationships and relevance to human health. Front. Chem. 2014, 2, 61. [Google Scholar] [CrossRef]
- Bode, B.P. Recent molecular advances in mammalian glutamine transport. J. Nutr. 2001, 131, 2475S–2485S; discussion 2486S–2477S. [Google Scholar] [CrossRef]
- Wang, Q.; Tiffen, J.; Bailey, C.G.; Lehman, M.L.; Ritchie, W.; Fazli, L.; Metierre, C.; Feng, Y.J.; Li, E.; Gleave, M.; et al. Targeting amino acid transport in metastatic castration-resistant prostate cancer: Effects on cell cycle, cell growth, and tumor development. J. Natl. Cancer Inst. 2013, 105, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.S.; Salji, M.J.; Rushworth, L.; Ntala, C.; Rodriguez Blanco, G.; Hedley, A.; Clark, W.; Peixoto, P.; Hervouet, E.; Renaude, E.; et al. SLFN5 Regulates LAT1-Mediated mTOR Activation in Castration-Resistant Prostate Cancer. Cancer Res. 2021, 81, 3664–3678. [Google Scholar] [CrossRef]
- Xu, M.; Sakamoto, S.; Matsushima, J.; Kimura, T.; Ueda, T.; Mizokami, A.; Kanai, Y.; Ichikawa, T. Up-Regulation of LAT1 during Antiandrogen Therapy Contributes to Progression in Prostate Cancer Cells. J. Urol. 2016, 195, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, H.; Kimura, T.; Yamaga, T.; Kosaka, T.; Suehiro, J.I.; Sakurai, H. Prostate Cancer Cells in Different Androgen Receptor Status Employ Different Leucine Transporters. Prostate 2017, 77, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Broer, A.; Gauthier-Coles, G.; Rahimi, F.; van Geldermalsen, M.; Dorsch, D.; Wegener, A.; Holst, J.; Broer, S. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J. Biol. Chem. 2019, 294, 4012–4026. [Google Scholar] [CrossRef]
- Gauthier-Coles, G.; Fairweather, S.J.; Broer, A.; Broer, S. Do Amino Acid Antiporters Have Asymmetric Substrate Specificity? Biomolecules 2023, 13, 301. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine transporters as pharmacological targets: From function to drug design. Asian J. Pharm. Sci. 2020, 15, 207–219. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Console, L.; Losso, M.A.; Indiveri, C. The Human SLC1A5 (ASCT2) Amino Acid Transporter: From Function to Structure and Role in Cell Biology. Front. Cell Dev. Biol. 2018, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Morcillo, M.; Grande-Garcia, A.; Ruiz-Ramos, A.; Del Cano-Ochoa, F.; Boskovic, J.; Ramon-Maiques, S. Structural Insight into the Core of CAD, the Multifunctional Protein Leading De Novo Pyrimidine Biosynthesis. Structure 2017, 25, 912–923 e915. [Google Scholar] [CrossRef]
- Cory, J.G.; Cory, A.H. Critical roles of glutamine as nitrogen donors in purine and pyrimidine nucleotide synthesis: Asparaginase treatment in childhood acute lymphoblastic leukemia. In Vivo 2006, 20, 587–589. [Google Scholar] [PubMed]
- Smith, J.L.; Zaluzec, E.J.; Wery, J.P.; Niu, L.; Switzer, R.L.; Zalkin, H.; Satow, Y. Structure of the allosteric regulatory enzyme of purine biosynthesis. Science 1994, 264, 1427–1433. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, B.; Butler, W.; Xu, H.; Song, N.; Chen, X.; Spencer Hauck, J.; Gao, X.; Zhang, H.; Groth, J.; et al. Targeting glutamine metabolism network for the treatment of therapy-resistant prostate cancer. Oncogene 2022, 41, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Galluccio, M.; Console, L.; Pochini, L.; Indiveri, C. The Human SLC7A5 (LAT1): The Intriguing Histidine/Large Neutral Amino Acid Transporter and Its Relevance to Human Health. Front. Chem. 2018, 6, 243. [Google Scholar] [CrossRef]
- Deitmer, J.W.; Broer, A.; Broer, S. Glutamine efflux from astrocytes is mediated by multiple pathways. J. Neurochem. 2003, 87, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Elgadi, K.M.; Meguid, R.A.; Qian, M.; Souba, W.W.; Abcouwer, S.F. Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol. Genom. 1999, 1, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Cassago, A.; Ferreira, A.P.; Ferreira, I.M.; Fornezari, C.; Gomes, E.R.; Greene, K.S.; Pereira, H.M.; Garratt, R.C.; Dias, S.M.; Ambrosio, A.L. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 1092–1097. [Google Scholar] [CrossRef]
- Martin-Rufian, M.; Tosina, M.; Campos-Sandoval, J.A.; Manzanares, E.; Lobo, C.; Segura, J.A.; Alonso, F.J.; Mates, J.M.; Marquez, J. Mammalian glutaminase Gls2 gene encodes two functional alternative transcripts by a surrogate promoter usage mechanism. PLoS ONE 2012, 7, e38380. [Google Scholar] [CrossRef]
- Katt, W.P.; Lukey, M.J.; Cerione, R.A. A tale of two glutaminases: Homologous enzymes with distinct roles in tumorigenesis. Future Med. Chem. 2017, 9, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017, 358, 941–946. [Google Scholar] [CrossRef]
- Liu, G.; Zhu, J.; Yu, M.; Cai, C.; Zhou, Y.; Yu, M.; Fu, Z.; Gong, Y.; Yang, B.; Li, Y.; et al. Glutamate dehydrogenase is a novel prognostic marker and predicts metastases in colorectal cancer patients. J. Transl. Med. 2015, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Christa, L.; Simon, M.T.; Flinois, J.P.; Gebhardt, R.; Brechot, C.; Lasserre, C. Overexpression of glutamine synthetase in human primary liver cancer. Gastroenterology 1994, 106, 1312–1320. [Google Scholar] [CrossRef]
- Peng, S.; Wang, Z.; Tang, P.; Wang, S.; Huang, Y.; Xie, Q.; Wang, Y.; Tan, X.; Tang, T.; Yan, X.; et al. PHF8-GLUL axis in lipid deposition and tumor growth of clear cell renal cell carcinoma. Sci. Adv. 2023, 9, eadf3566. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Li, Z.; Xiao, L.; Hu, W.; Zhang, L.; Xie, B.; Zhou, Q.; He, J.; Qiu, Y.; Wen, M.; et al. Glutamine Synthetase Promotes Radiation Resistance via Facilitating Nucleotide Metabolism and Subsequent DNA Damage Repair. Cell Rep. 2019, 28, 1136–1143.e4. [Google Scholar] [CrossRef]
- Lomelino, C.L.; Andring, J.T.; McKenna, R.; Kilberg, M.S. Asparagine synthetase: Function, structure, and role in disease. J. Biol. Chem. 2017, 292, 19952–19958. [Google Scholar] [CrossRef]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.M.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef]
- Hewton, K.G.; Johal, A.S.; Parker, S.J. Transporters at the Interface between Cytosolic and Mitochondrial Amino Acid Metabolism. Metabolites 2021, 11, 112. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Choi, W.; Chen, Y.; Zhang, Q.; Deng, H.; He, W.; Shi, Y. A proposed role for glutamine in cancer cell growth through acid resistance. Cell Res. 2013, 23, 724–727. [Google Scholar] [CrossRef]
- Bell, H.N.; Huber, A.K.; Singhal, R.; Korimerla, N.; Rebernick, R.J.; Kumar, R.; El-Derany, M.O.; Sajjakulnukit, P.; Das, N.K.; Kerk, S.A.; et al. Microenvironmental ammonia enhances T cell exhaustion in colorectal cancer. Cell Metab. 2023, 35, 134–149 e136. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Wang, H.; Dong, D.; Li, T.; Yu, Z.; Guo, J.; Zhou, W.; Li, D.; Yan, R.; Wang, L.; et al. Urea as a By-Product of Ammonia Metabolism Can Be a Potential Serum Biomarker of Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2021, 9, 650748. [Google Scholar] [CrossRef] [PubMed]
- Won, J.H.; Parekattil, S.J.; Davidson, S.D.; Luddy, J.S.; Choudhury, M.S.; Mallouh, C.; Tazaki, H.; Konno, S. Ammonium-chloride-induced prostatic hypertrophy in vitro: Urinary ammonia as a potential risk factor for benign prostatic hyperplasia. Urol. Res. 1999, 27, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Casimir, M.; Lasorsa, F.M.; Rubi, B.; Caille, D.; Palmieri, F.; Meda, P.; Maechler, P. Mitochondrial glutamate carrier GC1 as a newly identified player in the control of glucose-stimulated insulin secretion. J. Biol. Chem. 2009, 284, 25004–25014. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Aguilar, M.; Baines, C.P. Physiological and pathological roles of mitochondrial SLC25 carriers. Biochem. J. 2013, 454, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Boudker, O. Symport and antiport mechanisms of human glutamate transporters. Nat. Commun. 2023, 14, 2579. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Xia, P.; Dubrovska, A. CD98 heavy chain as a prognostic biomarker and target for cancer treatment. Front. Oncol. 2023, 13, 1251100. [Google Scholar] [CrossRef]
- Yap, L.P.; Sancheti, H.; Ybanez, M.D.; Garcia, J.; Cadenas, E.; Han, D. Determination of GSH, GSSG, and GSNO using HPLC with electrochemical detection. Methods Enzym. 2010, 473, 137–147. [Google Scholar]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef]
- Perry, R.R.; Mazetta, J.A.; Levin, M.; Barranco, S.C. Glutathione levels and variability in breast tumors and normal tissue. Cancer 1993, 72, 783–787. [Google Scholar] [CrossRef]
- Wong, D.Y.; Hsiao, Y.L.; Poon, C.K.; Kwan, P.C.; Chao, S.Y.; Chou, S.T.; Yang, C.S. Glutathione concentration in oral cancer tissues. Cancer Lett. 1994, 81, 111–116. [Google Scholar] [CrossRef]
- Chen, Y.J.; Lu, C.T.; Lee, T.Y.; Chen, Y.J. dbGSH: A database of S-glutathionylation. Bioinformatics 2014, 30, 2386–2388. [Google Scholar] [CrossRef]
- Shin, E.; Kim, B.; Kang, H.; Lee, H.; Park, J.; Kang, J.; Park, E.; Jo, S.; Kim, H.Y.; Lee, J.S.; et al. Mitochondrial glutamate transporter SLC25A22 uni-directionally export glutamate for metabolic rewiring in radioresistant glioblastoma. Int. J. Biol. Macromol. 2023, 253, 127511. [Google Scholar] [CrossRef]
- Holmes, E.W.; Wyngaarden, J.B.; Kelley, W.N. Human glutamine phosphoribosylpyrophosphate amidotransferase. Two molecular forms interconvertible by purine ribonucleotides and phosphoribosylpyrophosphate. J. Biol. Chem. 1973, 248, 6035–6040. [Google Scholar] [CrossRef]
- Yelamanchi, S.D.; Jayaram, S.; Thomas, J.K.; Gundimeda, S.; Khan, A.A.; Singhal, A.; Keshava Prasad, T.S.; Pandey, A.; Somani, B.L.; Gowda, H. A pathway map of glutamate metabolism. J. Cell Commun. Signal 2016, 10, 69–75. [Google Scholar] [CrossRef]
- Yang, J.; Vitery, M.D.C.; Chen, J.; Osei-Owusu, J.; Chu, J.; Qiu, Z. Glutamate-Releasing SWELL1 Channel in Astrocytes Modulates Synaptic Transmission and Promotes Brain Damage in Stroke. Neuron 2019, 102, 813–827.e6. [Google Scholar] [CrossRef]
- Schousboe, A.; Bak, L.K.; Waagepetersen, H.S. Astrocytic Control of Biosynthesis and Turnover of the Neurotransmitters Glutamate and GABA. Front. Endocrinol. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.Q.; Li, C.; Stanley, C.A.; Smith, T.J. Glutamate Dehydrogenase, a Complex Enzyme at a Crucial Metabolic Branch Point. Neurochem. Res. 2019, 44, 117–132. [Google Scholar] [CrossRef]
- Holecek, M. Roles of malate and aspartate in gluconeogenesis in various physiological and pathological states. Metabolism 2023, 145, 155614. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 2014, 56, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Duran, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef]
- Dieterich, I.A.; Lawton, A.J.; Peng, Y.; Yu, Q.; Rhoads, T.W.; Overmyer, K.A.; Cui, Y.; Armstrong, E.A.; Howell, P.R.; Burhans, M.S.; et al. Acetyl-CoA flux regulates the proteome and acetyl-proteome to maintain intracellular metabolic crosstalk. Nat. Commun. 2019, 10, 3929. [Google Scholar] [CrossRef]
- Mews, P.; Donahue, G.; Drake, A.M.; Luczak, V.; Abel, T.; Berger, S.L. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 2017, 546, 381–386. [Google Scholar] [CrossRef]
- Helenius, I.T.; Madala, H.R.; Yeh, J.J. An Asp to Strike Out Cancer? Therapeutic Possibilities Arising from Aspartate’s Emerging Roles in Cell Proliferation and Survival. Biomolecules 2021, 11, 1666. [Google Scholar] [CrossRef]
- Beier, A.K.; Ebersbach, C.; Siciliano, T.; Scholze, J.; Hofmann, J.; Honscheid, P.; Baretton, G.B.; Woods, K.; Guezguez, B.; Dubrovska, A.; et al. Targeting the glutamine metabolism to suppress cell proliferation in mesenchymal docetaxel-resistant prostate cancer. Oncogene 2024, 43, 2038–2050. [Google Scholar] [CrossRef]
- Tran, K.A.; Dillingham, C.M.; Sridharan, R. The role of alpha-ketoglutarate-dependent proteins in pluripotency acquisition and maintenance. J. Biol. Chem. 2019, 294, 5408–5419. [Google Scholar] [CrossRef] [PubMed]
- Gaete, D.; Rodriguez, D.; Watts, D.; Sormendi, S.; Chavakis, T.; Wielockx, B. HIF-Prolyl Hydroxylase Domain Proteins (PHDs) in Cancer-Potential Targets for Anti-Tumor Therapy? Cancers 2021, 13, 988. [Google Scholar] [CrossRef]
- Tran, T.Q.; Ishak Gabra, M.B.; Lowman, X.H.; Yang, Y.; Reid, M.A.; Pan, M.; O’Connor, T.R.; Kong, M. Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLoS Biol. 2017, 15, e2002810. [Google Scholar] [CrossRef]
- Traube, F.R.; Ozdemir, D.; Sahin, H.; Scheel, C.; Gluck, A.F.; Geserich, A.S.; Oganesian, S.; Kostidis, S.; Iwan, K.; Rahimoff, R.; et al. Redirected nuclear glutamate dehydrogenase supplies Tet3 with alpha-ketoglutarate in neurons. Nat. Commun. 2021, 12, 4100. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Gajwani, P.; Jousma, J.; Miyamoto, H.; Kwon, Y.; Jana, A.; Toth, P.T.; Yan, G.; Ong, S.G.; Rehman, J. Nuclear translocation of mitochondrial dehydrogenases as an adaptive cardioprotective mechanism. Nat. Commun. 2023, 14, 4360. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Tommasini-Ghelfi, S.; Murnan, K.; Kouri, F.M.; Mahajan, A.S.; May, J.L.; Stegh, A.H. Cancer-associated mutation and beyond: The emerging biology of isocitrate dehydrogenases in human disease. Sci. Adv. 2019, 5, eaaw4543. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 2019, 1, 404–415. [Google Scholar] [CrossRef]
- Chandel, N.S. NADPH-The Forgotten Reducing Equivalent. Cold Spring Harb. Perspect. Biol. 2021, 13, a040550. [Google Scholar] [CrossRef]
- Song, J.; Sun, H.; Zhang, S.; Shan, C. The Multiple Roles of Glucose-6-Phosphate Dehydrogenase in Tumorigenesis and Cancer Chemoresistance. Life 2022, 12, 271. [Google Scholar] [CrossRef]
- Aurora, A.B.; Khivansara, V.; Leach, A.; Gill, J.G.; Martin-Sandoval, M.; Yang, C.; Kasitinon, S.Y.; Bezwada, D.; Tasdogan, A.; Gu, W.; et al. Loss of glucose 6-phosphate dehydrogenase function increases oxidative stress and glutaminolysis in metastasizing melanoma cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2120617119. [Google Scholar] [CrossRef]
- Polat, I.H.; Tarrado-Castellarnau, M.; Benito, A.; Hernandez-Carro, C.; Centelles, J.; Marin, S.; Cascante, M. Glutamine Modulates Expression and Function of Glucose 6-Phosphate Dehydrogenase via NRF2 in Colon Cancer Cells. Antioxidants 2021, 10, 1349. [Google Scholar] [CrossRef]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bai, C.; Ruan, Y.; Liu, M.; Chu, Q.; Qiu, L.; Yang, C.; Li, B. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 2019, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Morotti, M.; Bridges, E.; Valli, A.; Choudhry, H.; Sheldon, H.; Wigfield, S.; Gray, N.; Zois, C.E.; Grimm, F.; Jones, D.; et al. Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 12452–12461. [Google Scholar] [CrossRef] [PubMed]
- Horie, T.; Fukasawa, K.; Iezaki, T.; Park, G.; Onishi, Y.; Ozaki, K.; Kanayama, T.; Hiraiwa, M.; Kitaguchi, Y.; Kaneda, K.; et al. Hypoxic Stress Upregulates the Expression of Slc38a1 in Brown Adipocytes via Hypoxia-Inducible Factor-1alpha. Pharmacology 2018, 101, 64–71. [Google Scholar] [CrossRef]
- Elorza, A.; Soro-Arnaiz, I.; Melendez-Rodriguez, F.; Rodriguez-Vaello, V.; Marsboom, G.; de Carcer, G.; Acosta-Iborra, B.; Albacete-Albacete, L.; Ordonez, A.; Serrano-Oviedo, L.; et al. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell 2012, 48, 681–691. [Google Scholar] [CrossRef]
- Wang, B.; Pei, J.; Xu, S.; Liu, J.; Yu, J. A glutamine tug-of-war between cancer and immune cells: Recent advances in unraveling the ongoing battle. J. Exp. Clin. Cancer Res. 2024, 43, 74. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.K.; Park, M.; Lee, S.; Yun, J.W.; Lee, J.; Kim, J.S.; Cho, S.J.; Jeon, H.J.; Lee, I.K.; Choi, Y.K.; et al. Inhibition of Glutamine Utilization Synergizes with Immune Checkpoint Inhibitor to Promote Antitumor Immunity. Mol. Cell 2020, 80, 592–606.e8. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; You, Z.; Shi, H.; Sun, Y.; Du, X.; Palacios, G.; Guy, C.; Yuan, S.; Chapman, N.M.; Lim, S.A.; et al. SLC38A2 and glutamine signalling in cDC1s dictate anti-tumour immunity. Nature 2023, 620, 200–208. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Rais, R.; Lemberg, K.M.; Tenora, L.; Arwood, M.L.; Pal, A.; Alt, J.; Wu, Y.; Lam, J.; Aguilar, J.M.H.; Zhao, L.; et al. Discovery of DRP-104, a tumor-targeted metabolic inhibitor prodrug. Sci. Adv. 2022, 8, eabq5925. [Google Scholar] [CrossRef]
- Praharaj, M.; Shen, F.; Lee, A.J.; Zhao, L.; Nirschl, T.R.; Theodros, D.; Singh, A.K.; Wang, X.; Adusei, K.M.; Lombardo, K.A.; et al. Metabolic Reprogramming of Tumor-Associated Macrophages Using Glutamine Antagonist JHU083 Drives Tumor Immunity in Myeloid-Rich Prostate and Bladder Cancers. Cancer Immunol. Res. 2024, 12, 854–875. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.; LeBoeuf, S.E.; Hao, Y.; New, C.; Blum, J.L.E.; Rashidfarrokhi, A.; Huang, S.M.; Bahamon, C.; Wu, W.L.; Karadal-Ferrena, B.; et al. Glutamine antagonist DRP-104 suppresses tumor growth and enhances response to checkpoint blockade in KEAP1 mutant lung cancer. Sci. Adv. 2024, 10, eadm9859. [Google Scholar] [CrossRef]
- Li, Q.; Zhong, X.; Yao, W.; Yu, J.; Wang, C.; Li, Z.; Lai, S.; Qu, F.; Fu, X.; Huang, X.; et al. Inhibitor of glutamine metabolism V9302 promotes ROS-induced autophagic degradation of B7H3 to enhance antitumor immunity. J. Biol. Chem. 2022, 298, 101753. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shen, H.; Gu, W.; Zheng, H.; Wang, Y.; Ma, G.; Du, J. Prediction of prognosis, immunogenicity and efficacy of immunotherapy based on glutamine metabolism in lung adenocarcinoma. Front. Immunol. 2022, 13, 960738. [Google Scholar] [CrossRef]
- Huang, M.; Xiong, D.; Pan, J.; Zhang, Q.; Sei, S.; Shoemaker, R.H.; Lubet, R.A.; Montuenga, L.M.; Wang, Y.; Slusher, B.S.; et al. Targeting Glutamine Metabolism to Enhance Immunoprevention of EGFR-Driven Lung Cancer. Adv. Sci. 2022, 9, e2105885. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, M.; Shen, F.; Lee, A.J.; Zhao, L.; Nirschl, T.; Wang, X.; Theodros, D.; Singh, A.K.; Williams, R.A.; Sena, L.A.; et al. Abstract 6432: Glutamine antagonist prodrug JHU083 reprograms immunosuppressive tumor-associated macrophages (TAMs) to drive tumor immunity in urologic cancers. Cancer Res. 2023, 83, 6432. [Google Scholar] [CrossRef]
- Notarangelo, G.; Spinelli, J.B.; Perez, E.M.; Baker, G.J.; Kurmi, K.; Elia, I.; Stopka, S.A.; Baquer, G.; Lin, J.R.; Golby, A.J.; et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8+ T cell function. Science 2022, 377, 1519–1529. [Google Scholar] [CrossRef]
- Best, S.A.; Gubser, P.M.; Sethumadhavan, S.; Kersbergen, A.; Negron Abril, Y.L.; Goldford, J.; Sellers, K.; Abeysekera, W.; Garnham, A.L.; McDonald, J.A.; et al. Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metab. 2022, 34, 874–887.e6. [Google Scholar] [CrossRef]
- Peitzsch, C.; Gorodetska, I.; Klusa, D.; Shi, Q.; Alves, T.C.; Pantel, K.; Dubrovska, A. Metabolic regulation of prostate cancer heterogeneity and plasticity. Semin. Cancer Biol. 2022, 82, 94–119. [Google Scholar] [CrossRef] [PubMed]
- Skvortsov, S.; Skvortsova, I.I.; Tang, D.G.; Dubrovska, A. Concise Review: Prostate Cancer Stem Cells: Current Understanding. Stem Cells 2018, 36, 1457–1474. [Google Scholar] [CrossRef]
- Thiruvalluvan, M.; Billet, S.; Bhowmick, N.A. Antagonizing Glutamine Bioavailability Promotes Radiation Sensitivity in Prostate Cancer. Cancers 2022, 14, 2491. [Google Scholar] [CrossRef]
- Wang, Q.; Hardie, R.-A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef]
- Zacharias, N.M.; McCullough, C.; Shanmugavelandy, S.; Lee, J.; Lee, Y.; Dutta, P.; McHenry, J.; Nguyen, L.; Norton, W.; Jones, L.W.; et al. Metabolic Differences in Glutamine Utilization Lead to Metabolic Vulnerabilities in Prostate Cancer. Sci. Rep. 2017, 7, 16159. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Petrashen, A.P.; Sanders, J.A.; Peterson, A.L.; Sedivy, J.M. SLC1A5 glutamine transporter is a target of MYC and mediates reduced mTORC1 signaling and increased fatty acid oxidation in long-lived Myc hypomorphic mice. Aging Cell 2019, 18, e12947. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Lin, C.; Rajapakshe, K.; Dong, J.; Shi, Y.; Tsouko, E.; Mukhopadhyay, R.; Jasso, D.; Dawood, W.; Coarfa, C.; et al. Glutamine Transporters Are Targets of Multiple Oncogenic Signaling Pathways in Prostate Cancer. Mol. Cancer Res. 2017, 15, 1017–1028. [Google Scholar] [CrossRef]
- Yue, M.; Jiang, J.; Gao, P.; Liu, H.; Qing, G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep. 2017, 21, 3819–3832. [Google Scholar] [CrossRef]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef]
- Jenkins, R.B.; Qian, J.; Lieber, M.M.; Bostwick, D.G. Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res. 1997, 57, 524–531. [Google Scholar] [PubMed]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Boufaied, N.; Hallal, T.; Feit, A.; de Polo, A.; Luoma, A.M.; Alahmadi, W.; Larocque, J.; Zadra, G.; Xie, Y.; et al. MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets. Nat. Commun. 2022, 13, 2559. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Myint, Z.W.; Sun, R.C.; Hensley, P.J.; James, A.C.; Wang, P.; Strup, S.E.; McDonald, R.J.; Yan, D.; St Clair, W.H.; Allison, D.B. Evaluation of Glutaminase Expression in Prostate Adenocarcinoma and Correlation with Clinicopathologic Parameters. Cancers 2021, 13, 2157. [Google Scholar] [CrossRef] [PubMed]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Craze, M.L.; Jewa, N.; Oldfield, A.; Cheung, H.; Toss, M.; Rakha, E.A.; Green, A.R. The Biological and Clinical Significance of Glutaminase in Luminal Breast Cancer. Cancers 2021, 13, 3963. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Gao, L.; Wu, G.; Shen, G.; Xie, S.; Wen, H.; Yang, J.; Zhou, Y.; Tu, Z.; Qian, W. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem. Biophys. Res. Commun. 2015, 456, 452–458. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Cho, N.Y.; Choi, M.; Kim, B.H.; Cho, Y.M.; Moon, K.C.; Kang, G.H. BRAF and KRAS mutations in prostatic adenocarcinoma. Int. J. Cancer 2006, 119, 1858–1862. [Google Scholar] [CrossRef]
- Xu, L.; Yin, Y.; Li, Y.; Chen, X.; Chang, Y.; Zhang, H.; Liu, J.; Beasley, J.; McCaw, P.; Zhang, H.; et al. A glutaminase isoform switch drives therapeutic resistance and disease progression of prostate cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2012748118. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Y.; Zhao, W.; Liu, H.; Tu, H.; Xia, Z.; Wang, R.; Tang, J.; Zhu, C.; Li, R.; et al. A Five Glutamine-Associated Signature Predicts Prognosis of Prostate Cancer and Links Glutamine Metabolism with Tumor Microenvironment. J. Clin. Med. 2023, 12, 2243. [Google Scholar] [CrossRef]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J. Clin. Investig. 2018, 128, 4472–4484. [Google Scholar] [CrossRef] [PubMed]
- Valencia, T.; Kim, J.Y.; Abu-Baker, S.; Moscat-Pardos, J.; Ahn, C.S.; Reina-Campos, M.; Duran, A.; Castilla, E.A.; Metallo, C.M.; Diaz-Meco, M.T.; et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 2014, 26, 121–135. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Augsten, M.; Rundqvist, H.; Bianchi, J.; Sarne, V.; Egevad, L.; Bykov, V.J.; Ostman, A.; Wiman, K.G. Human cancer-associated fibroblasts enhance glutathione levels and antagonize drug-induced prostate cancer cell death. Cell Death Dis. 2017, 8, e2848. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Cojoc, M.; Mabert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef]
- Cojoc, M.; Peitzsch, C.; Kurth, I.; Trautmann, F.; Kunz-Schughart, L.A.; Telegeev, G.D.; Stakhovsky, E.A.; Walker, J.R.; Simin, K.; Lyle, S.; et al. Aldehyde Dehydrogenase Is Regulated by beta-Catenin/TCF and Promotes Radioresistance in Prostate Cancer Progenitor Cells. Cancer Res. 2015, 75, 1482–1494. [Google Scholar] [CrossRef]
- Peitzsch, C.; Cojoc, M.; Hein, L.; Kurth, I.; Mabert, K.; Trautmann, F.; Klink, B.; Schrock, E.; Wirth, M.P.; Krause, M.; et al. An Epigenetic Reprogramming Strategy to Resensitize Radioresistant Prostate Cancer Cells. Cancer Res. 2016, 76, 2637–2651. [Google Scholar] [CrossRef] [PubMed]
- Gorodetska, I.; Offermann, A.; Puschel, J.; Lukiyanchuk, V.; Gaete, D.; Kurzyukova, A.; Freytag, V.; Haider, M.T.; Fjeldbo, C.S.; Di Gaetano, S.; et al. ALDH1A1 drives prostate cancer metastases and radioresistance by interplay with AR- and RAR-dependent transcription. Theranostics 2024, 14, 714–737. [Google Scholar] [CrossRef]
- Puschel, J.; Dubrovska, A.; Gorodetska, I. The Multifaceted Role of Aldehyde Dehydrogenases in Prostate Cancer Stem Cells. Cancers 2021, 13, 4703. [Google Scholar] [CrossRef]
- Le Grand, M.; Mukha, A.; Puschel, J.; Valli, E.; Kamili, A.; Vittorio, O.; Dubrovska, A.; Kavallaris, M. Interplay between MycN and c-Myc regulates radioresistance and cancer stem cell phenotype in neuroblastoma upon glutamine deprivation. Theranostics 2020, 10, 6411–6429. [Google Scholar] [CrossRef]
- Mukha, A.; Kahya, U.; Dubrovska, A. Targeting glutamine metabolism and autophagy: The combination for prostate cancer radiosensitization. Autophagy 2021, 17, 3879–3881. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S.; Majumdar, S.; Azabdaftari, G.; Attwood, K.; Scioneaux, R.; Subramani, D.; Manhardt, C.; Lorusso, G.D.; Willard, S.S.; Thompson, H.; et al. Serum glutamate levels correlate with Gleason score and glutamate blockade decreases proliferation, migration, and invasion and induces apoptosis in prostate cancer cells. Clin. Cancer Res. 2012, 18, 5888–5901. [Google Scholar] [CrossRef] [PubMed]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Davidson, S.M.; Wirth, G.J.; Fiske, B.; Mayers, J.R.; Schwab, M.; Bellinger, G.; Csibi, A.; et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013, 73, 4429–4438. [Google Scholar] [CrossRef]
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904. [Google Scholar] [CrossRef]
- Zhang, J.; Mao, S.; Guo, Y.; Wu, Y.; Yao, X.; Huang, Y. Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci. Rep. 2019, 39, BSR20181826. [Google Scholar] [CrossRef]
- Chen, J.; Guccini, I.; Di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 2018, 50, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Bok, R.A.; DeLos Santos, J.; Upadhyay, D.; DeLos Santos, R.; Agarwal, S.; Van Criekinge, M.; Vigneron, D.B.; Aggarwal, R.; Peehl, D.M.; et al. Resistance to Androgen Deprivation Leads to Altered Metabolism in Human and Murine Prostate Cancer Cell and Tumor Models. Metabolites 2021, 11, 139. [Google Scholar] [CrossRef]
- Cardoso, H.J.; Figueira, M.I.; Vaz, C.V.; Carvalho, T.M.A.; Bras, L.A.; Madureira, P.A.; Oliveira, P.J.; Sardao, V.A.; Socorro, S. Glutaminolysis is a metabolic route essential for survival and growth of prostate cancer cells and a target of 5alpha-dihydrotestosterone regulation. Cell Oncol. 2021, 44, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Linder, S.J.; Bernasocchi, T.; Martinez-Pastor, B.; Sullivan, K.D.; Galbraith, M.D.; Lewis, C.A.; Ferrer, C.M.; Boon, R.; Silveira, G.G.; Cho, H.M.; et al. Inhibition of the proline metabolism rate-limiting enzyme P5CS allows proliferation of glutamine-restricted cancer cells. Nat. Metab. 2023, 5, 2131–2147. [Google Scholar] [CrossRef]
- Beier, A.K.; Puhr, M.; Stope, M.B.; Thomas, C.; Erb, H.H.H. Metabolic changes during prostate cancer development and progression. J. Cancer Res. Clin. Oncol. 2023, 149, 2259–2270. [Google Scholar] [CrossRef]
- Eagle, H.; Washington, C.L.; Levy, M.; Cohen, L. The population-dependent requirement by cultured mammalian cells for metabolites which they can synthesize. II. Glutamic acid and glutamine; aspartic acid and asparagine. J. Biol. Chem. 1966, 241, 4994–4999. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Byun, J.-K.; Choi, Y.-K.; Park, K.-G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [CrossRef]
- Lyons, S.D.; Sant, M.E.; Christopherson, R.I. Cytotoxic mechanisms of glutamine antagonists in mouse L1210 leukemia. J. Biol. Chem. 1990, 265, 11377–11381. [Google Scholar] [CrossRef]
- Ahluwalia, G.S.; Grem, J.L.; Hao, Z.; Cooney, D.A. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol. Ther. 1990, 46, 243–271. [Google Scholar] [CrossRef] [PubMed]
- Broer, A.; Rahimi, F.; Broer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–13205. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832. [Google Scholar] [CrossRef]
- Moon, D.; Hauck, J.S.; Jiang, X.; Quang, H.; Xu, L.; Zhang, F.; Gao, X.; Wild, R.; Everitt, J.I.; Macias, E.; et al. Targeting glutamine dependence with DRP-104 inhibits proliferation and tumor growth of castration-resistant prostate cancer. Prostate 2024, 84, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.M.; McBryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.V.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 2007, 406, 407–414. [Google Scholar] [CrossRef]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Sappington, D.R.; Siegel, E.R.; Hiatt, G.; Desai, A.; Penney, R.B.; Jamshidi-Parsian, A.; Griffin, R.J.; Boysen, G. Glutamine drives glutathione synthesis and contributes to radiation sensitivity of A549 and H460 lung cancer cell lines. Biochim. Biophys. Acta 2016, 1860, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Xie, G.; Liu, C.; Zhou, J.; Chen, J.; Yu, S.; Li, J.; Pang, X.; Shi, H.; Liang, H. Knock-down of glutaminase 2 expression decreases glutathione, NADH, and sensitizes cervical cancer to ionizing radiation. Biochim. Biophys. Acta. 2013, 1833, 2996–3005. [Google Scholar] [CrossRef]
- Boysen, G.; Jamshidi-Parsian, A.; Davis, M.A.; Siegel, E.R.; Simecka, C.M.; Kore, R.A.; Dings, R.P.M.; Griffin, R.J. Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int. J. Radiat. Biol. 2019, 95, 436–442. [Google Scholar] [CrossRef]
- Rashmi, R.; Jayachandran, K.; Zhang, J.; Menon, V.; Muhammad, N.; Zahner, M.; Ruiz, F.; Zhang, S.; Cho, K.; Wang, Y.; et al. Glutaminase Inhibitors Induce Thiol-Mediated Oxidative Stress and Radiosensitization in Treatment-Resistant Cervical Cancers. Mol. Cancer Ther. 2020, 19, 2465–2475. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef]
- Wicker, C.A.; Hunt, B.G.; Krishnan, S.; Aziz, K.; Parajuli, S.; Palackdharry, S.; Elaban, W.R.; Wise-Draper, T.M.; Mills, G.B.; Waltz, S.E.; et al. Glutaminase inhibition with telaglenastat (CB-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett. 2021, 502, 180–188. [Google Scholar] [CrossRef]
- Eskandari, R.; Kim, N.; Mamakhanyan, A.; Saoi, M.; Zhang, G.; Berisaj, M.; Granlund, K.L.; Poot, A.J.; Cross, J.; Thompson, C.B.; et al. Hyperpolarized [5-(13)C,4,4-(2)H(2),5-(15)N]-L-glutamine provides a means of annotating in vivo metabolic utilization of glutamine. Proc. Natl. Acad. Sci. USA 2022, 119, e2120595119. [Google Scholar] [CrossRef]
- Lee, C.H.; Motzer, R.; Emamekhoo, H.; Matrana, M.; Percent, I.; Hsieh, J.J.; Hussain, A.; Vaishampayan, U.; Liu, S.; McCune, S.; et al. Telaglenastat plus Everolimus in Advanced Renal Cell Carcinoma: A Randomized, Double-Blinded, Placebo-Controlled, Phase II ENTRATA Trial. Clin. Cancer Res. 2022, 28, 3248–3255. [Google Scholar] [CrossRef]
- Tannir, N.M.; Agarwal, N.; Porta, C.; Lawrence, N.J.; Motzer, R.; McGregor, B.; Lee, R.J.; Jain, R.K.; Davis, N.; Appleman, L.J.; et al. Efficacy and Safety of Telaglenastat Plus Cabozantinib vs Placebo Plus Cabozantinib in Patients with Advanced Renal Cell Carcinoma: The CANTATA Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1411–1418. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Zhong, X.; He, Z.; Yin, L.; Fan, Y.; Tong, Y.; Kang, Y.; Bi, Q. Glutamine metabolism in tumor metastasis: Genes, mechanisms and the therapeutic targets. Heliyon 2023, 9, e20656. [Google Scholar] [CrossRef]
- Lam, E.T.; Su, L.-J.; Salzmann-Sullivan, M.; Nordeen, S.K.; Flaig, T.W. Preclinical evaluation of teleglenastat (CB-839) in prostate cancer. J. Clin. Oncol. 2023, 41, 378. [Google Scholar] [CrossRef]
- Tannir, N.M.; Agarwal, N.; Porta, C.; Lawrence, N.J.; Motzer, R.J.; Lee, R.J.; Jain, R.K.; Davis, N.B.; Appleman, L.J.; Goodman, O.B.; et al. CANTATA: Primary analysis of a global, randomized, placebo (Pbo)-controlled, double-blind trial of telaglenastat (CB-839) + cabozantinib versus Pbo + cabozantinib in advanced/metastatic renal cell carcinoma (mRCC) patients (pts) who progressed on immune checkpoint inhibitor (ICI) or anti-angiogenic therapies. J. Clin. Oncol. 2021, 39, 4501. [Google Scholar]
- Eads, J.R.; Krishnamurthi, S.S.; Saltzman, J.N.; Bajor, D.L.; Vinayak, S.; Barnholtz-Sloan, J.; Meropol, N.J.; Markowitz, S.D.; Wang, Z. Phase I clinical trial of the glutaminase inhibitor CB-839 plus capecitabine in patients with advanced solid tumors. J. Clin. Oncol. 2018, 36, 2562. [Google Scholar] [CrossRef]
- DeMichele, A.; Harding, J.J.; Telli, M.L.; Munster, P.N.; McKay, R.; Iliopoulos, O.; Orford, K.W.; Bennett, M.K.; Mier, J.W.; Owonikoko, T.K.; et al. Phase 1 study of CB-839, a small molecule inhibitor of glutaminase (GLS) in combination with paclitaxel (Pac) in patients (pts) with triple negative breast cancer (TNBC). J. Clin. Oncol. 2016, 34, 1011. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Lee, R.J.; Carthon, B.C.; Iliopoulos, O.; Mier, J.W.; Patel, M.R.; Tannir, N.M.; Owonikoko, T.K.; Haas, N.B.; Voss, M.H.; et al. CB-839, a glutaminase inhibitor, in combination with cabozantinib in patients with clear cell and papillary metastatic renal cell cancer (mRCC): Results of a phase I study. J. Clin. Oncol. 2019, 37, 549. [Google Scholar] [CrossRef]
- Kisner, D.L.; Catane, R.; Muggia, F.M. The rediscovery of DON (6-diazo-5-oxo-L-norleucine). Recent Results Cancer Res. 1980, 74, 258–263. [Google Scholar] [PubMed]
- Rais, R.; Jančařík, A.; Tenora, L.; Nedelcovych, M.; Alt, J.; Englert, J.; Rojas, C.; Le, A.; Elgogary, A.; Tan, J.; et al. Discovery of 6-Diazo-5-oxo-l-norleucine (DON) Prodrugs with Enhanced CSF Delivery in Monkeys: A Potential Treatment for Glioblastoma. J. Med. Chem. 2016, 59, 8621–8633. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Doroshow, D.B.; Seiwert, T.Y.; Gibson, M.K.; Velcheti, V.; Lisberg, A.E.; Patel, S.A.; Scheffler, M.; Lafleur, F.; Dugan, M.H.; et al. Phase 1 and phase 2a, first-in-human (FIH) study, of DRP-104, a broad glutamine antagonist, in adult patients with advanced solid tumors. J. Clin. Oncol. 2021, 39, TPS3149. [Google Scholar] [CrossRef]
- Daemen, A.; Liu, B.; Song, K.; Kwong, M.; Gao, M.; Hong, R.; Nannini, M.; Peterson, D.; Liederer, B.M.; de la Cruz, C.; et al. Pan-Cancer Metabolic Signature Predicts Co-Dependency on Glutaminase and De Novo Glutathione Synthesis Linked to a High-Mesenchymal Cell State. Cell Metab. 2018, 28, 383–399.e9. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens Health 2019, 37, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Naito, S. Pro-survival and anti-apoptotic properties of androgen receptor signaling by oxidative stress promote treatment resistance in prostate cancer. Endocr. Relat. Cancer 2012, 19, R243–R253. [Google Scholar] [CrossRef] [PubMed]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef]
- Santer, F.R.; Erb, H.H.; McNeill, R.V. Therapy escape mechanisms in the malignant prostate. Semin. Cancer Biol. 2015, 35, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.N.; Avery, V.M.; Carrasco-Pozo, C. Metabolic Roles of Androgen Receptor and Tip60 in Androgen-Dependent Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 6622. [Google Scholar] [CrossRef]
- Kahya, U.; Koseer, A.S.; Dubrovska, A. Amino Acid Transporters on the Guard of Cell Genome and Epigenome. Cancers 2021, 13, 125. [Google Scholar] [CrossRef]
- Cornford, P.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer. Part II-2020 Update: Treatment of Relapsing and Metastatic Prostate Cancer. Eur. Urol. 2021, 79, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Baunacke, M.; Erb, H.H.H.; Füssel, S.; Erdmann, K.; Putz, J.; Borkowetz, A. Systemic Triple Therapy in Metastatic Hormone-Sensitive Prostate Cancer (mHSPC): Ready for Prime Time or Still to Be Explored? Cancers 2021, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Ebersbach, C.; Beier, A.-M.K.; Thomas, C.; Erb, H.H.H. Impact of STAT Proteins in Tumor Progress and Therapy Resistance in Advanced and Metastasized Prostate Cancer. Cancers 2021, 13, 4854. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, A.; Gerke, T.; Penney, K.L.; Lis, R.T.; Stack, E.C.; Pértega-Gomes, N.; Zadra, G.; Tyekucheva, S.; Giovannucci, E.L.; Mucci, L.A.; et al. MYC Overexpression at the Protein and mRNA Level and Cancer Outcomes among Men Treated with Radical Prostatectomy for Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2018, 27, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhu, Y.; Shao, X.; Cai, A.; Dong, B.; Xue, W.; Gao, H. Distinct Metabolic Signatures of Hormone-Sensitive and Castration-Resistant Prostate Cancer Revealed by a 1H NMR-Based Metabolomics of Biopsy Tissue. J. Proteome Res. 2020, 19, 3741–3749. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Zhang, C.; Martincuks, A.; Herrmann, A.; Yu, H. STAT proteins in cancer: Orchestration of metabolism. Nat. Rev. Cancer 2023, 23, 115–134. [Google Scholar] [CrossRef]
- Kaushik, A.K.; Vareed, S.K.; Basu, S.; Putluri, V.; Putluri, N.; Panzitt, K.; Brennan, C.A.; Chinnaiyan, A.M.; Vergara, I.A.; Erho, N.; et al. Metabolomic Profiling Identifies Biochemical Pathways Associated with Castration-Resistant Prostate Cancer. J. Proteome Res. 2014, 13, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Vayalil, P.K.; Landar, A. Mitochondrial oncobioenergetic index: A potential biomarker to predict progression from indolent to aggressive prostate cancer. Oncotarget. 2015, 6, 43065. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Chen, Y.H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Chen, Y.-H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.-N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Bumbaca, B.; Li, W. Taxane resistance in castration-resistant prostate cancer: Mechanisms and therapeutic strategies. Acta Pharm. Sin. B 2018, 8, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.C.; Han, J.M. Amino Acid Metabolism in Cancer Drug Resistance. Cells 2022, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.K.; Lau, P.M.; Kwan, Y.W.; Kong, S.K. Mitochondrial Fuel Dependence on Glutamine Drives Chemo-Resistance in the Cancer Stem Cells of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3315. [Google Scholar] [CrossRef] [PubMed]
- Puhr, M.; Hoefer, J.; Schafer, G.; Erb, H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-mesenchymal transition leads to docetaxel resistance in prostate cancer and is mediated by reduced expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yang, F.-H.; Zhang, W.-T.; Guo, Y.-D.; Ye, L.; Yao, X.-D. Mesenchymal stem cells desensitize castration-resistant prostate cancer to docetaxel chemotherapy via inducing TGF-β1-mediated cell autophagy. Cell Biosci. 2021, 11, 7. [Google Scholar] [CrossRef]
- Marín-Aguilera, M.; Codony-Servat, J.; Reig, Ò.; Lozano, J.J.; Fernández, P.L.; Pereira, M.V.; Jiménez, N.; Donovan, M.; Puig, P.; Mengual, L.; et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol. Cancer Ther. 2014, 13, 1270–1284. [Google Scholar] [CrossRef]
- Hanrahan, K.; O’Neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The role of epithelial–mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Mol. Oncol. 2017, 11, 251–265. [Google Scholar] [CrossRef]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial–mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.Y.; Yang, M.H. Metabolic Reprogramming and Epithelial-Mesenchymal Plasticity: Opportunities and Challenges for Cancer Therapy. Front. Oncol. 2020, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C. Overcoming docetaxel resistance in prostate cancer: A perspective review. Ther. Adv. Med. Oncol. 2012, 4, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Linke, D.; Donix, L.; Peitzsch, C.; Erb, H.H.H.; Dubrovska, A.; Pfeifer, M.; Thomas, C.; Fuessel, S.; Erdmann, K. Comprehensive Evaluation of Multiple Approaches Targeting ABCB1 to Resensitize Docetaxel-Resistant Prostate Cancer Cell Lines. Int. J. Mol. Sci. 2022, 24, 666. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, T.; Shiina, H.; Urakami, S.; Kikuno, N.; Yoneda, T.; Shigeno, K.; Igawa, M. Bcl-2 expression as a predictive marker of hormone-refractory prostate cancer treated with taxane-based chemotherapy. Clin. Cancer Res. 2006, 12, 6116–6124. [Google Scholar] [CrossRef]
- Ye, Q.F.; Zhang, Y.C.; Peng, X.Q.; Long, Z.; Ming, Y.Z.; He, L.Y. Silencing Notch-1 induces apoptosis and increases the chemosensitivity of prostate cancer cells to docetaxel through Bcl-2 and Bax. Oncol. Lett. 2012, 3, 879–884. [Google Scholar] [PubMed]
- Rudin, C.M.; Yang, Z.; Schumaker, L.M.; VanderWeele, D.J.; Newkirk, K.; Egorin, M.J.; Zuhowski, E.G.; Cullen, K.J. Inhibition of Glutathione Synthesis Reverses Bcl-2-mediated Cisplatin Resistance1. Cancer Res. 2003, 63, 312–318. [Google Scholar]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, C.; Nadiminty, N.; Lou, W.; Tummala, R.; Evans, C.P.; Gao, A.C. Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Mol. Cancer Ther 2013, 12, 1829–1836. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Genes/Pathways | Models | Selected Key Findings Related to the Topic | Refs. |
|---|---|---|---|---|
| 1 | GLS, MYC, miR-23a/b, glutaminolysis | PCa PC3 cells and lymphoma cells | 1. MYC upregulates GLS expression by transcriptional repression of miR-23a and miR-23b; 2. Glutamine withdrawal or GLS knockdown reduces ATP and GSH levels, increases ROS, and inhibits cell proliferation. | [114] |
| 2 | GLS (GAC and KGA isoforms) | PCa cell lines PC3 and DU145; breast and lung cancer cell lines | 1. GAC but not KGA is localized in mitochondria; 2. The catalytic efficiency of GAC depends on the presence of inorganic phosphate. | [29] |
| 3 | MYC, POX/PRODH, interconnection of Gln/Pro metabolic pathways | PCa PC3 cells and lymphoma cells | 1. MYC suppresses POX/PRODH expression by upregulating miR-23b* (miR-23b [114] and miR-23b* are processed from the same transcript but differently regulated). 2. MYC induces Pro biosynthesis from Gln. | [39] |
| 4 | Glutamine and glucose mediated anaplerosis, mitochondrial complex I | PCa cell lines LNCaP and PC3; the transgenic adenocarcinoma of the mouse prostate (TRAMP) model | Metformin therapy enhances glutamine anaplerosis and synergizes with glutamine metabolism inhibition in cancer cells. | [137] |
| 5 | TXNIP, GLS, glucose uptake, glutaminolysis | PC3 cells, BPH, and PCa patient tissue samples | 1. GLS is highly expressed in PCa tissues compared to BPH; GLS levels correlate with Gleason scores and TNM stages in patients with PCa; 2. Gln and GLS positively regulate glucose uptake by inhibiting TXNIP expression. | [118] |
| 6 | ASTC2-mediated Gln uptake, mTORC1 pathway, E2F-regulated cell cycle genes, FA synthesis | PCa cell lines LNCaP, PC3 and DU145; PC3 xenograft murine model; PCa patient tissue samples | 1. ASCT2 expression level is increased in PCa tissues; 2. ASCT2 expression is AR-dependent; 3. ASCT2 mediates Gln uptake in PCa cells; 3. Chemical inhibition of ASCT2 decreases basal OCR and FA synthesis; 4. ASCT2 expression is essential for tumor cell growth in vitro and in vivo. | [10] |
| 7 | MYC, miR-205, oxidative metabolism | PCa cells PC3 and docetaxel-resistant PC3-DR cells; CAFs | Docetaxel-resistant PCa cells acquire Gln addiction | [138] |
| 8 | MYC, AR, and mTORC1 signaling pathways, SLC1A4, SLC1A5 | PCa cell lines LNCaP and VCaP | MYC, AR and mTORC1 oncogenic pathways regulates the expression levels of Gln transporters. | [108] |
| 9 | ALKBH, DNA-repair and apoptotic pathways | PC3 cells; MEF; multiple cancer cell lines; PC3 xenograft murine model | 1. Gln metabolism regulates the DNA alkylation damage repair by regulation of the α-KG-dependent ALKBH; 2. Combination of DON or CB-839 and alkylating agent MMS inhibits tumor growth in vivo. | [72] |
| 10 | Gln and glucose catabolism, mTORC1 pathway, AMPK, GLS (GAC and KGA isoforms), GLS2 | PCa cell lines PC3, PC3M, and non-transformed cells RWPE-2 and RWPE-1 | Metastatic PCa cells have increased Gln utilization and high sensitivity to GLS and mTORC1 inhibition. | [106] |
| 11 | GLS, WNT/β-catenin, cell cycle and apoptosis pathways | PCa cell lines 22Rv1, DU145, PC-3, LNCaP, and non-transformed RWPE-1 cells | GLS knockdown suppresses WNT/β-catenin pathway, inhibits cell proliferation, and induces apoptosis and cell cycle arrest. | [139] |
| 12 | RAS, RASAL3, TCA cycle, mitochondrial bioenergetics, PCa neuroendocrine differentiation signaling | PCa cell lines 22Rv1, C4-2B; prostatic fibroblasts derived from the patients with PCa and from murine prostates; BPH cells | 1. Epigenetic silencing of the RASAL3 gene in human prostatic CAFs results in oncogenic RAS activity and Gln synthesis and secretion; 2. CAF-derived Gln is utilized by PCa cells and induces neuroendocrine differentiation; 3. ADT promotes epigenetic silencing of RASAL3 in CAFs; 4. A high level of Gln in the blood of the patients with PCa treated with ADT correlates with therapy resistance. | [124] |
| 13 | PDHA1, PTEN, lipogenic genes and metabolic pathways | PCa cell lines 22Rv1, LNCaP, PC3, and DU145, PNT2C2; 22Rv1 xenograft murine model; transgenic murine models for prostate-specific deletion of PTEN and PDHA1 | 1. Gln plays an important role in de novo lipogenesis; 2. Knockdown of PDHA1 decreases the incorporation of Gln and glucose carbon into lipids and cholesterol; 3. PTEN-negative prostate cells have increased Gln carbon incorporation into citrate, fumarate, and malate compared to normal epithelial cells. | [140] |
| 14 | Glutaminolysis, TCA cycle, DNA damage signaling, autophagy, MYC, GLS, CSC regulation | PCa cell lines DU145, LNCaP, PC3 and their radioresistant (RR) derivatives; patient-derived cell cultures of PCa and BPH; LNCaP and DU145 xenograft murine models; blood plasma samples of PCa patients | 1. Inhibition of Gln metabolism increases oxidative stress, DNA damage and PCa sensitivity to radiotherapy; 2. Activation of ATG-mediated autophagy abrogates the radiosensitizing effect of Gln metabolism inhibition; 3. Gln metabolism regulates CSC populations by the α-KG-dependent epigenetic reprogramming; 4. A high expression of MYC and GLS genes and a high blood level of Gln correlate with a poor prognosis in PCa patients treated with radiotherapy. | [6] |
| 15 | Glutaminolysis, TCA cycle | PCa cell lines LNCaP and PC3; the transgenic adenocarcinoma of the mouse prostate (TRAMP) model | 1. CRPC cells possess Gln addiction; 2. The upregulation of glutaminolysis and Gln anaplerosis into the TCA cycle are characteristic of castration-resistant PCa. | [141] |
| 16 | ASCT2, glutaminolysis, glycolytic and lipid metabolic pathways | PCa cell lines DU145, LNCaP, and PC3; rat model | 1. CRPC cells are more Gln dependent than androgen-sensitive PCa cells; 2. DHT induce GLS and ASCT2 expression in androgen-sensitive PCa cells; 3. Inhibition of GLS with BPTES decreased PCa migration and induce cell death; 4. Anti-androgen treatment increases PCa cell sensitivity to GLS inhibition with BPTES; 5. GLS inhibition with BPTES affects glycolytic and lipid metabolism in a cell line-dependent manner. | [142] |
| 17 | Gln carbon and nitrogen metabolic pathways, pyrimidine synthesis, CAD, GLS, PI3K-AKT-mTOR pathway | PCa cell lines DU145, LNCaP, PC3, C4-2, C4-2MDVR and non-transformed RWPE-1 cells; PC3 and C4-2MDVR xenograft murine models; PCa patient tissue samples | 1. Gln-derived nitrogen and carbon are both required for pyrimidine synthesis in PCa cells, whereas Gln is not a main carbon source for purine synthesis; 2. CAD, a key enzyme for pyrimidine synthesis, is upregulated in PCa tissues; 3. A combination of CAD knockdown and GLS inhibition with CD-839 or knockdown has a superior inhibitory effect on PCa in vitro and in vivo than inhibition of a single protein. | [21] |
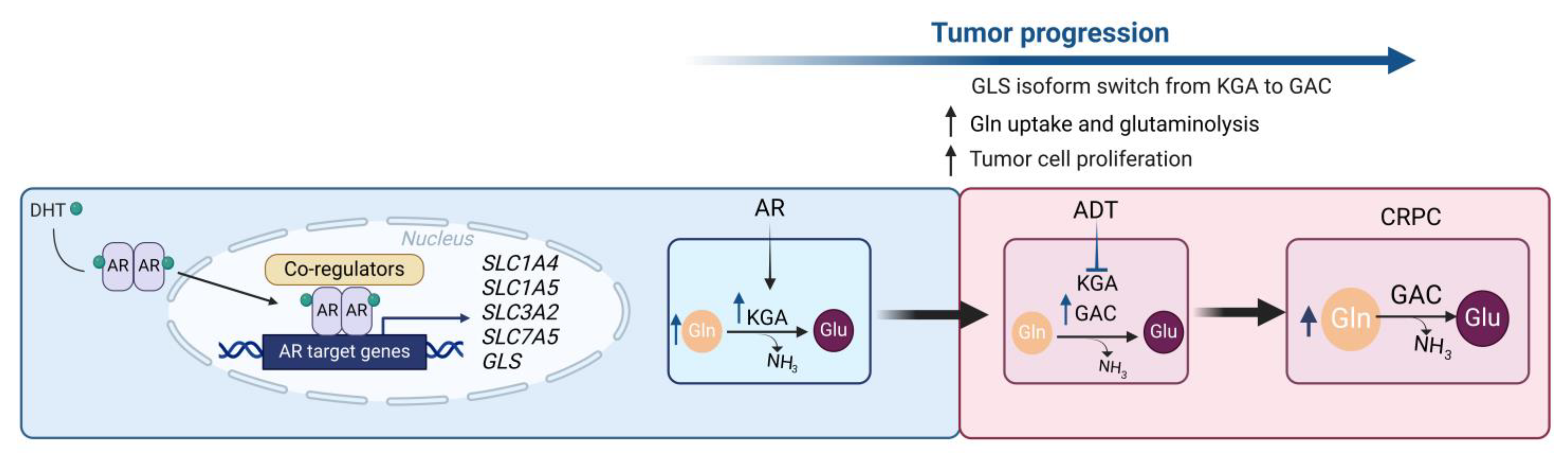
| 18 | GLS (GAC and KGA isoforms), glutaminolysis, AR and c-MYC signaling | PCa cell lines LNCaP, PC3, C4-2, C4-2MDVR; PCa patient tissue samples; LNCaP and PC3 xenograft murine models | 1. ADT inhibits glutaminolysis; 2. Advanced PCa and CRPC cells are highly dependent on Gln; 3. GLS isoforms are differently expressed at the different stages of PCa: KGA is highly expressed in hormone-naive PCa cells, whereas GAC is highly expressed in advanced PCa and CRPC; 4. GAC and KGA expression levels inversely correlate in PCa tissues; 5. GLS isoform switch is associated with PCa resistance to ADT; 6. Advanced PCa and CRPC cells are more sensitive to GLS inhibition with CB-839 in vitro and in vivo than androgen-dependent PCa; 7. GLS isoform switch is regulated by MYC and AR. | [121] |
| 19 | L-ASP, Gln transporters, Asn catabolism, cell cycle and DNA repair signaling | PCa cell lines 22Rv1, PC3, ARCaPM and its radiation-resistant derivative ARCaPM-IR; CAF; ARCaPM/CAF xenograft murine models | 1. Gln is conditionally essential for PCa cells; 2. L-ASP sensitizes PCa cells to radiotherapy through depletion of Gln; 3. Both L-ASP and Gln depletion induce cell cycle arrest and inhibit DNA repair; 4. CAF-induced PCa radioresistance can be decreased in vitro and in vivo by L-ASP. | [104] |
| 20 | Pro and Gln biosynthesis pathways, P5CS, ALDH18A1; TCA cycle, pyrimidine synthesis | PCa cell lines VCAP, 22Rv1, LNCaP and PC3; multiple cancer cell lines; gastric cancer patient tissue samples | 1. α-KG, Asn, and nucleotides are the key metabolites for cell survival under Gln deprivation; 2. A lowering of P5CS (ALDH18A1) expression is a common adaptation to Gln deprivation in different types of tumor cells, including PCa; 3. P5CS inhibition promotes Gln de novo synthesis. | [143] |
| 21 | GLS, glutaminolysis, mitochondrial bioenergetics, cell cycle and viability regulation | Docetaxel-sensitive and -resistant PCa cell lines PC3 and DU145; PCa patient tissue samples | 1. Gln deprivation and GLS inhibition with CB-839 reduces mitochondrial functions and induces apoptosis in chemotherapy-resistant and sensitive PCa cells; 2. Docetaxel-resistant PCa cells are more sensitive to the Gln metabolism inhibition than parental cells; 3. GLS expression is elevated in PCa and correlates with clinical outcomes. | [144] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erb, H.H.H.; Polishchuk, N.; Stasyk, O.; Kahya, U.; Weigel, M.M.; Dubrovska, A. Glutamine Metabolism and Prostate Cancer. Cancers 2024, 16, 2871. https://doi.org/10.3390/cancers16162871
Erb HHH, Polishchuk N, Stasyk O, Kahya U, Weigel MM, Dubrovska A. Glutamine Metabolism and Prostate Cancer. Cancers. 2024; 16(16):2871. https://doi.org/10.3390/cancers16162871
Chicago/Turabian StyleErb, Holger H. H., Nikita Polishchuk, Oleh Stasyk, Uğur Kahya, Matthias M. Weigel, and Anna Dubrovska. 2024. "Glutamine Metabolism and Prostate Cancer" Cancers 16, no. 16: 2871. https://doi.org/10.3390/cancers16162871
APA StyleErb, H. H. H., Polishchuk, N., Stasyk, O., Kahya, U., Weigel, M. M., & Dubrovska, A. (2024). Glutamine Metabolism and Prostate Cancer. Cancers, 16(16), 2871. https://doi.org/10.3390/cancers16162871