P18: Novel Anticancer Peptide from Induced Tumor-Suppressing Cells Targeting Breast Cancer and Bone Metastasis

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Peptides P11 to P22 and P18 Analogs
2.3. MTT and EdU Assays
2.4. Scratch Assay
2.5. Transwell Invasion Assay
2.6. Western Blot Analysis
2.7. Fluorescence Resonance Energy Transfer (FRET)-Based GTPase Activity Assay
2.8. Ex Vivo Cancer Tissue Assay
2.9. Bone Microenvironment Assay
2.10. Molecular Docking Analysis
2.11. Statistical Analysis
3. Results
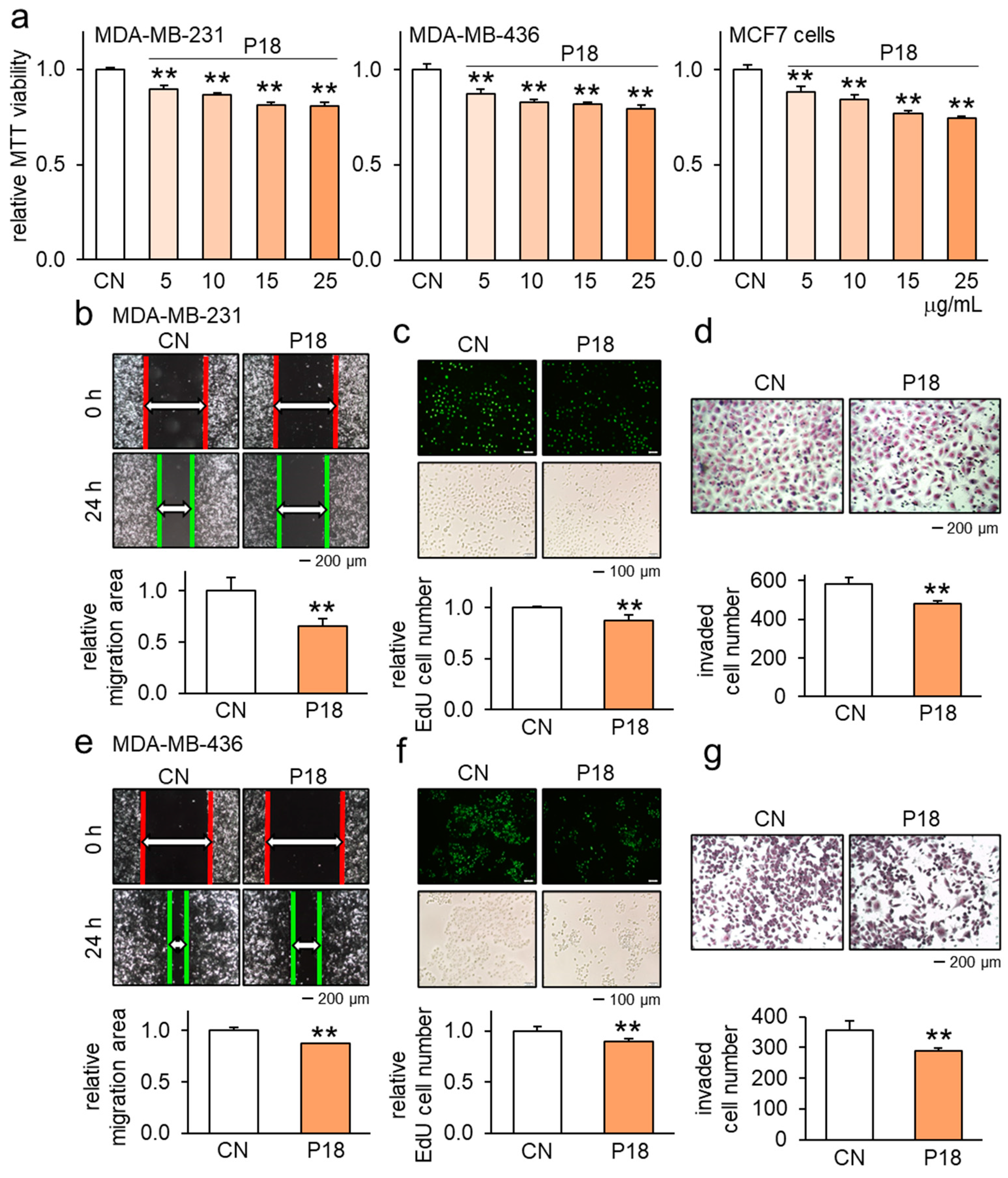
3.1. Anticancer Effects of P18 on Breast Cancer Cells
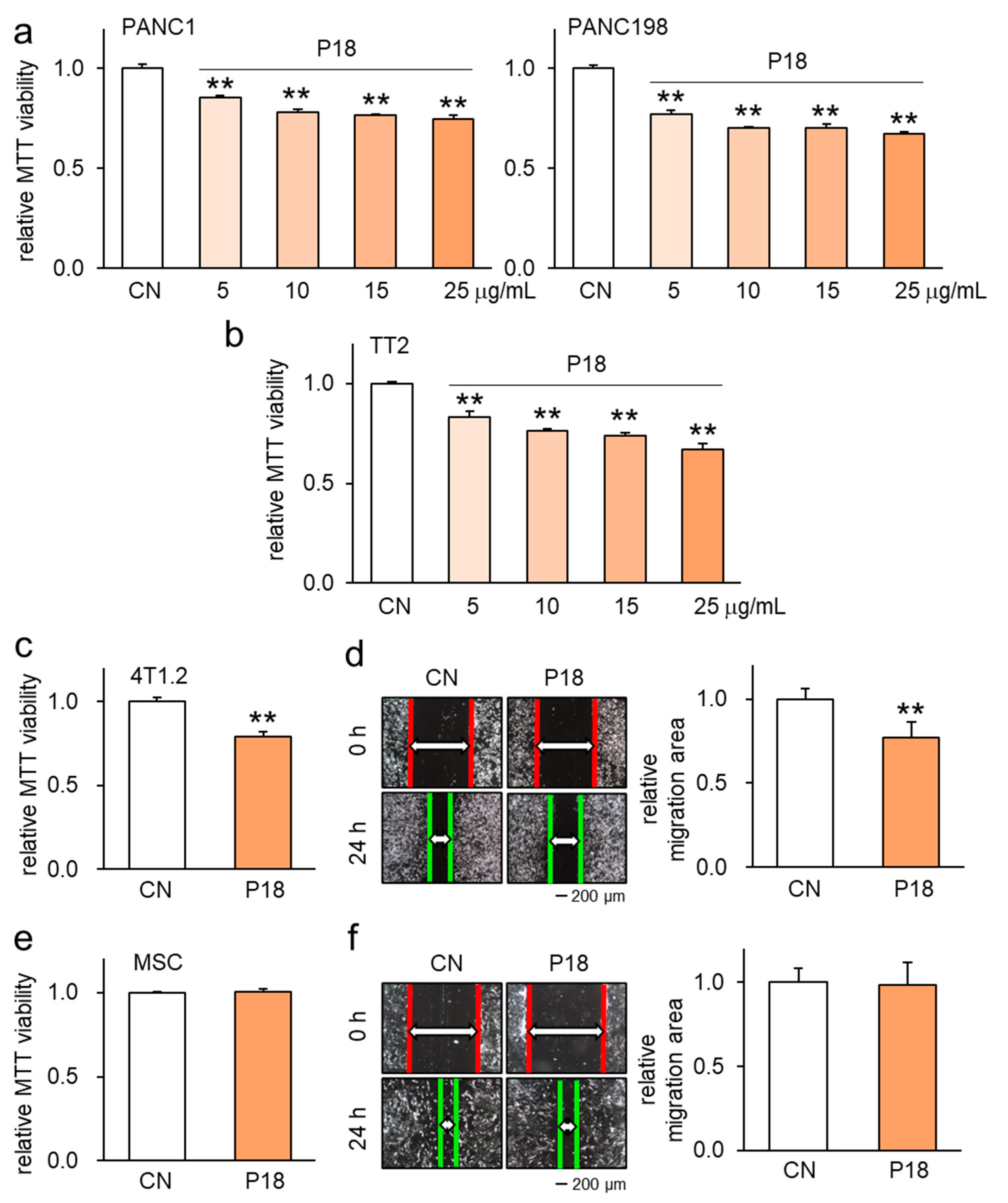
3.2. Antitumor Effect of P18 on Other Tumor Cell Lines but Not MSCs
3.3. Enhanced Antitumor Effects in Combination with Chemotherapeutic Agents
3.4. Combinatorial Effects with Other ACPs as well as Anti-Osteolytic Effects
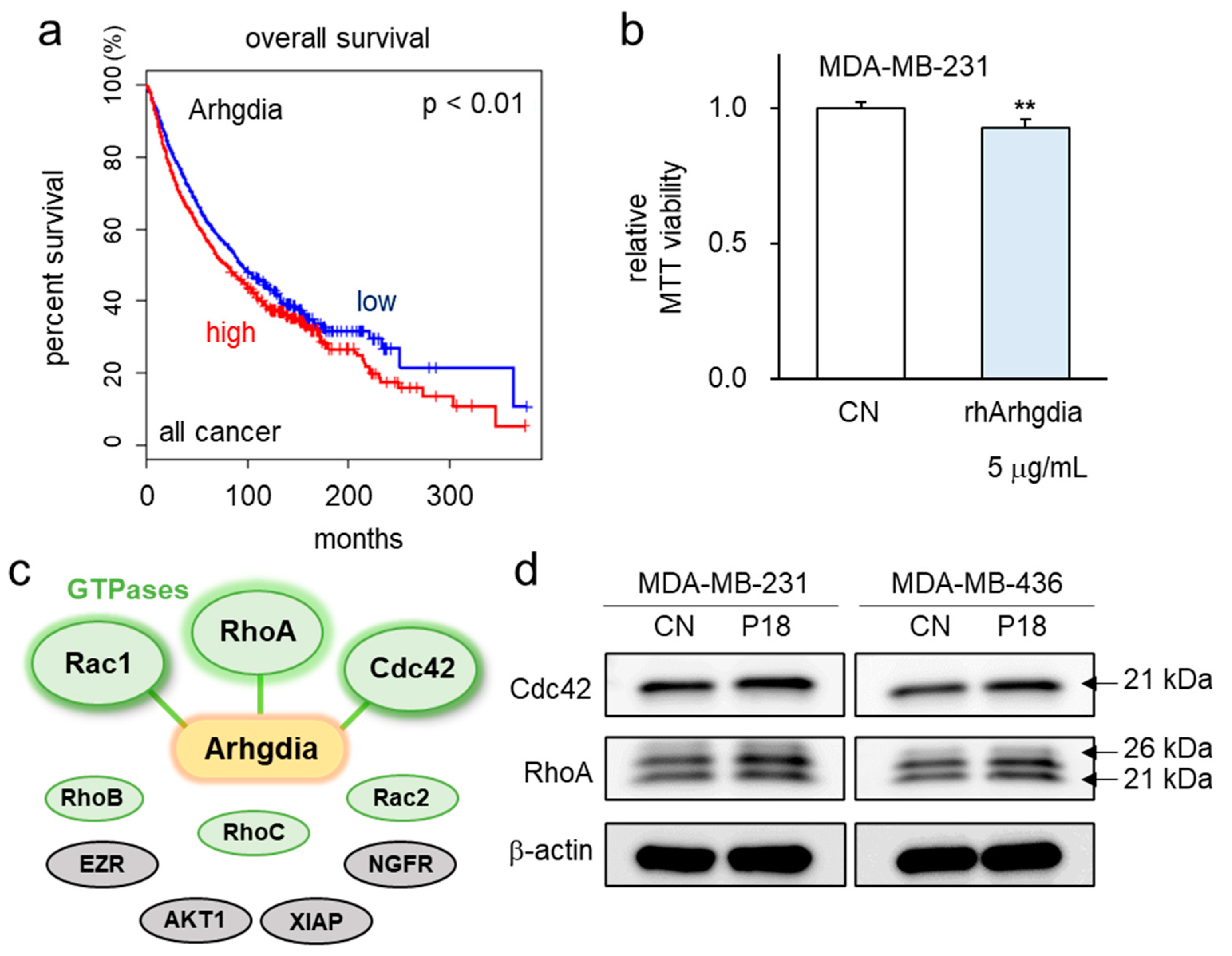
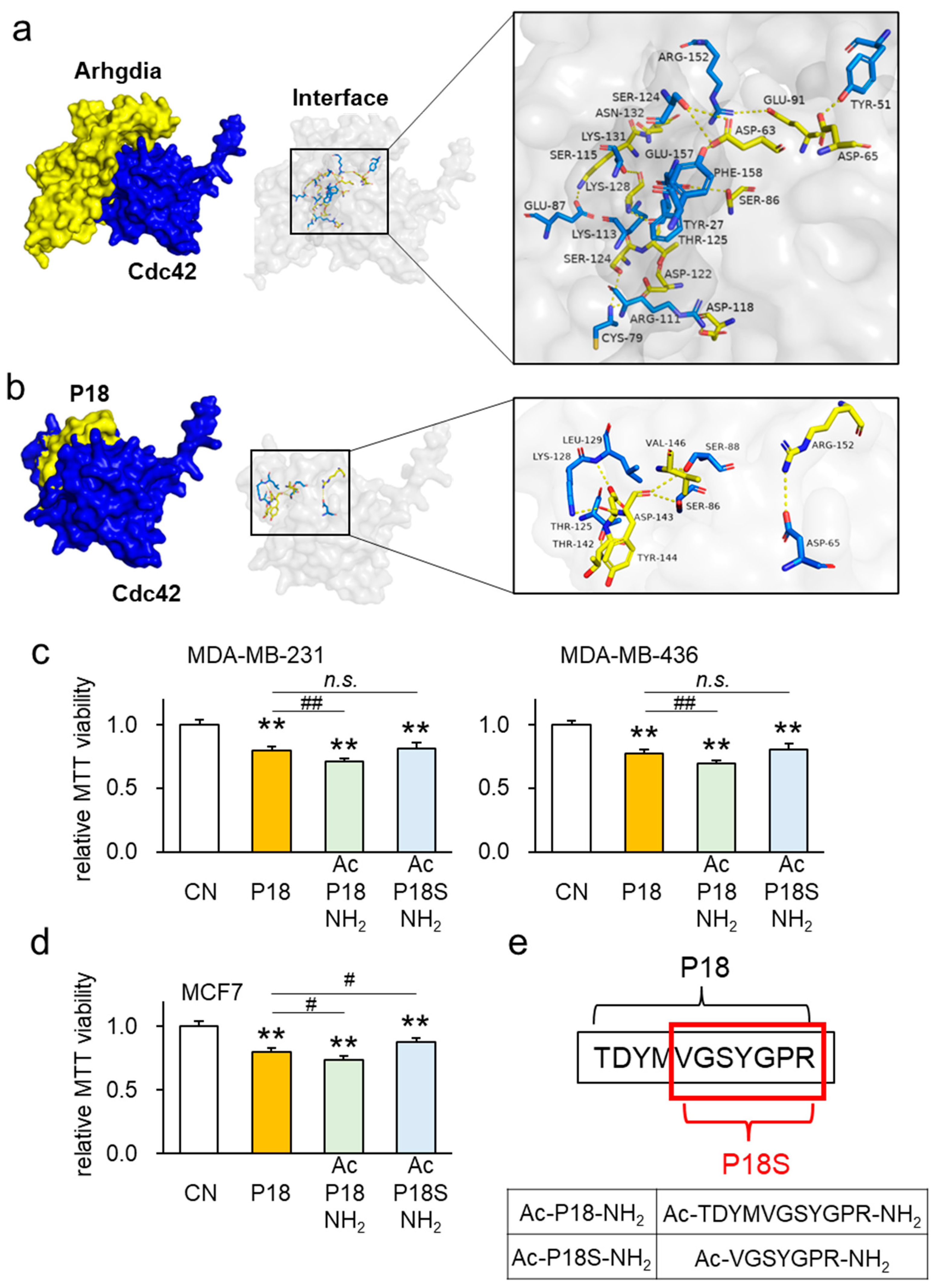
3.5. P18-Containing Protein Arhgdia and Its Binding Partners
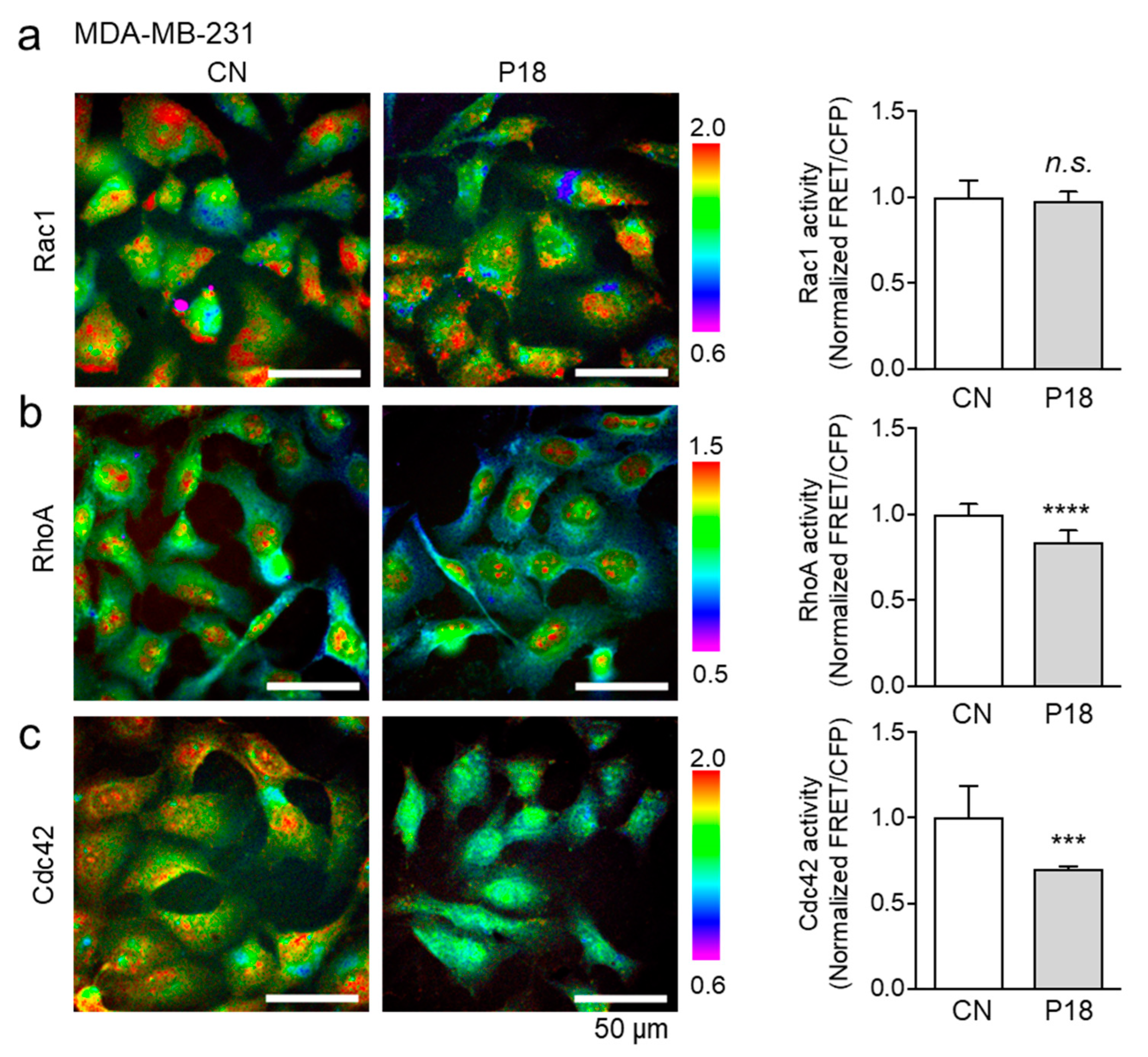
3.6. Effects of P18 on GTPase Activities of Rac1, RhoA, and Cdc42
3.7. Interaction between Arhgdia and Cdc42
3.8. Reduction in Oncoproteins by P18 in Ex Vivo Bone Culture
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Houthuijzen, J.M.; de Bruijn, R.; van der Burg, E.; Drenth, A.P.; Wientjens, E.; Filipovic, T.; Bullock, E.; Brambillasca, C.S.; Pulver, E.M.; Nieuwland, M.; et al. CD26-negative and CD26-positive tissue-resident fibroblasts contribute to functionally distinct CAF subpopulations in breast cancer. Nat. Commun. 2023, 14, 183. [Google Scholar] [CrossRef]
- Barrios, C.H. Global challenges in breast cancer detection and treatment. Breast 2022, 62 (Suppl. 1), S3–S6. [Google Scholar] [CrossRef]
- Debela, D.T.; Muzazu, S.G.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef]
- Marazzi, F.; Orlandi, A.; Manfrida, S.; Masiello, V.; Di Leone, A.; Massaccesi, M.; Moschella, F.; Franceschini, G.; Bria, E.; Gambacorta, M.A.; et al. Diagnosis and Treatment of Bone Metastases in Breast Cancer: Radiotherapy, Local Approach and Systemic Therapy in a Guide for Clinicians. Cancers 2020, 12, 2390. [Google Scholar] [CrossRef]
- Zhou, H.X.; Pang, X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef]
- Quemé-Peña, M.; Juhász, T.; Kohut, G.; Ricci, M.; Singh, P.; Szigyártó, I.C.; Papp, Z.I.; Fülöp, L.; Beke-Somfai, T. Membrane Association Modes of Natural Anticancer Peptides: Mechanistic Details on Helicity, Orientation, and Surface Coverage. Int. J. Mol. Sci. 2021, 22, 8613. [Google Scholar] [CrossRef]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef]
- Yewdell, J.W. MHC Class I Immunopeptidome: Past, Present, and Future. Mol. Cell. Proteom. 2022, 21, 100230. [Google Scholar] [CrossRef]
- Wang, X.; Ai, X.; Zhu, Z.; Zhang, M.; Pan, F.; Yang, Z.; Wang, O.; Zhao, L.; Zhao, L. Pancreatic lipase inhibitory effects of peptides derived from sesame proteins: In silico and in vitro analyses. Int. J. Biol. Macromol. 2022, 222, 1531–1537. [Google Scholar] [CrossRef]
- Wang, X.; Yu, Z.; Liu, W.; Tang, H.; Yi, D.; Wei, M. Recent progress on MHC-I epitope prediction in tumor immunotherapy. Am. J. Cancer Res. 2021, 11, 2401–2416. [Google Scholar]
- Li, K.; Sun, X.; Zha, R.; Liu, S.; Feng, Y.; Sano, T.; Aryal, U.K.; Sudo, A.; Li, B.Y.; Yokota, H. Counterintuitive production of tumor-suppressive secretomes from Oct4- and c-Myc-overexpressing tumor cells and MSCs. Theranostics 2022, 12, 3084–3103. [Google Scholar] [CrossRef] [PubMed]
- Gähwiler, E.K.N.; Motta, S.E.; Martin, M.; Nugraha, B.; Hoerstrup, S.P.; Emmert, M.Y. Human iPSCs and Genome Editing Technologies for Precision Cardiovascular Tissue Engineering. Front. Cell Dev. Biol. 2021, 9, 639699. [Google Scholar] [CrossRef]
- Cui, C.P.; Huo, Q.J.; Xiong, X.; Li, K.X.; Ma, P.; Qiang, G.F.; Pandya, P.H.; Saadatzadeh, M.R.; Bijangi Vishehsaraei, K.; Kacena, M.A.; et al. Anticancer peptides from induced tumor-suppressing cells for inhibiting osteosarcoma cells. Am. J. Cancer Res. 2023, 13, 4057–4072. [Google Scholar]
- Cui, C.; Huo, Q.; Xiong, X.; Li, K.; Fishel, M.L.; Li, B.; Yokota, H. Anticancer Peptides Derived from Aldolase A and Induced Tumor-Suppressing Cells Inhibit Pancreatic Ductal Adenocarcinoma Cells. Pharmaceutics 2023, 15, 2447. [Google Scholar] [CrossRef]
- Rossi, M.; Cappadone, C.; Picone, G.; Bisi, A.; Farruggia, G.; Belluti, F.; Blasi, P.; Gobbi, S.; Malucelli, E. Natural-like Chalcones with Antitumor Activity on Human MG63 Osteosarcoma Cells. Molecules 2022, 27, 3751. [Google Scholar] [CrossRef]
- Miura, K. Imaging technologies for the detection of multiple stains in proteomics. Proteomics 2003, 3, 1097–1108. [Google Scholar] [CrossRef]
- Yoshizaki, H.; Ohba, Y.; Kurokawa, K.; Itoh, R.E.; Nakamura, T.; Mochizuki, N.; Nagashima, K.; Matsuda, M. Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J. Cell Biol. 2003, 162, 223–232. [Google Scholar] [CrossRef]
- Itoh, R.E.; Kurokawa, K.; Ohba, Y.; Yoshizaki, H.; Mochizuki, N.; Matsuda, M. Activation of Rac and Cdc42 video imaged by fluorescent resonance energy transfer-based single-molecule probes in the membrane of living cells. Mol. Cell. Biol. 2002, 22, 6582–6591. [Google Scholar] [CrossRef]
- Wan, Q.; Cho, E.; Yokota, H.; Na, S. Rac1 and Cdc42 GTPases regulate shear stress-driven β-catenin signaling in osteoblasts. Biochem. Biophys. Res. Commun. 2013, 433, 502–507. [Google Scholar] [CrossRef]
- Palazzi, L.; Pasquato, A.; Vicario, M.; Roulin, A.; Polverino de Laureto, P.; Cendron, L. C-terminal tails mimicking bioactive intermediates cause different plasma degradation patterns and kinetics in neuropeptides γ-MSH, α-MSH, and neurotensin. J. Pept. Sci. 2020, 26, e3279. [Google Scholar] [CrossRef]
- Yang, Q.; Lei, X.; He, J.; Peng, Y.; Zhang, Y.; Ling, R.; Wu, C.; Zhang, G.; Zheng, B.; Chen, X.; et al. N4-Acetylcytidine Drives Glycolysis Addiction in Gastric Cancer via NAT10/SEPT9/HIF-1α Positive Feedback Loop. Adv. Sci. 2023, 10, e2300898. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Dastmalchi, S. Designing Novel Teduglutide Analogues with Improved Binding Affinity: An In Silico Peptide Engineering Approach. Curr. Comput.-Aided Drug Des. 2021, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, B.; Luan, J.; Sun, Y.; Wang, S.; Li, W.; Chen, L.; Wang, H.; Gao, Y.; Wang, J. Structural requirement of RARγ agonism through computational aspects. J. Mol. Model. 2023, 29, 108. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, X.; Sha, H.; Zhang, L.; Bian, X.; Han, X.; Liu, B. Tumor-penetrating peptide fused to a pro-apoptotic peptide facilitates effective gastric cancer therapy. Oncol. Rep. 2017, 37, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shi, W.; Li, J.; Liao, C.; Yang, L.; Huang, W.; Qian, H. Synthesis and biological evaluation of novel peptides based on antimicrobial peptides as potential agents with antitumor and multidrug resistance-reversing activities. Chem. Biol. Drug Des. 2017, 90, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Kepp, O.; Cerrato, G.; Pol, J.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Peptide-based vaccines in anticancer therapy. Oncoimmunology 2018, 7, e1511506. [Google Scholar] [CrossRef] [PubMed]
- Quandt, J.; Schlude, C.; Bartoschek, M.; Will, R.; Cid-Arregui, A.; Schölch, S.; Reissfelder, C.; Weitz, J.; Schneider, M.; Wiemann, S.; et al. Long-peptide vaccination with driver gene mutations in p53 and Kras induces cancer mutation-specific effector as well as regulatory T cell responses. Oncoimmunology 2018, 7, e1500671. [Google Scholar] [CrossRef] [PubMed]
- Dominari, A.; Hathaway Iii, D.; Pandav, K.; Matos, W.; Biswas, S.; Reddy, G.; Thevuthasan, S.; Khan, M.A.; Mathew, A.; Makkar, S.S.; et al. Thymosin alpha 1: A comprehensive review of the literature. World J. Virol. 2020, 9, 67–78. [Google Scholar] [CrossRef]
- Garaci, E.; Pica, F.; Rasi, G.; Favalli, C. Thymosin alpha 1 in the treatment of cancer: From basic research to clinical application. Int. J. Immunopharmacol. 2000, 22, 1067–1076. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, C.; Huo, Q.; Xiong, X.; Na, S.; Mitsuda, M.; Minami, K.; Li, B.; Yokota, H. P18: Novel Anticancer Peptide from Induced Tumor-Suppressing Cells Targeting Breast Cancer and Bone Metastasis. Cancers 2024, 16, 2230. https://doi.org/10.3390/cancers16122230
Cui C, Huo Q, Xiong X, Na S, Mitsuda M, Minami K, Li B, Yokota H. P18: Novel Anticancer Peptide from Induced Tumor-Suppressing Cells Targeting Breast Cancer and Bone Metastasis. Cancers. 2024; 16(12):2230. https://doi.org/10.3390/cancers16122230
Chicago/Turabian StyleCui, Changpeng, Qingji Huo, Xue Xiong, Sungsoo Na, Masaru Mitsuda, Kazumasa Minami, Baiyan Li, and Hiroki Yokota. 2024. "P18: Novel Anticancer Peptide from Induced Tumor-Suppressing Cells Targeting Breast Cancer and Bone Metastasis" Cancers 16, no. 12: 2230. https://doi.org/10.3390/cancers16122230
APA StyleCui, C., Huo, Q., Xiong, X., Na, S., Mitsuda, M., Minami, K., Li, B., & Yokota, H. (2024). P18: Novel Anticancer Peptide from Induced Tumor-Suppressing Cells Targeting Breast Cancer and Bone Metastasis. Cancers, 16(12), 2230. https://doi.org/10.3390/cancers16122230