
Transcriptomic Analysis Reveals Early Alterations Associated with Intrinsic Resistance to Targeted Therapy in Lung Adenocarcinoma Cell Lines

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Targeted Next-Generation Sequencing to Confirm Mutations in Lung Adenocarcinoma Cell Lines
2.3. Therapeutic Compounds
2.4. Dose–Response Curves to TKIs in EGFR-Mutant Lung Adenocarcinoma Cell Lines
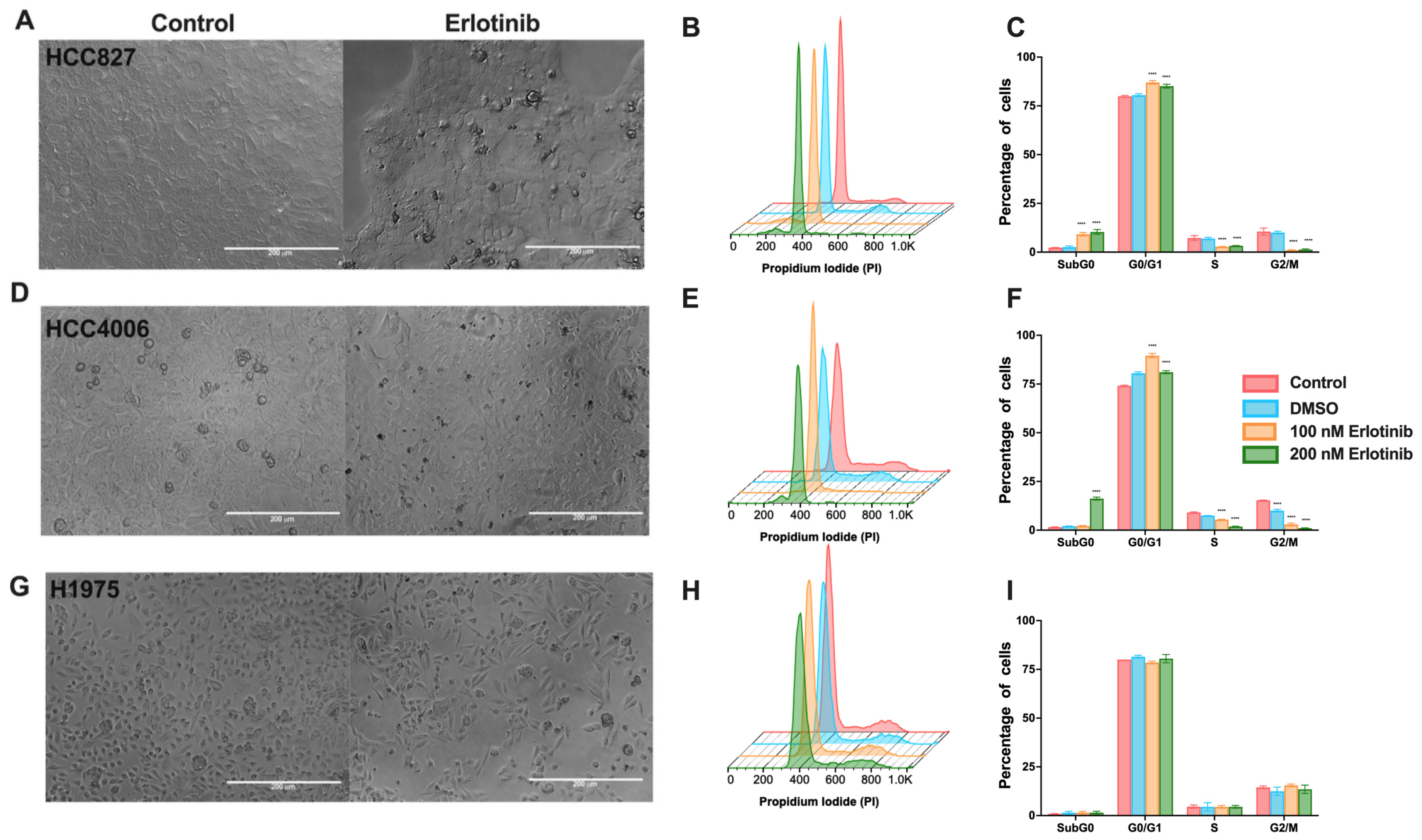
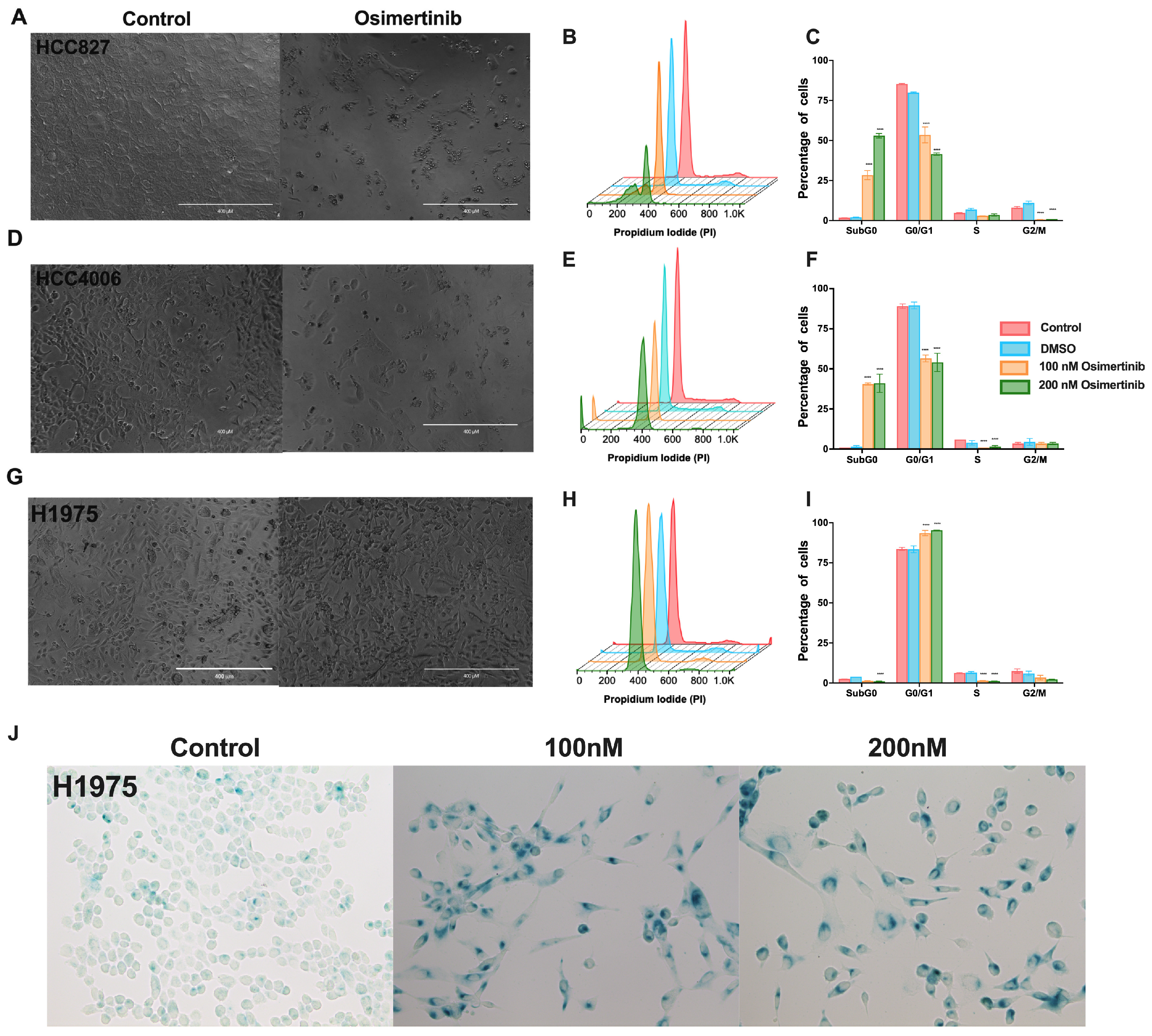
2.5. Cell-Cycle Analysis
2.6. Detection of Beta-Galactosidase Positive DTP Cells
2.7. Quantification of Viable, Apoptotic, and Necrotic Cells
2.8. Transcriptome Sequencing
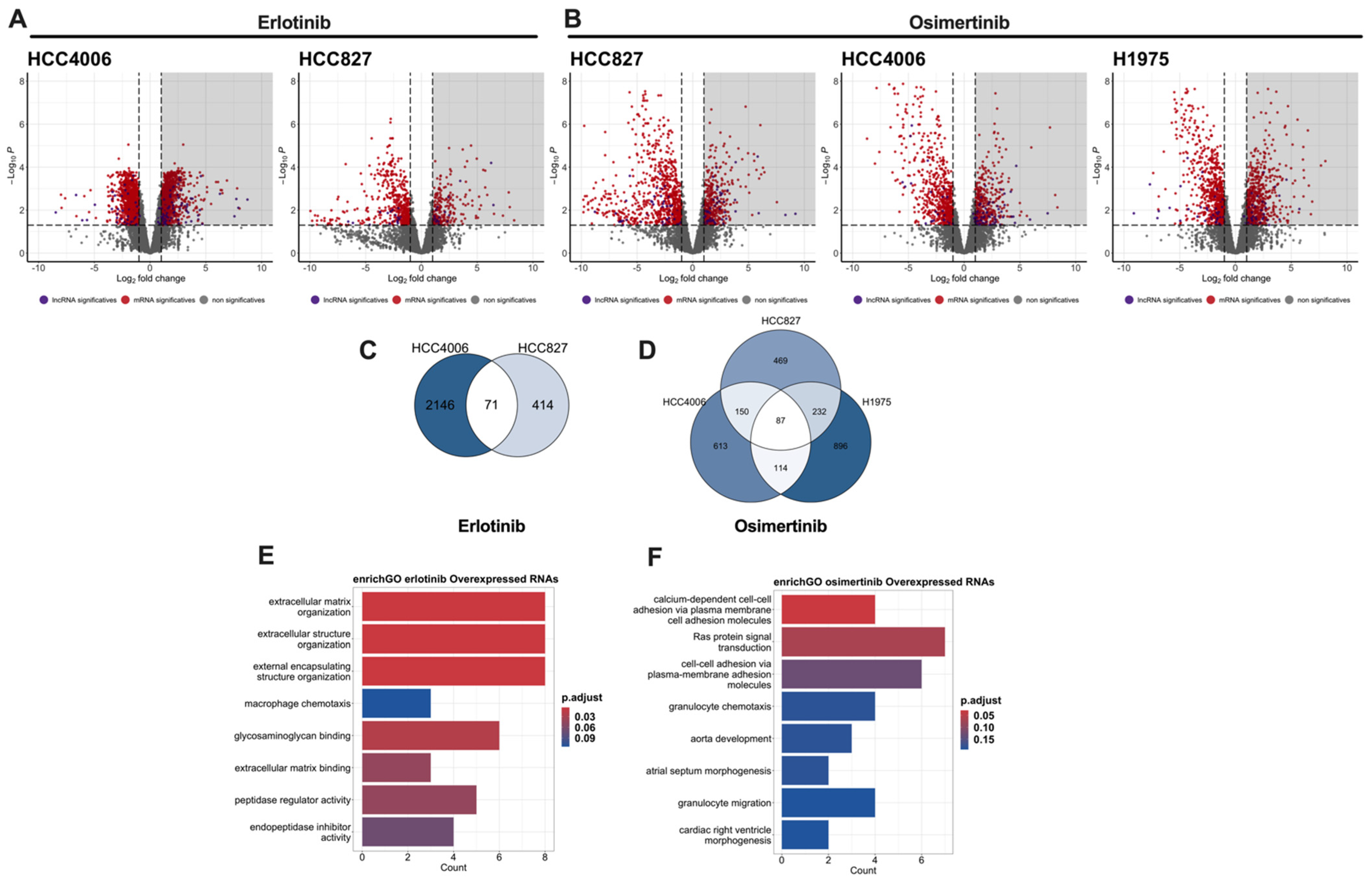
2.9. Bioinformatics Analysis of DTP Cell RNA-Seq Data
2.10. Survival Analysis
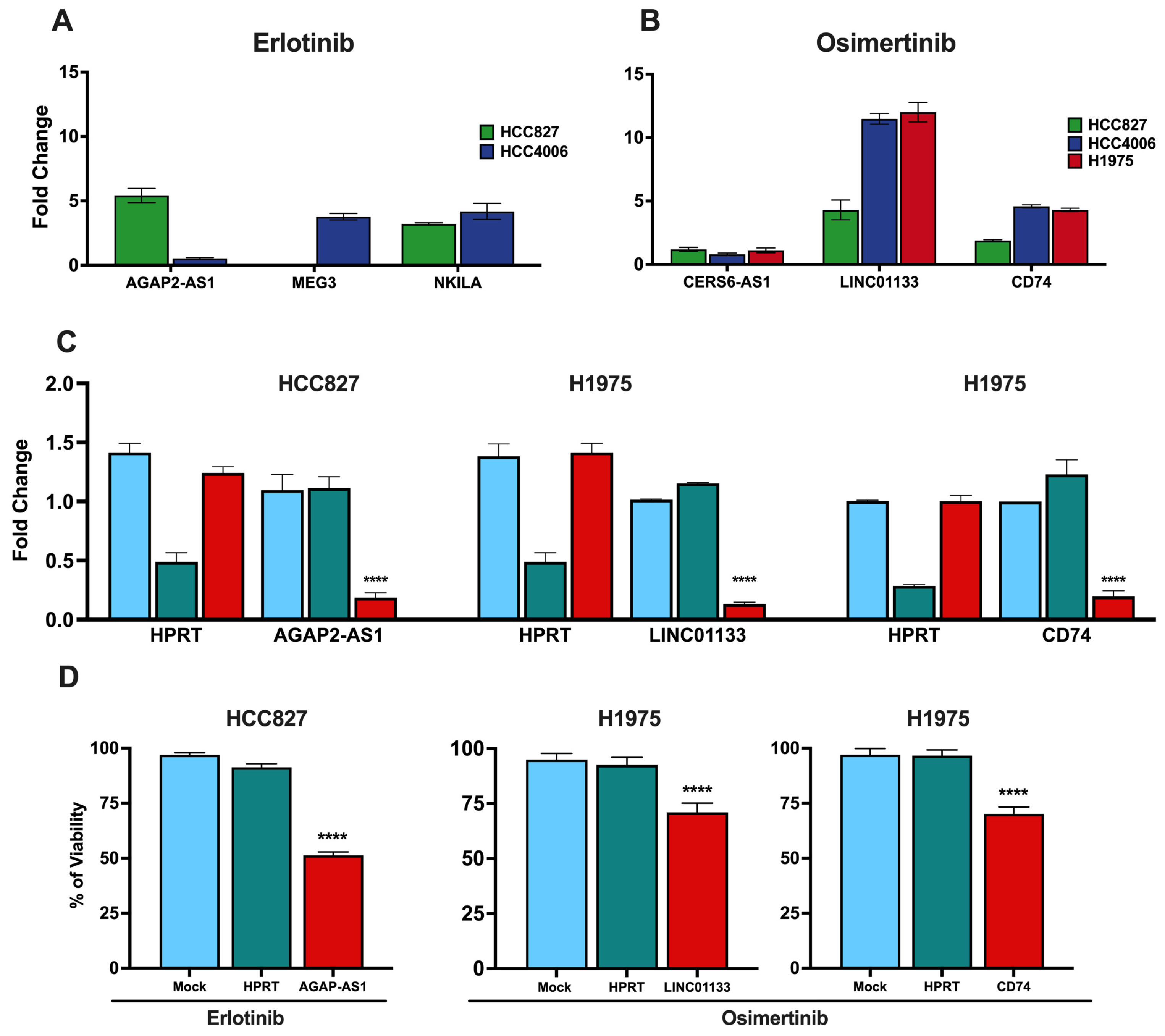
2.11. RT–qPCR Validation
2.12. lncRNA Knockdown
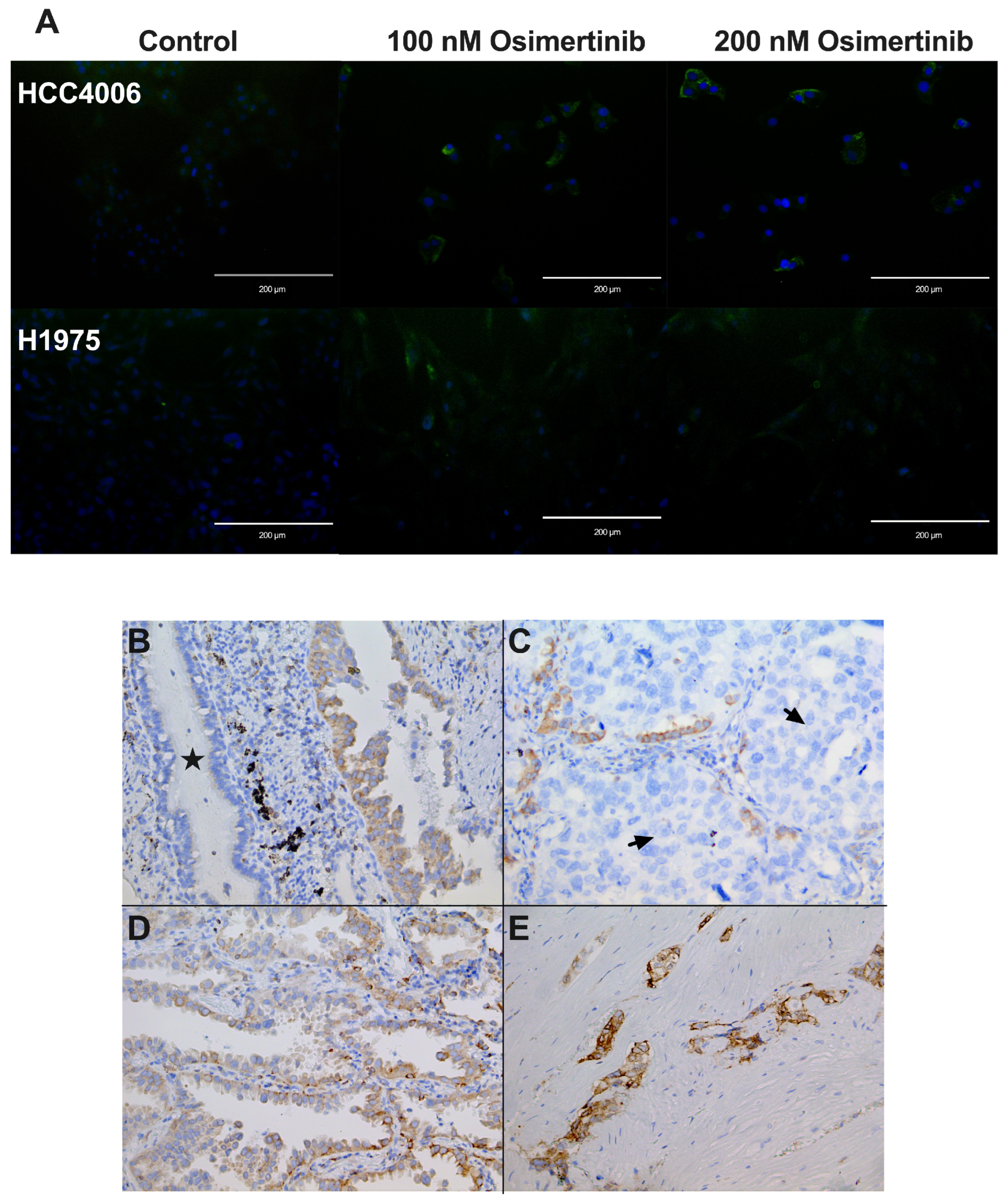
2.13. CD74 Localization in DTP Cells
2.14. Tissue Samples of Lung Adenocarcinoma
2.15. Validation of lncRNA in Lung Adenocarcinoma Biopsies
2.16. Immunohistochemical Staining of CD74
2.17. Statistical Analysis
2.18. Data Availability Statement
3. Results
3.1. Effects of TKIs on EGFR-Mutated Lung Adenocarcinoma Cell Lines
3.2. Osimertinib Induces β-Galactosidase Expression in Residual Cells
3.3. NGS-Based Mutation Analysis in Lung Adenocarcinoma Cell Lines
3.4. Transcriptomic Sequencing of Drug-Tolerant Persister Cells
3.5. Survival Analysis
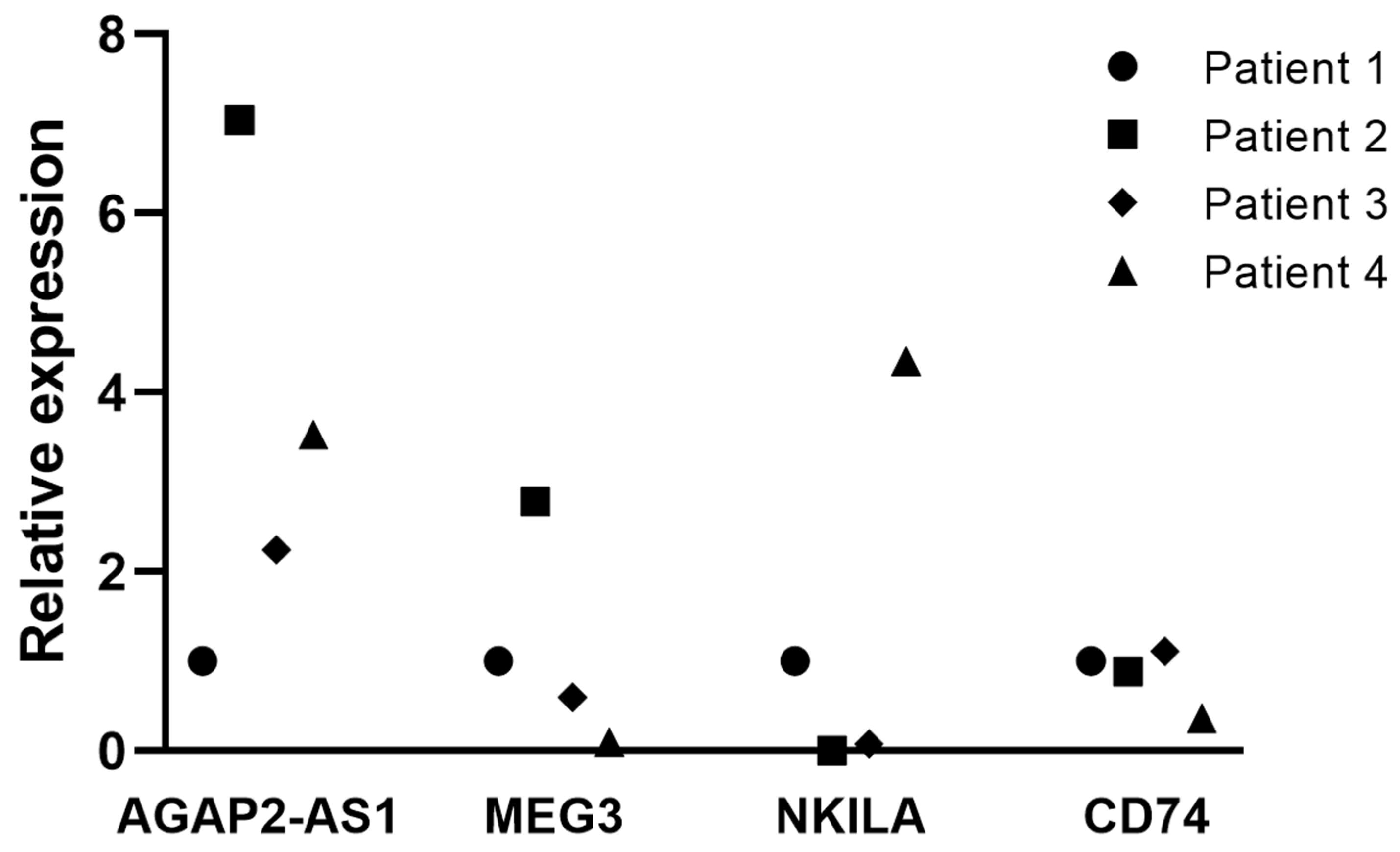
3.6. Validation of Clinically Relevant DTP-Associated lncRNAs and CD74
3.7. Knockdown of lncRNAs and CD74 Associated with the DTP State
3.8. Tissue Expression of Clinically Relevant lncRNAs Associated with the DTP State
3.9. Cellular Localization of CD74 in Cell Lines
3.10. CD74 Staining in lung Adenocarcinoma Biopsies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Tfayli, A.; Mohty, R. EGFR Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer: Treatment Paradigm, Current Evidence, and Challenges. Tumori J. 2020, 107, 376–384. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Nie, N.; Li, J.; Zhang, J.; Dai, J.; Liu, Z.; Ding, Z.; Wang, Y.; Zhu, M.; Hu, C.; Han, R.; et al. First-Line Osimertinib in Patients with EGFR-Mutated Non-Small Cell Lung Cancer: Effectiveness, Resistance Mechanisms, and Prognosis of Different Subsequent Treatments. Clin. Med. Insights Oncol. 2022, 16, 117955492211347. [Google Scholar] [CrossRef]
- Gharwan, H.; Groninger, H. Kinase Inhibitors and Monoclonal Antibodies in Oncology: Clinical Implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. [Google Scholar] [CrossRef]
- Shaban, N.; Kamashev, D.; Emelianova, A.; Buzdin, A. Targeted Inhibitors of EGFR: Structure, Biology, Biomarkers, and Clinical Applications. Cells 2023, 13, 47. [Google Scholar] [CrossRef]
- Pirker, R. Third-Generation Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Advanced Nonsmall Cell Lung Cancer. Curr. Opin. Oncol. 2016, 28, 115–121. [Google Scholar] [CrossRef]
- Sun, J.-M.; Park, K. Can We Define the Optimal Sequence of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors for the Treatment of Epidermal Growth Factor Receptor-Mutant Nonsmall Cell Lung Cancer? Curr. Opin. Oncol. 2017, 29, 89–96. [Google Scholar] [CrossRef]
- Tiefenbacher, A.; Pirker, R. Systemic Treatment of Advanced Non-Small Cell Lung Cancer: Controversies and Perspectives. Mag. Eur. Med. Oncol. 2018, 11, 112–115. [Google Scholar] [CrossRef]
- Wang, J.; Wang, B.; Chu, H.; Yao, Y. Intrinsic Resistance to EGFR Tyrosine Kinase Inhibitors in Advanced Non-Small-Cell Lung Cancer with Activating EGFR Mutations. OncoTargets Ther. 2016, 9, 3711. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef]
- Gomatou, G.; Syrigos, N.; Kotteas, E. Osimertinib Resistance: Molecular Mechanisms and Emerging Treatment Options. Cancers 2023, 15, 841. [Google Scholar] [CrossRef]
- Hinohara, K.; Polyak, K. Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Sullivan, I.; Planchard, D. Next-Generation EGFR Tyrosine Kinase Inhibitors for Treating EGFR-Mutant Lung Cancer beyond First Line. Front. Med. 2017, 3, 76. [Google Scholar] [CrossRef]
- Hammerlindl, H.; Schaider, H. Tumor Cell-Intrinsic Phenotypic Plasticity Facilitates Adaptive Cellular Reprogramming Driving Acquired Drug Resistance. J. Cell Commun. Signal. 2017, 12, 133–141. [Google Scholar] [CrossRef]
- De Conti, G.; Dias, M.H.; Bernards, R. Fighting Drug Resistance through the Targeting of Drug-Tolerant Persister Cells. Cancers 2021, 13, 1118. [Google Scholar] [CrossRef]
- Chhouri, H.; Alexandre, D.; Grumolato, L. Mechanisms of Acquired Resistance and Tolerance to EGFR Targeted Therapy in Non-Small Cell Lung Cancer. Cancers 2023, 15, 504. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Gao, X.; Liu, Q.; Chen, X.; Chen, S.; Yang, J.; Liu, Q.; Cheng, Y. Screening of Tumor Grade-related mRNAs and lncRNAs for Esophagus Squamous Cell Carcinoma. J. Clin. Lab. Anal. 2021, 35, e23797. [Google Scholar] [CrossRef]
- Yano, S.; Yamada, T.; Takeuchi, S.; Tachibana, K.; Minami, Y.; Yatabe, Y.; Mitsudomi, T.; Tanaka, H.; Kimura, T.; Kudoh, S.; et al. Hepatocyte Growth Factor Expression in EGFR Mutant Lung Cancer with Intrinsic and Acquired Resistance to Tyrosine Kinase Inhibitors in a Japanese Cohort. J. Thorac. Oncol. 2011, 6, 2011–2017. [Google Scholar] [CrossRef]
- Hsiao, S.-H.; Liu, H.E.; Lee, H.-L.; Lin, C.-L.; Chen, W.-Y.; Wu, Z.-H.; Lin, S.-E.; Chiang, L.-L.; Chung, C.-L. Distinct Clinical Outcomes of Non-Small Cell Lung Cancer Patients with Epidermal Growth Factor Receptor (EGFR) Mutations Treated with EGFR Tyrosine Kinase Inhibitors: Non-Responders versus Responders. PLoS ONE 2013, 8, e83266. [Google Scholar] [CrossRef]
- Swayden, M.; Chhouri, H.; Anouar, Y.; Grumolato, L. Tolerant/Persister Cancer Cells and the Path to Resistance to Targeted Therapy. Cells 2020, 9, 2601. [Google Scholar] [CrossRef]
- Recasens, A.; Munoz, L. Targeting Cancer Cell Dormancy. Trends Pharmacol. Sci. 2019, 40, 128–141. [Google Scholar] [CrossRef]
- Ding, D.; Zhang, J.; Luo, Z.; Wu, H.; Lin, Z.; Liang, W.; Xue, X. Analysis of the lncRNA–miRNA–mRNA Network Reveals a Potential Regulatory Mechanism of EGFR-TKI Resistance in NSCLC. Front. Genet. 2022, 13, 851391. [Google Scholar] [CrossRef]
- Mikubo, M.; Inoue, Y.; Liu, G.; Tsao, M.-S. Mechanism of Drug Tolerant Persister Cancer Cells: The Landscape and Clinical Implication for Therapy. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 1798–1809. [Google Scholar] [CrossRef]
- Vishwanathan, K.; So, K.; Thomas, K.; Bramley, A.; English, S.; Collier, J. Absolute Bioavailability of Osimertinib in Healthy Adults. Clin. Pharmacol. Drug Dev. 2018, 8, 198–207. [Google Scholar] [CrossRef]
- Le Louedec, F.; Puisset, F.; Chatelut, E.; Tod, M. Considering the Oral Bioavailability of Protein Kinase Inhibitors: Essential in Assessing the Extent of Drug–Drug Interaction and Improving Clinical Practice. Clin. Pharmacokinet. 2023, 62, 55–66. [Google Scholar] [CrossRef]
- Anders, S. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 19 September 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A Powerful Link between Biological Databases and Microarray Data Analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Nicholson, A.G.; Tsao, M.S.; Beasley, M.B.; Borczuk, A.C.; Brambilla, E.; Cooper, W.A.; Dacic, S.; Jain, D.; Kerr, K.M.; Lantuejoul, S.; et al. The 2021 WHO Classification of Lung Tumors: Impact of Advances Since 2015. J. Thorac. Oncol. 2022, 17, 362–387. [Google Scholar] [CrossRef]
- Leonce, C.; Saintigny, P.; Ortiz-Cuaran, S. Cell-Intrinsic Mechanisms of Drug Tolerance to Systemic Therapies in Cancer. Mol. Cancer Res. 2022, 20, 11–29. [Google Scholar] [CrossRef]
- Aguiar, P.N.; Haaland, B.; Park, W.; San Tan, P.; del Giglio, A.; de Lima Lopes, G. Cost-Effectiveness of Osimertinib in the First-Line Treatment of Patients with EGFR -Mutated Advanced Non–Small Cell Lung Cancer. JAMA Oncol. 2018, 4, 1080. [Google Scholar] [CrossRef]
- Kashima, Y.; Shibahara, D.; Suzuki, A.; Muto, K.; Kobayashi, I.S.; Plotnick, D.; Udagawa, H.; Izumi, H.; Shibata, Y.; Tanaka, K.; et al. Single-Cell Analyses Reveal Diverse Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors in Lung Cancer. Cancer Res. 2021, 81, 4835–4848. [Google Scholar] [CrossRef]
- Liu, L.; Lizaso, A.; Mao, X.; Yang, N.; Zhang, Y. Rechallenge with Erlotinib in Osimertinib-Resistant Lung Adenocarcinoma Mediated by Driver Gene Loss: A Case Report. Transl. Lung Cancer Res. 2020, 9, 144–147. [Google Scholar] [CrossRef]
- Fu, K.; Xie, F.; Wang, F.; Fu, L. Therapeutic Strategies for EGFR-Mutated Non-Small Cell Lung Cancer Patients with Osimertinib Resistance. J. Hematol. Amp Oncol. 2022, 15, 173. [Google Scholar] [CrossRef]
- Meador, C.B.; Hata, A.N. Acquired Resistance to Targeted Therapies in NSCLC: Updates and Evolving Insights. Pharmacol. Ther. 2020, 210, 107522. [Google Scholar] [CrossRef]
- Ashrafi, A.; Akter, Z.; Modareszadeh, P.; Modareszadeh, P.; Berisha, E.; Alemi, P.S.; Chacon Castro, M.D.C.; Deese, A.R.; Zhang, L. Current Landscape of Therapeutic Resistance in Lung Cancer and Promising Strategies to Overcome Resistance. Cancers 2022, 14, 4562. [Google Scholar] [CrossRef]
- Al Bakir, M.; Huebner, A.; Martínez-Ruiz, C.; Grigoriadis, K.; Watkins, T.B.K.; Pich, O.; Moore, D.A.; Veeriah, S.; Ward, S.; Laycock, J.; et al. The Evolution of Non-Small Cell Lung Cancer Metastases in TRACERx. Nature 2023, 616, 534–542. [Google Scholar] [CrossRef]
- Mondal, P.; Meeran, S.M. Emerging Role of Non-Coding RNAs in Resistance to Platinum-Based Anti-Cancer Agents in Lung Cancer. Front. Pharmacol. 2023, 14, 1105484. [Google Scholar] [CrossRef]
- Gupta, S.; Verma, V.; Dwarakanath, B.S. Emerging Concepts in Cancer Therapy: Mechanisms of Resistance. Cancer Rep. 2022, 5, e1715. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Dominguez, R.; Aguilar-Cazares, D.; Perez-Medina, M.; Avila-Rios, S.; Soto-Nava, M.; Mendez-Tenorio, A.; Islas-Vazquez, L.; Benito-Lopez, J.J.; Galicia-Velasco, M.; Lopez-Gonzalez, J.S. Transcriptional Signature of Early Cisplatin Drug-Tolerant Persister Cells in Lung Adenocarcinoma. Front. Oncol. 2023, 13, 1208403. [Google Scholar] [CrossRef]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12. [Google Scholar] [CrossRef]
- Fan, K.-J.; Liu, Y.; Yang, B.; Tian, X.-D.; Li, C.-R.; Wang, B. Prognostic and Diagnostic Significance of Long Non-Coding RNA AGAP2-AS1 Levels in Patients with Non-Small Cell Lung Cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2392–2396. [Google Scholar] [PubMed]
- Gao, L.; Zhao, A.; Wang, X. Upregulation of lncRNA AGAP2-AS1 Is an Independent Predictor of Poor Survival in Patients with Clear Cell Renal Carcinoma. Oncol. Lett. 2020, 19, 3993–4001. [Google Scholar] [CrossRef] [PubMed]
- Zang, C.; Nie, F.; Wang, Q.; Sun, M.; Li, W.; He, J.; Zhang, M.; Lu, K. Long Non-Coding RNA LINC01133 Represses KLF2, P21 and E-Cadherin Transcription through Binding with EZH2, LSD1 in Non Small Cell Lung Cancer. Oncotarget 2016, 7, 11696–11707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, N.; Chen, X. A Novel Long Noncoding RNA LINC01133 Is Upregulated in Lung Squamous Cell Cancer and Predicts Survival. Tumor Biol. 2015, 36, 7465–7471. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Li, X.; Song, Z.; Zhu, X.; Zhao, S. Long Non-Coding RNA AGAP2-AS1 Exerts Oncogenic Properties in Glioblastoma by Epigenetically Silencing TFPI2 through EZH2 and LSD1. Aging 2019, 11, 3811–3823. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Niu, X.; Zhao, R.; Liu, J.; Ren, W.; Dai, H.; Chen, J.; Yan, J.; Li, B.; Shao, Y.; et al. Long Non-Coding RNA AGAP2-AS1 Promotes Cell Proliferation and Invasion through Regulating miR-193a-3p/LOXL4 Axis in Laryngeal Squamous Cell Carcinoma. Cell Cycle 2022, 21, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guo, S.; Zhang, M.; Li, L.; Wang, F.; Song, B. Long Non-coding RNA AGAP2-AS1 Accelerates Cell Proliferation, Migration, Invasion and the EMT Process in Colorectal Cancer via Regulating the miR-4,668-3p/SRSF1 Axis. J. Gene Med. 2020, 22, e3250. [Google Scholar] [CrossRef] [PubMed]
- McClelland, M.; Zhao, L.; Carskadon, S.; Arenberg, D. Expression of CD74, the Receptor for Macrophage Migration Inhibitory Factor, in Non-Small Cell Lung Cancer. Am. J. Pathol. 2009, 174, 638–646. [Google Scholar] [CrossRef]
- Vargas, J.; Pantouris, G. Analysis of CD74 Occurrence in Oncogenic Fusion Proteins. Int. J. Mol. Sci. 2023, 24, 15981. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Medina, M.; Lopez-Gonzalez, J.S.; Benito-Lopez, J.J.; Ávila-Ríos, S.; Soto-Nava, M.; Matias-Florentino, M.; Méndez-Tenorio, A.; Galicia-Velasco, M.; Chavez-Dominguez, R.; Meza-Toledo, S.E.; et al. Transcriptomic Analysis Reveals Early Alterations Associated with Intrinsic Resistance to Targeted Therapy in Lung Adenocarcinoma Cell Lines. Cancers 2024, 16, 2490. https://doi.org/10.3390/cancers16132490
Perez-Medina M, Lopez-Gonzalez JS, Benito-Lopez JJ, Ávila-Ríos S, Soto-Nava M, Matias-Florentino M, Méndez-Tenorio A, Galicia-Velasco M, Chavez-Dominguez R, Meza-Toledo SE, et al. Transcriptomic Analysis Reveals Early Alterations Associated with Intrinsic Resistance to Targeted Therapy in Lung Adenocarcinoma Cell Lines. Cancers. 2024; 16(13):2490. https://doi.org/10.3390/cancers16132490
Chicago/Turabian StylePerez-Medina, Mario, Jose S. Lopez-Gonzalez, Jesus J. Benito-Lopez, Santiago Ávila-Ríos, Maribel Soto-Nava, Margarita Matias-Florentino, Alfonso Méndez-Tenorio, Miriam Galicia-Velasco, Rodolfo Chavez-Dominguez, Sergio E. Meza-Toledo, and et al. 2024. "Transcriptomic Analysis Reveals Early Alterations Associated with Intrinsic Resistance to Targeted Therapy in Lung Adenocarcinoma Cell Lines" Cancers 16, no. 13: 2490. https://doi.org/10.3390/cancers16132490
APA StylePerez-Medina, M., Lopez-Gonzalez, J. S., Benito-Lopez, J. J., Ávila-Ríos, S., Soto-Nava, M., Matias-Florentino, M., Méndez-Tenorio, A., Galicia-Velasco, M., Chavez-Dominguez, R., Meza-Toledo, S. E., & Aguilar-Cazares, D. (2024). Transcriptomic Analysis Reveals Early Alterations Associated with Intrinsic Resistance to Targeted Therapy in Lung Adenocarcinoma Cell Lines. Cancers, 16(13), 2490. https://doi.org/10.3390/cancers16132490