
Targeting Tyro3, Axl, and MerTK Receptor Tyrosine Kinases Significantly Sensitizes Triple-Negative Breast Cancer to CDK4/6 Inhibition

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Small-Molecule Inhibitors
2.2. Cell Culture, Cell Viability Detection, Reagents, and Clonogenic Assay
2.3. Immunoblotting and Antibody Array (ELISA)
2.4. Caspase-3/7 Assay and qRT-PCR
2.5. Mass Spectrometry Sample Preparation
2.6. Basic pH Reversed-Phase Separation (BPRP)
2.7. Mass Spectrometry Data Collection (LC-MS3)—Total Proteome
2.8. Total Proteome Data Analysis
2.9. Tissue Microarray (TMA) and IHC Staining
2.10. Animal Studies
2.11. Immune Profiling and Flow Cytometry
2.12. Bioinformatical Analyses
2.13. Statistics and Synergy Calculations
3. Results
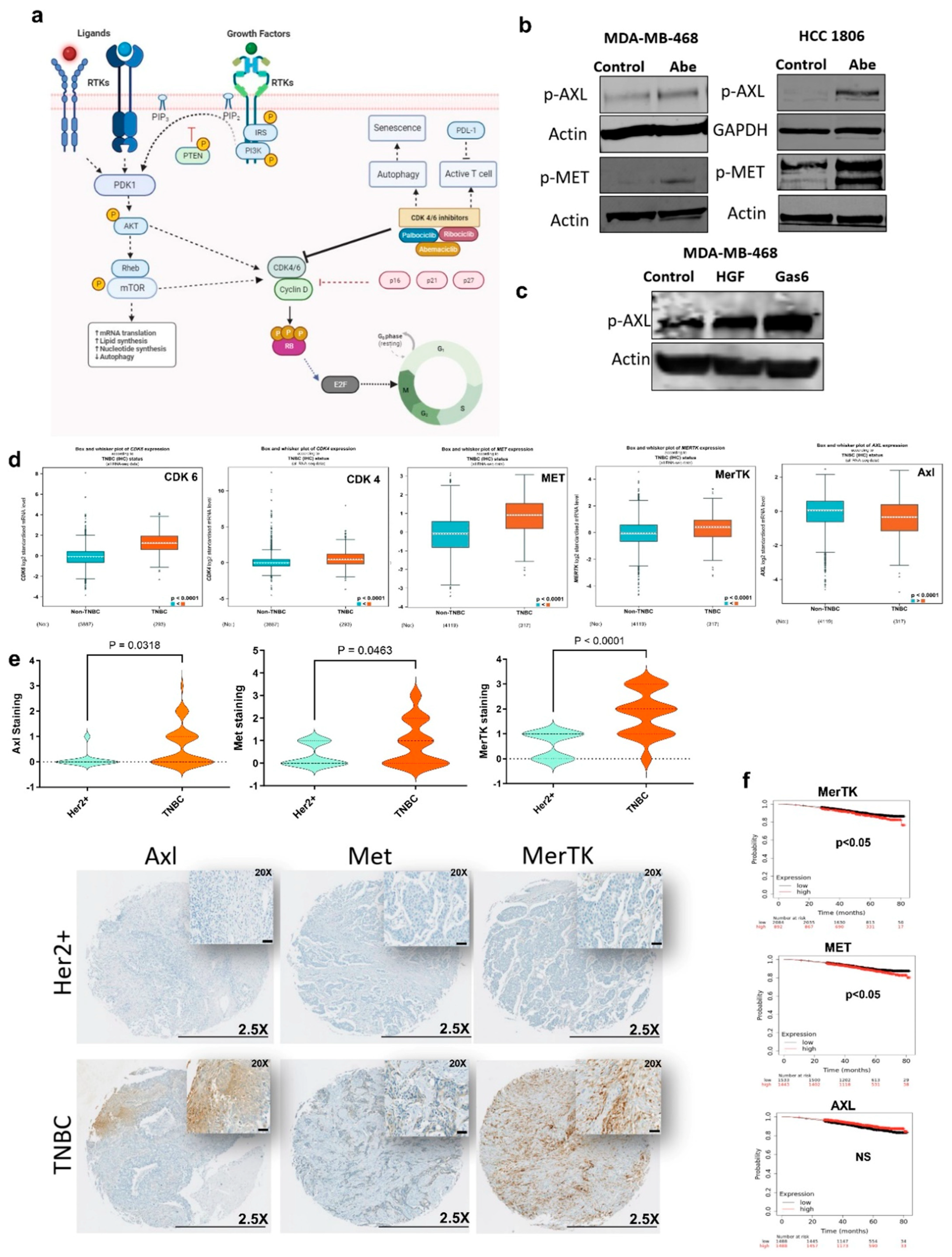
3.1. Certain Receptor Tyrosine Kinases (RTKs) Are Hyperactivated in Response to CDK4/6 Inhibition in TNBC
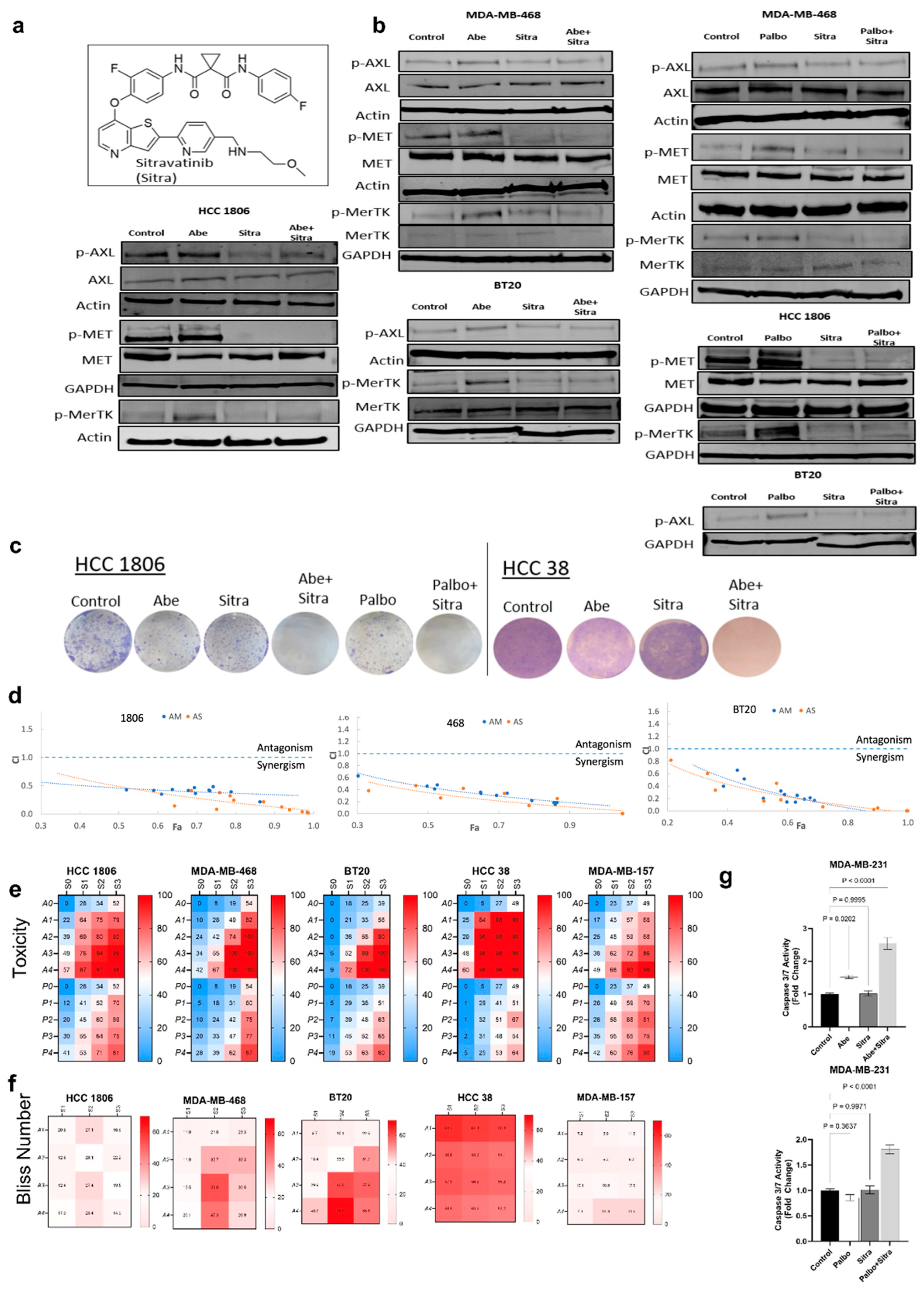
3.2. Simultaneous TAM/Met and CDK4/6 Inhibition Has Synergistic Activity against TNBC
3.3. Combined TAM/Met and CDK4/6 Inhibition Exhibits Enhanced Cytotoxicity against Drug-Resistant HER2+ Breast Cancer
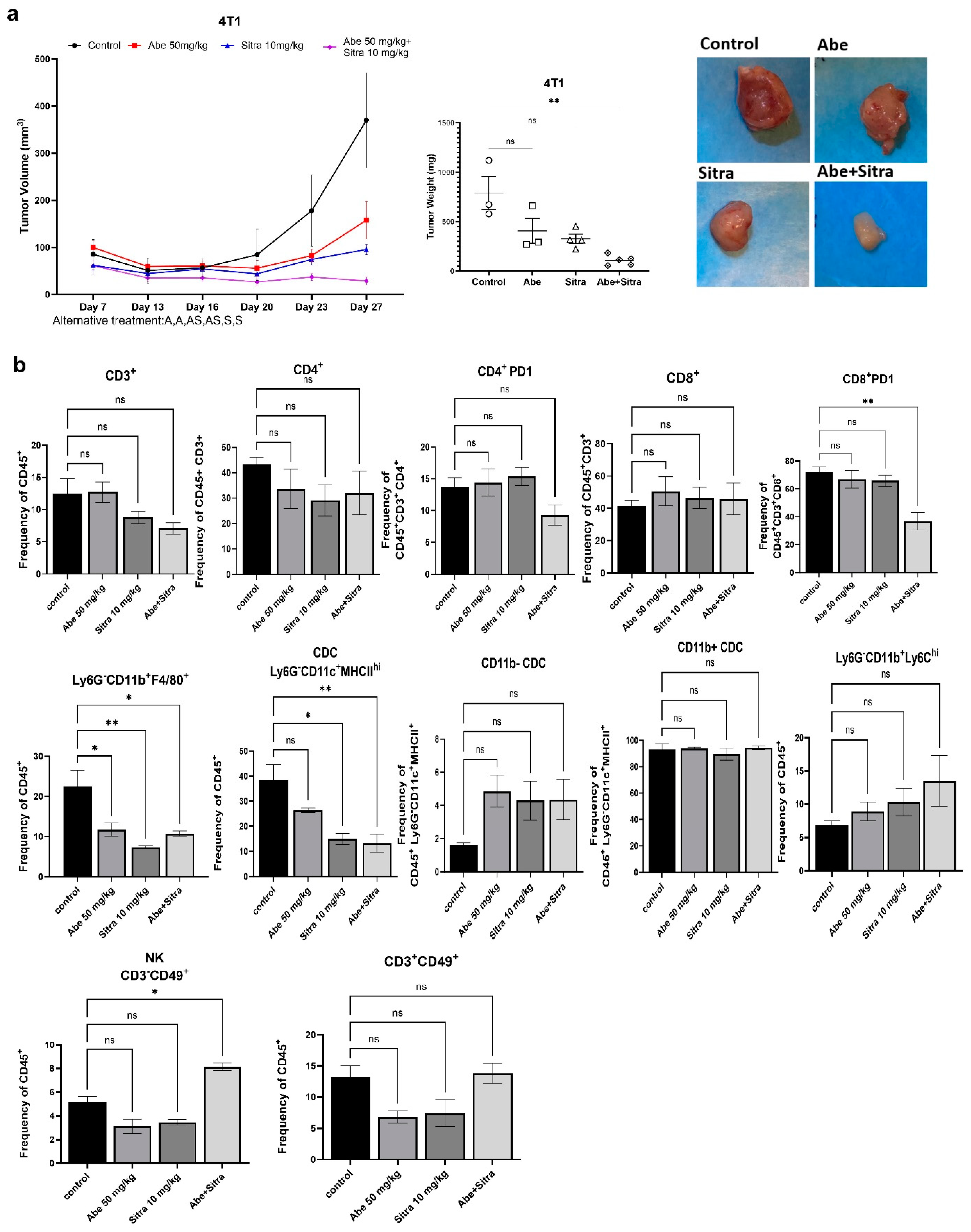
3.4. The Combination of TAM/Met and CDK4/6 Inhibition Is Highly Effective against TNBC and Drug-Resistant HER2+ Breast Cancer In Vivo
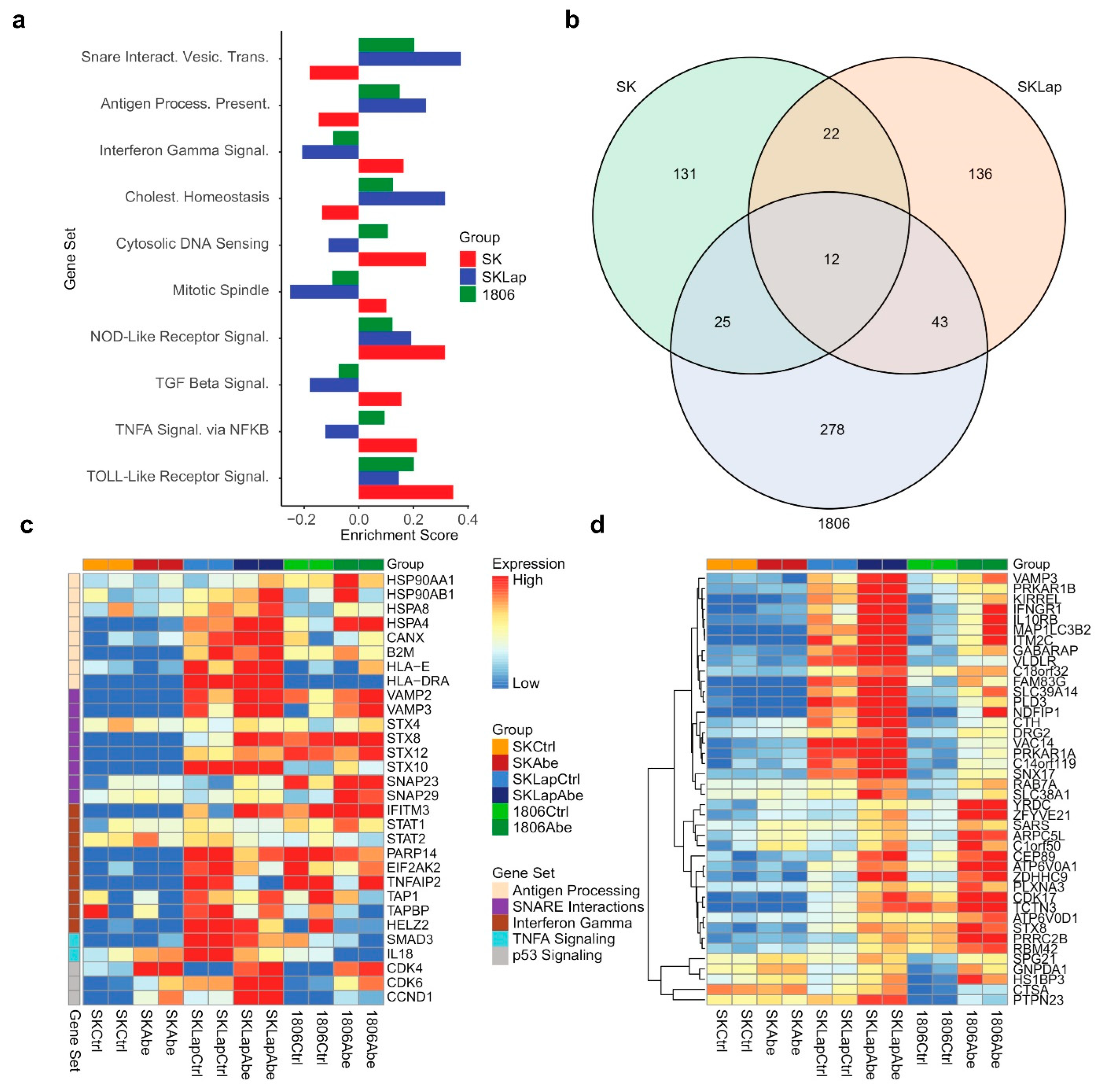
3.5. The Combined Treatment Reverses Immunosuppressive Tumor Microenvironment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guan, X.; Fan, Z.; Ching, L.M.; Li, Y.; Wang, X.; Cao, W.M.; Liu, D.X. Non-Invasive Biomarkers for Early Detection of Breast Cancer. Cancers 2020, 12, 2767. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, L.; Sanchez Cendra, A.; Sanchez Cendra, C.; Roberts Cervantes, E.D.; Espinosa, J.C.; Pekarek, T.; Fraile-Martinez, O.; Garcia-Montero, C.; Rodriguez-Slocker, A.M.; Jimenez-Alvarez, L.; et al. Exploring Biomarkers in Breast Cancer: Hallmarks of Diagnosis, Treatment, and Follow-Up in Clinical Practice. Medicina 2024, 60, 168. [Google Scholar] [CrossRef] [PubMed]
- Marra, A.; Chandarlapaty, S.; Modi, S. Management of patients with advanced-stage HER2-positive breast cancer: Current evidence and future perspectives. Nat. Rev. Clin. Oncol. 2024, 21, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Afghahi, A.; Sledge, G.W., Jr. Targeted Therapy for Cancer in the Genomic Era. Cancer J. 2015, 21, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Natarajan, E.; Balaji Raghavendran, H.R.; Markandan, U.D. Molecular Classification, Treatment, and Genetic Biomarkers in Triple-Negative Breast Cancer: A Review. Technol. Cancer Res. Treat. 2023, 22, 15330338221145246. [Google Scholar] [CrossRef] [PubMed]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, S142–S150. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Abramson, V.; Tan, T.; Dent, R. The Evolution of Triple-Negative Breast Cancer: From Biology to Novel Therapeutics. Am. Soc. Clin. Oncol. Educ. Book. 2016, 35, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Kagihara, J.A.; Andress, M.; Diamond, J.R. Nab-paclitaxel and atezolizumab for the treatment of PD-L1-positive, metastatic triple-negative breast cancer: Review and future directions. Expert. Rev. Precis. Med. Drug Dev. 2020, 5, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients with Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Fornier, M.; Fumoleau, P. The paradox of triple negative breast cancer: Novel approaches to treatment. Breast J. 2012, 18, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Royce, M.; Osgood, C.; Mulkey, F.; Bloomquist, E.; Pierce, W.F.; Roy, A.; Kalavar, S.; Ghosh, S.; Philip, R.; Rizvi, F.; et al. FDA Approval Summary: Abemaciclib with Endocrine Therapy for High-Risk Early Breast Cancer. J. Clin. Oncol. 2022, 40, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination with Fulvestrant in Women with HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortes, J.; Dieras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR(+)/HER2(-) Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Gao, J.; Wang, M.; Li, M. Potential Prospect of CDK4/6 Inhibitors in Triple-Negative Breast Cancer. Cancer Manag. Res. 2021, 13, 5223–5237. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Palencia, M.; Reza Herrera, L.; Parida, P.K.; Ghosh, S.; Kim, K.; Das, N.M.; Gonzalez-Ericsson, P.I.; Sanders, M.E.; Mobley, B.C.; Diegeler, S.; et al. AXL/WRNIP1 mediates replication stress response and promotes therapy resistance and metachronous metastasis in HER2+ breast cancer. Cancer Res. 2024, 84, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Bardol, T.; Eslami, S.Z.; Masmoudi, D.; Alexandre, M.; Duboys de Labarre, M.; Bobrie, A.; D’Hondt, V.; Guiu, S.; Kurma, K.; Cayrefourcq, L.; et al. First evidence of AXL expression on circulating tumor cells in metastatic breast cancer patients: A proof-of-concept study. Cancer Med. 2023, 13, e6843. [Google Scholar] [CrossRef]
- Goyette, M.A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2023, 42, 113604. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, J.; Zarrer, J.; Gensch, V.; Riecken, K.; Berenbrok, N.; Luu, T.V.; Beitzen-Heineke, A.; Vargas-Delgado, M.E.; Pantel, K.; Bokemeyer, C.; et al. Regulation of bone homeostasis by MERTK and TYRO3. Nat. Commun. 2022, 13, 7689. [Google Scholar] [CrossRef] [PubMed]
- Davra, V.; Kumar, S.; Geng, K.; Calianese, D.; Mehta, D.; Gadiyar, V.; Kasikara, C.; Lahey, K.C.; Chang, Y.J.; Wichroski, M.; et al. Axl and Mertk Receptors Cooperate to Promote Breast Cancer Progression by Combined Oncogenic Signaling and Evasion of Host Antitumor Immunity. Cancer Res. 2021, 81, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Penninger, J.M. The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy. Cancers 2016, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Olmez, I.; Shen, W.; McDonald, H.; Ozpolat, B. Dedifferentiation of patient-derived glioblastoma multiforme cell lines results in a cancer stem cell-like state with mitogen-independent growth. J. Cell. Mol. Med. 2015, 19, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Son, M.Y.; Seol, B.; Han, Y.M.; Cho, Y.S. Comparative receptor tyrosine kinase profiling identifies a novel role for AXL in human stem cell pluripotency. Hum. Mol. Genet. 2014, 23, 1802–1816. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.N.; Potter, J.A.; Aldo, P.; Kwon, J.Y.; Pitruzzello, M.; Tong, M.; Guller, S.; Rothlin, C.V.; Mor, G.; Abrahams, V.M. Viral Infection Sensitizes Human Fetal Membranes to Bacterial Lipopolysaccharide by MERTK Inhibition and Inflammasome Activation. J. Immunol. 2017, 199, 2885–2895. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Carbonell, W.S.; Hu, Y.L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Perea, J.; Yu, Q.; Gygi, S.P.; Paulo, J.A. Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res. 2018, 17, 2226–2236. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, Z.; Bomgarden, R.D.; Pike, I.; Kuhn, K.; Rogers, J.C.; Roberts, T.M.; Gygi, S.P.; Paulo, J.A. TMTpro-18plex: The Expanded and Complete Set of TMTpro Reagents for Sample Multiplexing. J. Proteome Res. 2021, 20, 2964–2972. [Google Scholar] [CrossRef] [PubMed]
- Schweppe, D.K.; Eng, J.K.; Yu, Q.; Bailey, D.; Rad, R.; Navarrete-Perea, J.; Huttlin, E.L.; Erickson, B.K.; Paulo, J.A.; Gygi, S.P. Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. J. Proteome Res. 2020, 19, 2026–2034. [Google Scholar] [CrossRef]
- Rad, R.; Li, J.; Mintseris, J.; O’Connell, J.; Gygi, S.P.; Schweppe, D.K. Improved Monoisotopic Mass Estimation for Deeper Proteome Coverage. J. Proteome Res. 2021, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef]
- Olmez, I.; Brenneman, B.; Xiao, A.; Serbulea, V.; Benamar, M.; Zhang, Y.; Manigat, L.; Abbas, T.; Lee, J.; Nakano, I.; et al. Combined CDK4/6 and mTOR Inhibition Is Synergistic against Glioblastoma via Multiple Mechanisms. Clin. Cancer Res. 2017, 23, 6958–6968. [Google Scholar] [CrossRef]
- Katsuta, E.; Oshi, M.; Rashid, O.M.; Takabe, K. Generating a Murine Orthotopic Metastatic Breast Cancer Model and Performing Murine Radical Mastectomy. J. Vis. Exp. 2018, 141, e57849. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: https://www.r-project.org/ (accessed on 1 March 2022).
- Fitzgerald, J.B.; Schoeberl, B.; Nielsen, U.B.; Sorger, P.K. Systems biology and combination therapy in the quest for clinical efficacy. Nat. Chem. Biol. 2006, 2, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Wetzker, R.; Bohmer, F.D. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat. Rev. Mol. Cell Biol. 2003, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Jezequel, P.; Gouraud, W.; Ben Azzouz, F.; Guerin-Charbonnel, C.; Juin, P.P.; Lasla, H.; Campone, M. bc-GenExMiner 4.5: New mining module computes breast cancer differential gene expression analyses. Database 2021, 2021, baab007. [Google Scholar] [CrossRef] [PubMed]
- Oliva, M.; Chepeha, D.; Araujo, D.V.; Diaz-Mejia, J.J.; Olson, P.; Prawira, A.; Spreafico, A.; Bratman, S.V.; Shek, T.; de Almeida, J.; et al. Antitumor immune effects of preoperative sitravatinib and nivolumab in oral cavity cancer: SNOW window-of-opportunity study. J. Immunother. Cancer 2021, 9, e003476. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Goswami, S.; Thall, P.F.; Wang, X.; Yuan, Y.; Jonasch, E.; Gao, J.; Campbell, M.T.; Shah, A.Y.; Corn, P.G.; et al. A phase 1-2 trial of sitravatinib and nivolumab in clear cell renal cell carcinoma following progression on antiangiogenic therapy. Sci. Transl. Med. 2022, 14, eabm6420. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.; Cho, B.C.; Heist, R.; Bazhenova, L.; Werner, T.; Goel, S.; Kim, D.W.; Adkins, D.; Carvajal, R.D.; Alva, A.; et al. First-in-human phase 1/1b study to evaluate sitravatinib in patients with advanced solid tumors. Investig. New Drugs 2022, 40, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Berz, D.; Gadgeel, S.M.; Iams, W.T.; Bruno, D.S.; Blakely, C.M.; Spira, A.I.; Patel, M.R.; Waterhouse, D.M.; Richards, D.A.; et al. MRTX-500 Phase 2 Trial: Sitravatinib with Nivolumab in Patients with Nonsquamous NSCLC Progressing on or after Checkpoint Inhibitor Therapy or Chemotherapy. J. Thorac. Oncol. 2023, 18, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; de Marinis, F.; Dumoulin, D.; Reynolds, C.; Theelen, W.; Percent, I.; Gutierrez Calderon, V.; Johnson, M.L.; Madroszyk-Flandin, A.; Garon, E.B.; et al. SAPPHIRE: Phase III study of sitravatinib plus nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. Ann. Oncol. 2024, 35, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Sung, T.; Jessen, B.A.; Thibault, S.; Finkelstein, M.B.; Khan, N.K.; Sacaan, A.I. Mechanistic Investigation of Bone Marrow Suppression Associated with Palbociclib and its Differentiation from Cytotoxic Chemotherapies. Clin. Cancer Res. 2016, 22, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Mills, C.E.; Subramanian, K.; Chen, C.; Chung, M.; Boswell, S.A.; Everley, R.A.; Liu, C.; Walmsley, C.S.; Juric, D.; et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem. Biol. 2019, 26, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. g:Profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef] [PubMed]
- Andreu, Z.; Yanez-Mo, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef] [PubMed]
- Wiedemeyer, W.R.; Dunn, I.F.; Quayle, S.N.; Zhang, J.; Chheda, M.G.; Dunn, G.P.; Zhuang, L.; Rosenbluh, J.; Chen, S.; Xiao, Y.; et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc. Natl. Acad. Sci. USA 2010, 107, 11501–11506. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.L.; Thangavel, C.; McClendon, A.K.; Reed, C.A.; Knudsen, E.S. Therapeutic CDK4/6 inhibition in breast cancer: Key mechanisms of response and failure. Oncogene 2010, 29, 4018–4032. [Google Scholar] [CrossRef]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Kim, S.Y.; Kim, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, S.; Liu, Y.; Dean, D.C. Mutation of the Rb1 pathway leads to overexpression of mTor, constitutive phosphorylation of Akt on serine 473, resistance to anoikis, and a block in c-Raf activation. Mol. Cell Biol. 2009, 29, 5710–5717. [Google Scholar] [CrossRef] [PubMed]
- Minuti, G.; Landi, L. MET deregulation in breast cancer. Ann. Transl. Med. 2015, 3, 181. [Google Scholar] [CrossRef]
- Aehnlich, P.; Powell, R.M.; Peeters, M.J.W.; Rahbech, A.; Thor Straten, P. TAM Receptor Inhibition-Implications for Cancer and the Immune System. Cancers 2021, 13, 1195. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Engelman, J.A. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci. Signal. 2013, 6, re6. [Google Scholar] [CrossRef] [PubMed]
- McNeill, R.S.; Canoutas, D.A.; Stuhlmiller, T.J.; Dhruv, H.D.; Irvin, D.M.; Bash, R.E.; Angus, S.P.; Herring, L.E.; Simon, J.M.; Skinner, K.R.; et al. Combination therapy with potent PI3K and MAPK inhibitors overcomes adaptive kinome resistance to single agents in preclinical models of glioblastoma. Neuro-Oncology 2017, 19, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.J.; Ghosh, D.; Gujja, S. Signaling Circuits and Regulation of Immune Suppression by Ovarian Tumor-Associated Macrophages. Vaccines 2015, 3, 448–466. [Google Scholar] [CrossRef] [PubMed]
- Gomez, V.; Eykyn, T.R.; Mustapha, R.; Flores-Borja, F.; Male, V.; Barber, P.R.; Patsialou, A.; Green, R.; Panagaki, F.; Li, C.W.; et al. Breast cancer-associated macrophages promote tumorigenesis by suppressing succinate dehydrogenase in tumor cells. Sci. Signal. 2020, 13, eaax4585. [Google Scholar] [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demirsoy, S.; Tran, H.; Liu, J.; Li, Y.; Yang, S.; Aregawi, D.; Glantz, M.J.; Jacob, N.K.; Walter, V.; Schell, T.D.; et al. Targeting Tyro3, Axl, and MerTK Receptor Tyrosine Kinases Significantly Sensitizes Triple-Negative Breast Cancer to CDK4/6 Inhibition. Cancers 2024, 16, 2253. https://doi.org/10.3390/cancers16122253
Demirsoy S, Tran H, Liu J, Li Y, Yang S, Aregawi D, Glantz MJ, Jacob NK, Walter V, Schell TD, et al. Targeting Tyro3, Axl, and MerTK Receptor Tyrosine Kinases Significantly Sensitizes Triple-Negative Breast Cancer to CDK4/6 Inhibition. Cancers. 2024; 16(12):2253. https://doi.org/10.3390/cancers16122253
Chicago/Turabian StyleDemirsoy, Seyma, Ha Tran, Joseph Liu, Yunzhan Li, Shengyu Yang, Dawit Aregawi, Michael J. Glantz, Naduparambil K. Jacob, Vonn Walter, Todd D. Schell, and et al. 2024. "Targeting Tyro3, Axl, and MerTK Receptor Tyrosine Kinases Significantly Sensitizes Triple-Negative Breast Cancer to CDK4/6 Inhibition" Cancers 16, no. 12: 2253. https://doi.org/10.3390/cancers16122253
APA StyleDemirsoy, S., Tran, H., Liu, J., Li, Y., Yang, S., Aregawi, D., Glantz, M. J., Jacob, N. K., Walter, V., Schell, T. D., & Olmez, I. (2024). Targeting Tyro3, Axl, and MerTK Receptor Tyrosine Kinases Significantly Sensitizes Triple-Negative Breast Cancer to CDK4/6 Inhibition. Cancers, 16(12), 2253. https://doi.org/10.3390/cancers16122253