

Dendritic Cells in Shaping Anti-Tumor T Cell Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
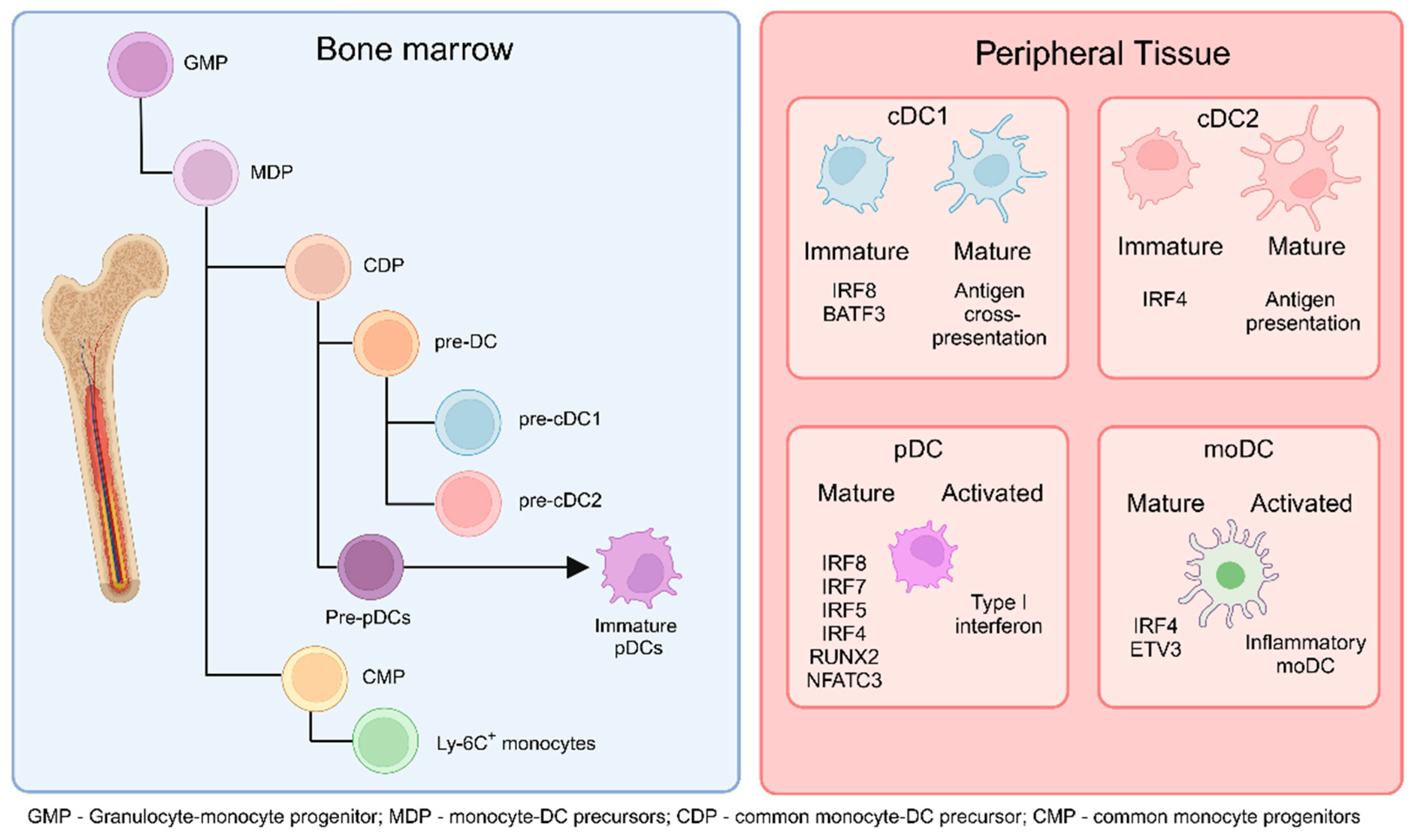
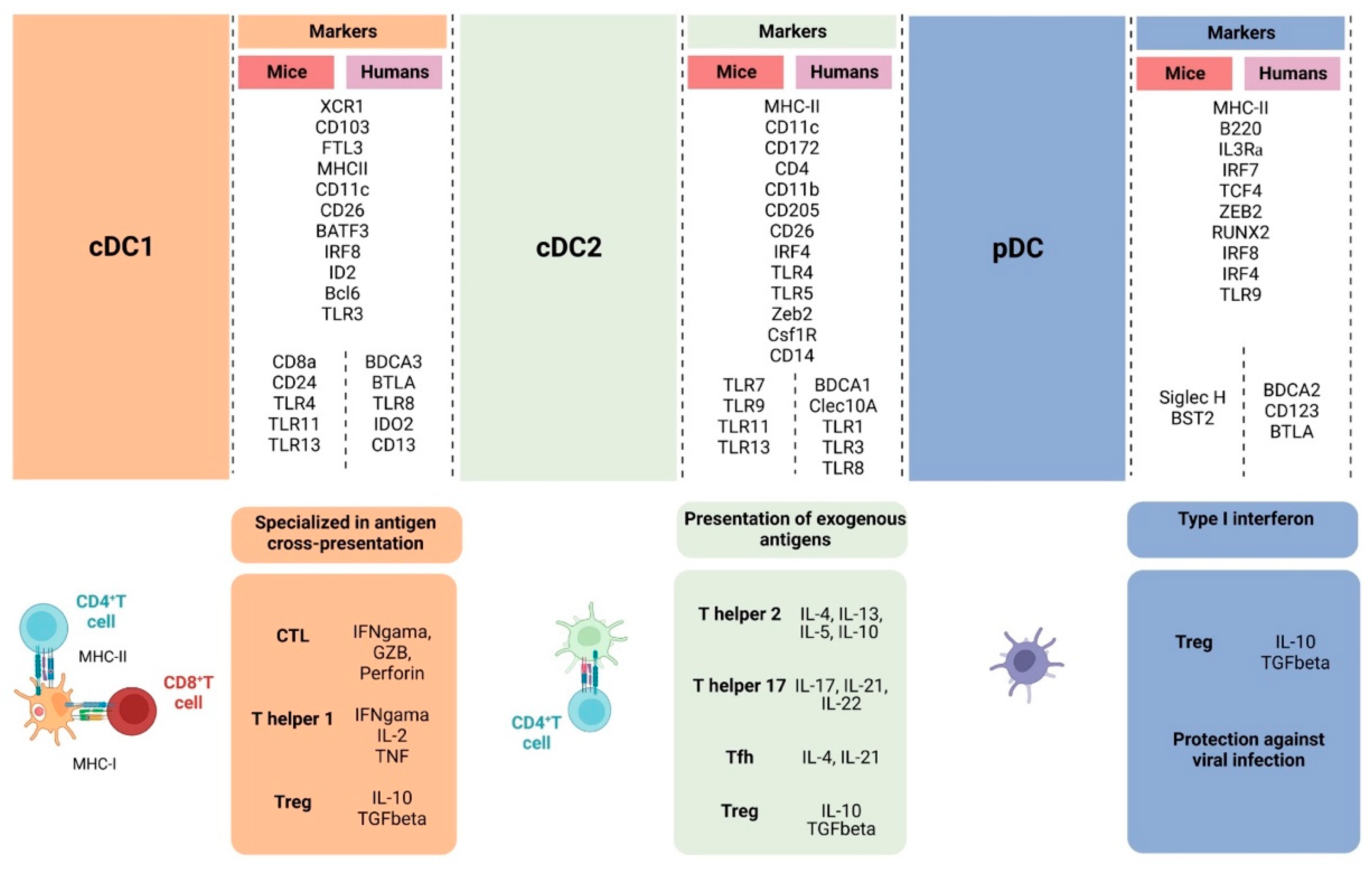
2. Subsets of Dendritic Cells
2.1. Conventional Dendritic Cells
2.2. Plasmacytoid Dendritic Cells
2.3. Monocyte-Derived Dendritic Cells
3. Functions of Dendritic Cells
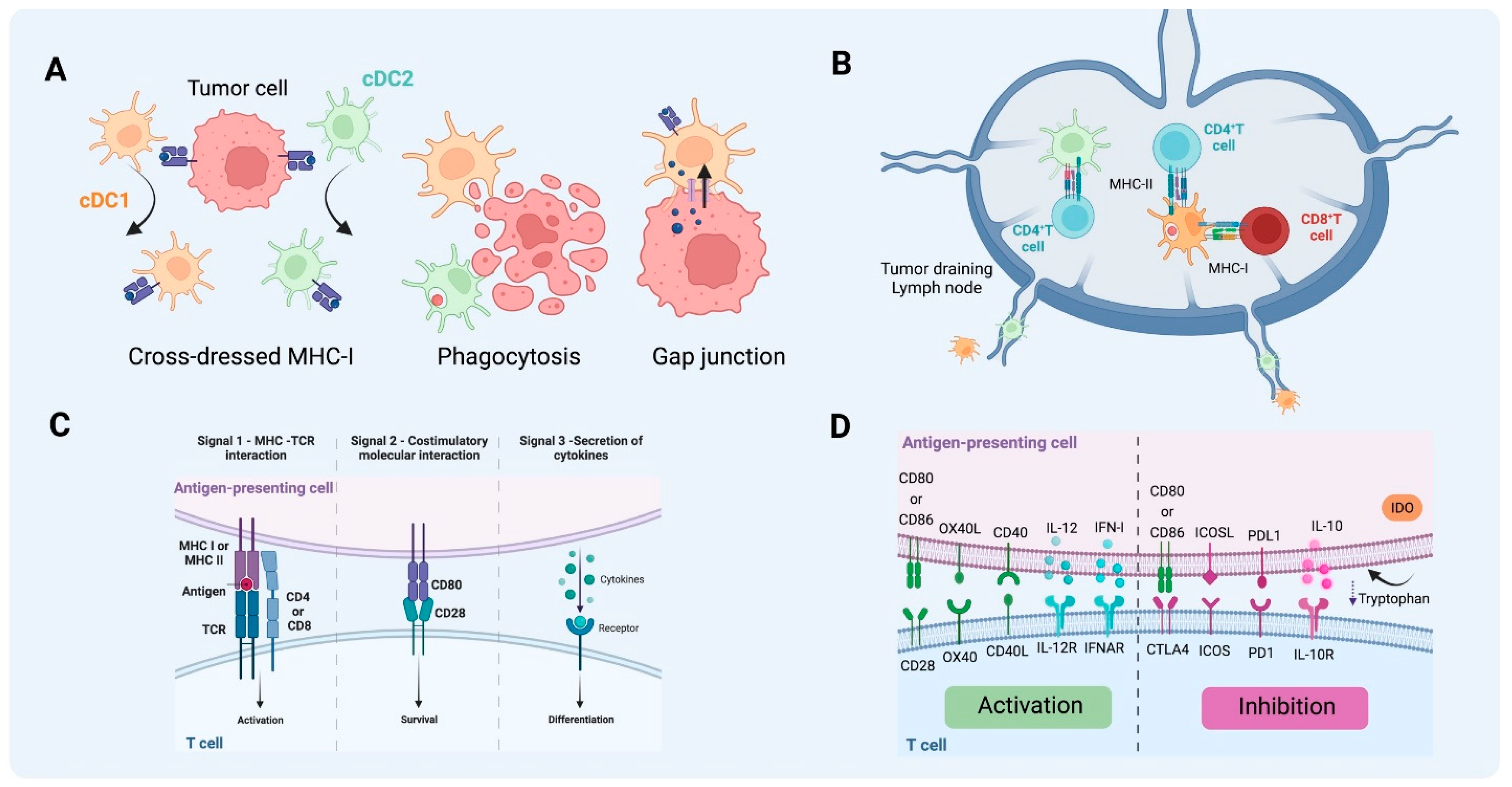
3.1. Antigen Uptake
3.2. Antigen Processing and Presentation
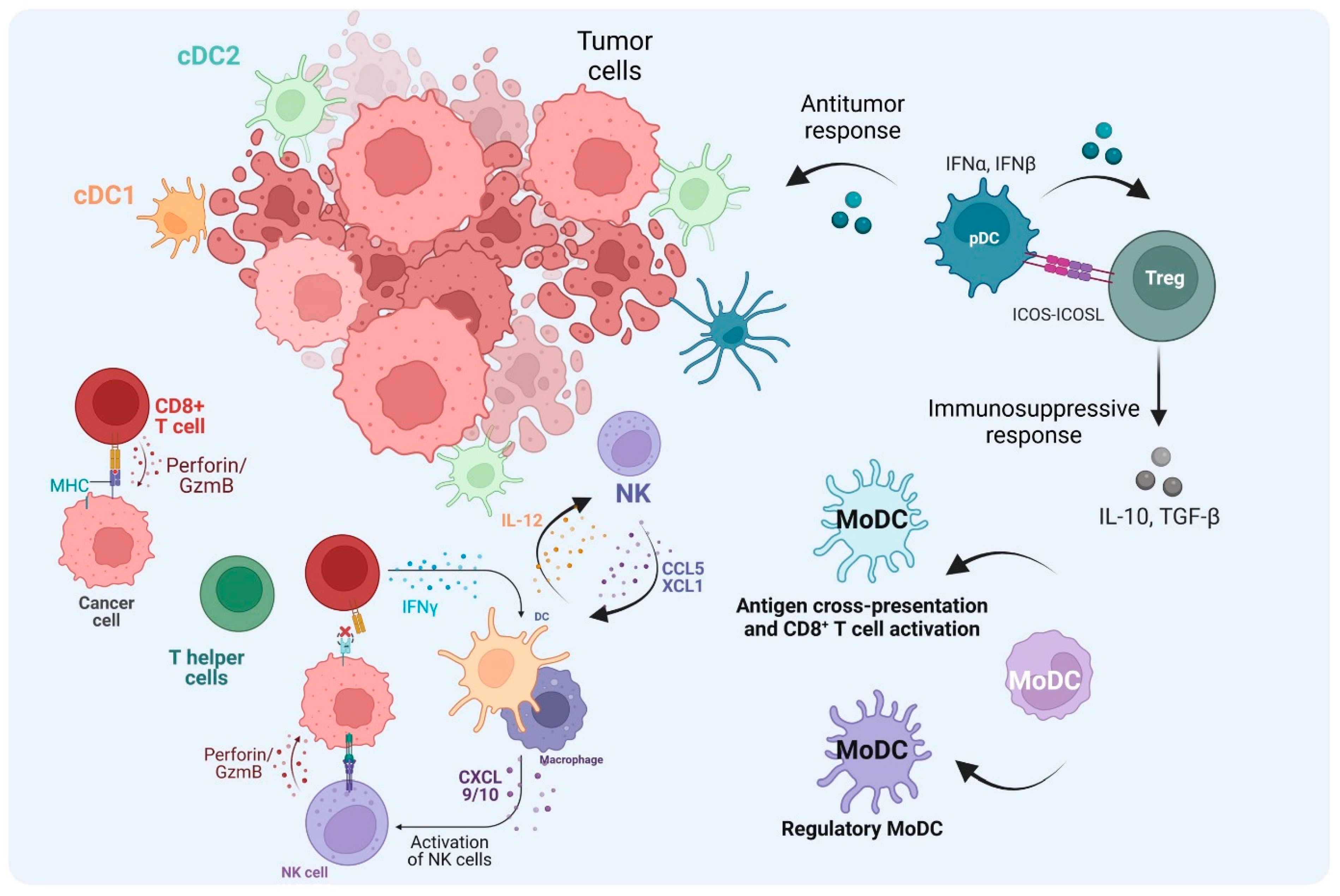
4. Dendritic Cells Shape T Cell Response in the TME
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [PubMed]
- Kayama, H.; Takeda, K. Regulation of intestinal homeostasis by innate and adaptive immunity. Int. Immunol. 2012, 24, 673–680. [Google Scholar] [CrossRef]
- Puhr, S.; Lee, J.; Zvezdova, E.; Zhou, Y.J.; Liu, K. Dendritic cell development-history, advances, and open questions. Semin. Immunol. 2015, 27, 388–396. [Google Scholar] [CrossRef]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, I.; Jeon, D.; Moseman, J.E.; Muralidhar, A.; Potluri, H.K.; McNeel, D.G. Role of b cells as antigen presenting cells. Front. Immunol. 2022, 13, 954936. [Google Scholar] [CrossRef] [PubMed]
- Wylie, B.; Macri, C.; Mintern, J.D.; Waithman, J. Dendritic cells and cancer: From biology to therapeutic intervention. Cancers 2019, 11, 521. [Google Scholar] [CrossRef]
- Belz, G.T.; Nutt, S.L. Transcriptional programming of the dendritic cell network. Nat. Rev. Immunol. 2012, 12, 101–113. [Google Scholar] [CrossRef]
- Satpathy, A.T.; Wu, X.; Albring, J.C.; Murphy, K.M. Re(de)fining the dendritic cell lineage. Nat. Immunol. 2012, 13, 1145–1154. [Google Scholar] [CrossRef]
- Hasegawa, H.; Matsumoto, T. Mechanisms of tolerance induction by dendritic cells in vivo. Front. Immunol. 2018, 9, 350. [Google Scholar] [CrossRef]
- Ferris, S.T.; Durai, V.; Wu, R.; Theisen, D.J.; Ward, J.P.; Bern, M.D.; Davidson, J.T.; Bagadia, P.; Liu, T.; Briseno, C.G.; et al. Cdc1 prime and are licensed by CD4+ t cells to induce anti-tumour immunity. Nature 2020, 584, 624–629. [Google Scholar] [CrossRef]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4+ t cell immunity. Cell 2019, 177, 556–571.e516. [Google Scholar] [CrossRef] [PubMed]
- Duong, E.; Fessenden, T.B.; Lutz, E.; Dinter, T.; Yim, L.; Blatt, S.; Bhutkar, A.; Wittrup, K.D.; Spranger, S. Type i interferon activates mhc class i-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ t cell immunity. Immunity 2022, 55, 308–323.e309. [Google Scholar] [CrossRef]
- de Mingo Pulido, A.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 regulates CD103+ dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell 2018, 33, 60–74.e66. [Google Scholar] [CrossRef]
- Gardner, A.; de Mingo Pulido, A.; Hanggi, K.; Bazargan, S.; Onimus, A.; Kasprzak, A.; Conejo-Garcia, J.R.; Rejniak, K.A.; Ruffell, B. TIM-3 blockade enhances IL-12-dependent antitumor immunity by promoting CD8+ t cell and XCR1+ dendritic cell spatial co-localization. J. Immunother. Cancer 2022, 10, e003571. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36–44. [Google Scholar] [CrossRef]
- Tesone, A.J.; Svoronos, N.; Allegrezza, M.J.; Conejo-Garcia, J.R. Pathological mobilization and activities of dendritic cells in tumor-bearing hosts: Challenges and opportunities for immunotherapy of cancer. Front. Immunol. 2013, 4, 435. [Google Scholar] [CrossRef]
- Tran Janco, J.M.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-infiltrating dendritic cells in cancer pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Gutkin, D.W.; Han, B.; Ma, Y.; Keskinov, A.A.; Shurin, M.R.; Shurin, G.V. Origin and pharmacological modulation of tumor-associated regulatory dendritic cells. Int. J. Cancer 2014, 134, 2633–2645. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. Er stress sensor xbp1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 2016, 161, 1527–1538. [Google Scholar] [CrossRef]
- Gulubova, M.V.; Ananiev, J.R.; Vlaykova, T.I.; Yovchev, Y.; Tsoneva, V.; Manolova, I.M. Role of dendritic cells in progression and clinical outcome of colon cancer. Int. J. Color. Dis. 2011, 27, 159–169. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic pd-l1 and braf inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Baird, J.R.; Tesone, A.J.; Rutkowski, M.R.; Scarlett, U.K.; Camposeco-Jacobs, A.L.; Anadon-Arnillas, J.; Harwood, N.M.; Korc, M.; Fiering, S.N.; et al. Reprogramming tumor-associated dendritic cells in vivo using mirna mimetics triggers protective immunity against ovarian cancer. Cancer Res. 2012, 72, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.C.; Xu, K.; Banchereau, R.; Marches, F.; Yu, C.I.; Martinek, J.; Anguiano, E.; Pedroza-Gonzalez, A.; Snipes, G.J.; O’Shaughnessy, J.; et al. Reprogramming tumor-infiltrating dendritic cells for CD103+ CD8+ mucosal t-cell differentiation and breast cancer rejection. Cancer Immunol. Res. 2014, 2, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.P.; Duncan, B.; Larabee, S.; Krauss, A.; Davis, J.P.; Cui, Y.; Kim, S.Y.; Guimond, M.; Bachovchin, W.; Fry, T.J. Val-boropro accelerates t cell priming via modulation of dendritic cell trafficking resulting in complete regression of established murine tumors. PLoS ONE 2013, 8, e58860. [Google Scholar] [CrossRef]
- Schultze, J.L.; Aschenbrenner, A.C. Systems immunology allows a new view on human dendritic cells. Semin. Cell Dev. Biol. 2019, 86, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Ueno, H.; Klechevsky, E.; Schmitt, N.; Ni, L.; Flamar, A.L.; Zurawski, S.; Zurawski, G.; Palucka, K.; Banchereau, J.; Oh, S. Targeting human dendritic cell subsets for improved vaccines. Semin. Immunol. 2011, 23, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Shortman, K.; Heath, W.R. The CD8+ dendritic cell subset. Immunol. Rev. 2010, 234, 18–31. [Google Scholar] [CrossRef]
- Guilliams, M.; Dutertre, C.A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef]
- Heger, L.; Balk, S.; Luhr, J.J.; Heidkamp, G.F.; Lehmann, C.H.K.; Hatscher, L.; Purbojo, A.; Hartmann, A.; Garcia-Martin, F.; Nishimura, S.I.; et al. Clec10a is a specific marker for human CD1c+ dendritic cells and enhances their toll-like receptor 7/8-induced cytokine secretion. Front. Immunol. 2018, 9, 744. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Reis e Sousa, C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef]
- Persson, E.K.; Uronen-Hansson, H.; Semmrich, M.; Rivollier, A.; Hagerbrand, K.; Marsal, J.; Gudjonsson, S.; Hakansson, U.; Reizis, B.; Kotarsky, K.; et al. Irf4 transcription-factor-dependent CD103+CD11b+ dendritic cells drive mucosal t helper 17 cell differentiation. Immunity 2013, 38, 958–969. [Google Scholar] [CrossRef]
- Bajana, S.; Roach, K.; Turner, S.; Paul, J.; Kovats, S. Irf4 promotes cutaneous dendritic cell migration to lymph nodes during homeostasis and inflammation. J. Immunol. 2012, 189, 3368–3377. [Google Scholar] [CrossRef]
- Alcantara-Hernandez, M.; Leylek, R.; Wagar, L.E.; Engleman, E.G.; Keler, T.; Marinkovich, M.P.; Davis, M.M.; Nolan, G.P.; Idoyaga, J. High-dimensional phenotypic mapping of human dendritic cells reveals interindividual variation and tissue specialization. Immunity 2017, 47, 1037–1050.e1036. [Google Scholar] [CrossRef]
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallee, V.P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional basis of mouse and human dendritic cell heterogeneity. Cell 2019, 179, 846–863.e824. [Google Scholar] [CrossRef]
- Mansouri, S.; Katikaneni, D.S.; Gogoi, H.; Pipkin, M.; Machuca, T.N.; Emtiazjoo, A.M.; Jin, L. Lung IFNAR1(hi) TNFR2+ cDC2 promotes lung regulatory t cells induction and maintains lung mucosal tolerance at steady state. Mucosal Immunol. 2020, 13, 595–608. [Google Scholar] [CrossRef]
- Maier, B.; Leader, A.M.; Chen, S.T.; Tung, N.; Chang, C.; LeBerichel, J.; Chudnovskiy, A.; Maskey, S.; Walker, L.; Finnigan, J.P.; et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 2020, 580, 257–262. [Google Scholar] [CrossRef]
- Leader, A.M.; Grout, J.A.; Maier, B.B.; Nabet, B.Y.; Park, M.D.; Tabachnikova, A.; Chang, C.; Walker, L.; Lansky, A.; Le Berichel, J.; et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 2021, 39, 1594–1609.e1512. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M.; Merad, M. Expanding dendritic cell nomenclature in the single-cell era. Nat. Rev. Immunol. 2022, 22, 67–68. [Google Scholar] [CrossRef]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 2019, 50, 1317–1334.e1310. [Google Scholar] [CrossRef]
- Cheng, S.; Li, Z.; Gao, R.; Xing, B.; Gao, Y.; Yang, Y.; Qin, S.; Zhang, L.; Ouyang, H.; Du, P.; et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 2021, 184, 792–809.e723. [Google Scholar] [CrossRef]
- Minohara, K.; Imai, M.; Matoba, T.; Wing, J.B.; Shime, H.; Odanaka, M.; Uraki, R.; Kawakita, D.; Toyama, T.; Takahashi, S.; et al. Mature dendritic cells enriched in regulatory molecules may control regulatory t cells and the prognosis of head and neck cancer. Cancer Sci. 2023, 114, 1256–1269. [Google Scholar] [CrossRef]
- Dixon, K.O.; Tabaka, M.; Schramm, M.A.; Xiao, S.; Tang, R.; Dionne, D.; Anderson, A.C.; Rozenblatt-Rosen, O.; Regev, A.; Kuchroo, V.K. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature 2021, 595, 101–106. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Cherian, J.; Moschos, S.J.; Tawbi, H.A.; Shuai, Y.; Gooding, W.E.; Sander, C.; Kirkwood, J.M. Safety and efficacy of combination immunotherapy with interferon alfa-2b and tremelimumab in patients with stage iv melanoma. J. Clin. Oncol. 2012, 30, 322–328. [Google Scholar] [CrossRef]
- Gardner, A.; Ruffell, B. Dendritic cells and cancer immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Ye, Y.; Gaugler, B.; Mohty, M.; Malard, F. Plasmacytoid dendritic cell biology and its role in immune-mediated diseases. Clin. Transl. Immunol. 2020, 9, e1139. [Google Scholar] [CrossRef]
- Musumeci, A.; Lutz, K.; Winheim, E.; Krug, A.B. What makes a pDC: Recent advances in understanding plasmacytoid dc development and heterogeneity. Front. Immunol. 2019, 10, 1222. [Google Scholar] [CrossRef]
- Anderson, D.A., 3rd; Dutertre, C.A.; Ginhoux, F.; Murphy, K.M. Genetic models of human and mouse dendritic cell development and function. Nat. Rev. Immunol. 2021, 21, 101–115. [Google Scholar] [CrossRef]
- Oshi, M.; Newman, S.; Tokumaru, Y.; Yan, L.; Matsuyama, R.; Kalinski, P.; Endo, I.; Takabe, K. Plasmacytoid dendritic cell (pDC) infiltration correlate with tumor infiltrating lymphocytes, cancer immunity, and better survival in triple negative breast cancer (TNBC) more strongly than conventional dendritic cell (cDC). Cancers 2020, 12, 3342. [Google Scholar] [CrossRef]
- Yang, L.; Li, S.; Chen, L.; Zhang, Y. Emerging roles of plasmacytoid dendritic cell crosstalk in tumor immunity. Cancer Biol. Med. 2023, 20, 728–747. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, M.; Mok, W.H.; Radford, K.J. Human dendritic cell subsets and function in health and disease. Cell Mol. Life Sci. 2015, 72, 4309–4325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, P.; Prestwood, T.R.; Zhang, H.; Carmi, Y.; Tolentino, L.L.; Wu, N.; Choi, O.; Winer, D.A.; Strober, S.; et al. Human regulatory dendritic cells develop from monocytes in response to signals from regulatory and helper t cells. Front. Immunol. 2020, 11, 1982. [Google Scholar] [CrossRef] [PubMed]
- Briseno, C.G.; Haldar, M.; Kretzer, N.M.; Wu, X.; Theisen, D.J.; Kc, W.; Durai, V.; Grajales-Reyes, G.E.; Iwata, A.; Bagadia, P.; et al. Distinct transcriptional programs control cross-priming in classical and monocyte-derived dendritic cells. Cell Rep. 2016, 15, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Schetters, S.T.T.; Rodriguez, E.; Kruijssen, L.J.W.; Crommentuijn, M.H.W.; Boon, L.; Van den Bossche, J.; Den Haan, J.M.M.; Van Kooyk, Y. Monocyte-derived apcs are central to the response of pd1 checkpoint blockade and provide a therapeutic target for combination therapy. J. Immunother. Cancer 2020, 8, e000588. [Google Scholar] [CrossRef] [PubMed]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef]
- Heath, W.R.; Carbone, F.R. Cross-presentation in viral immunity and self-tolerance. Nat. Rev. Immunol. 2001, 1, 126–134. [Google Scholar] [CrossRef]
- Mellins, E.D.; Stern, L.J. HLA-DM and HLA-DO, key regulators of MHC-ii processing and presentation. Curr. Opin. Immunol. 2014, 26, 115–122. [Google Scholar] [CrossRef]
- Caronni, N.; Piperno, G.M.; Simoncello, F.; Romano, O.; Vodret, S.; Yanagihashi, Y.; Dress, R.; Dutertre, C.A.; Bugatti, M.; Bourdeley, P.; et al. TIM4 expression by dendritic cells mediates uptake of tumor-associated antigens and anti-tumor responses. Nat. Commun. 2021, 12, 2237. [Google Scholar] [CrossRef]
- Maschalidi, S.; Ravichandran, K.S. Phagocytosis: Sweet repulsions via the glycocalyx. Curr. Biol. 2021, 31, R20–R22. [Google Scholar] [CrossRef]
- Segura, E.; Amigorena, S. Cross-presentation in mouse and human dendritic cells. Adv. Immunol. 2015, 127, 1–31. [Google Scholar]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of t cell immunity in melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef]
- Bevan, M.J. Cross-priming for a secondary cytotoxic response to minor h antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 1976, 143, 1283–1288. [Google Scholar] [CrossRef]
- Embgenbroich, M.; Burgdorf, S. Current concepts of antigen cross-presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef]
- Ackerman, A.L.; Giodini, A.; Cresswell, P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity 2006, 25, 607–617. [Google Scholar] [CrossRef]
- Kropshofer, H.; Hammerling, G.J.; Vogt, A.B. The impact of the non-classical MHC proteins HLA-DM and HLA-DO on loading of MHC class II molecules. Immunol. Rev. 1999, 172, 267–278. [Google Scholar] [CrossRef]
- Pierre, P.; Turley, S.J.; Gatti, E.; Hull, M.; Meltzer, J.; Mirza, A.; Inaba, K.; Steinman, R.M.; Mellman, I. Developmental regulation of mhc class ii transport in mouse dendritic cells. Nature 1997, 388, 787–792. [Google Scholar] [CrossRef]
- Thery, C.; Amigorena, S. The cell biology of antigen presentation in dendritic cells. Curr. Opin. Immunol. 2001, 13, 45–51. [Google Scholar] [CrossRef]
- Zhang, T.; Aipire, A.; Li, Y.; Guo, C.; Li, J. Antigen cross-presentation in dendric cells: From bench to bedside. Biomed. Pharmacother. 2023, 168, 115758. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.M.; Chan, A.; Rock, K.L. Pathways of mhc i cross-presentation of exogenous antigens. Semin. Immunol. 2023, 66, 101729. [Google Scholar] [CrossRef]
- Yang, X.; Gieni, R.S.; Mosmann, T.R.; HayGlass, K.T. Chemically modified antigen preferentially elicits induction of Th1-like cytokine synthesis patterns in vivo. J. Exp. Med. 1993, 178, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Toth, I. Recent advances in peptide-based subunit nanovaccines. Nanomedicine 2014, 9, 2657–2669. [Google Scholar] [CrossRef]
- Burgdorf, S.; Kautz, A.; Bohnert, V.; Knolle, P.A.; Kurts, C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 t cell activation. Science 2007, 316, 612–616. [Google Scholar] [CrossRef]
- Brewer, J.M.; Pollock, K.G.; Tetley, L.; Russell, D.G. Vesicle size influences the trafficking, processing, and presentation of antigens in lipid vesicles. J. Immunol. 2004, 173, 6143–6150. [Google Scholar] [CrossRef]
- MacNabb, B.W.; Tumuluru, S.; Chen, X.; Godfrey, J.; Kasal, D.N.; Yu, J.; Jongsma, M.L.M.; Spaapen, R.M.; Kline, D.E.; Kline, J. Dendritic cells can prime anti-tumor CD8+ t cell responses through major histocompatibility complex cross-dressing. Immunity 2022, 55, 982–997.e988. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, F.; Spranger, S. Mhc-dressing on dendritic cells: Boosting anti-tumor immunity via unconventional tumor antigen presentation. Semin. Immunol. 2023, 66, 101710. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral t cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef]
- Shiao, S.L.; Ganesan, A.P.; Rugo, H.S.; Coussens, L.M. Immune microenvironments in solid tumors: New targets for therapy. Genes. Dev. 2011, 25, 2559–2572. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Shen, Z.; Zhou, S.; Wang, Y.; Li, R.L.; Zhong, C.; Liang, C.; Sun, Y. Higher intratumoral infiltrated Foxp3+ treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 1585–1595. [Google Scholar] [CrossRef]
- Preston, C.C.; Maurer, M.J.; Oberg, A.L.; Visscher, D.W.; Kalli, K.R.; Hartmann, L.C.; Goode, E.L.; Knutson, K.L. The ratios of CD8+ t cells to CD4+CD25+ Foxp3+ and Foxp3- t cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS ONE 2013, 8, e80063. [Google Scholar] [CrossRef]
- Sarkar, T.; Dhar, S.; Chakraborty, D.; Pati, S.; Bose, S.; Panda, A.K.; Basak, U.; Chakraborty, S.; Mukherjee, S.; Guin, A.; et al. Foxp3/hat1 axis controls treg infiltration in the tumor microenvironment by inducing ccr4 expression in breast cancer. Front. Immunol. 2022, 13, 740588. [Google Scholar] [CrossRef]
- Villar, J.; Segura, E. Decoding the heterogeneity of human dendritic cell subsets. Trends Immunol. 2020, 41, 1062–1071. [Google Scholar] [CrossRef]
- Del Prete, A.; Sozio, F.; Barbazza, I.; Salvi, V.; Tiberio, L.; Laffranchi, M.; Gismondi, A.; Bosisio, D.; Schioppa, T.; Sozzani, S. Functional role of dendritic cell subsets in cancer progression and clinical implications. Int. J. Mol. Sci. 2020, 21, 3930. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-residing batf3 dendritic cells are required for effector t cell trafficking and adoptive t cell therapy. Cancer Cell 2017, 31, 711–723.e714. [Google Scholar] [CrossRef] [PubMed]
- Iwanowycz, S.; Ngoi, S.; Li, Y.; Hill, M.; Koivisto, C.; Parrish, M.; Guo, B.; Li, Z.; Liu, B. Type 2 dendritic cells mediate control of cytotoxic t cell resistant tumors. JCI Insight 2021, 6, e145885. [Google Scholar] [CrossRef] [PubMed]
- Kasmani, M.Y.; Zander, R.; Chung, H.K.; Chen, Y.; Khatun, A.; Damo, M.; Topchyan, P.; Johnson, K.E.; Levashova, D.; Burns, R.; et al. Clonal lineage tracing reveals mechanisms skewing CD8+ t cell fate decisions in chronic infection. J. Exp. Med. 2023, 220, e20220679. [Google Scholar] [CrossRef]
- Dahling, S.; Mansilla, A.M.; Knopper, K.; Grafen, A.; Utzschneider, D.T.; Ugur, M.; Whitney, P.G.; Bachem, A.; Arampatzi, P.; Imdahl, F.; et al. Type 1 conventional dendritic cells maintain and guide the differentiation of precursors of exhausted t cells in distinct cellular niches. Immunity 2022, 55, 656–670.e658. [Google Scholar] [CrossRef]
- Prokhnevska, N.; Cardenas, M.A.; Valanparambil, R.M.; Sobierajska, E.; Barwick, B.G.; Jansen, C.; Reyes Moon, A.; Gregorova, P.; delBalzo, L.; Greenwald, R.; et al. CD8+ t cell activation in cancer comprises an initial activation phase in lymph nodes followed by effector differentiation within the tumor. Immunity 2023, 56, 107–124.e105. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Chen, S.; Eisenbarth, S.C. Dendritic cell regulation of t helper cells. Annu. Rev. Immunol. 2021, 39, 759–790. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Tato, A.; Leon, B.; Lund, F.E.; Randall, T.D. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8+ t cell responses to influenza. Nat. Immunol. 2010, 11, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Durand, M.; Amigorena, S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J. Exp. Med. 2013, 210, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Chen, Q.; Soncin, I.; Ng, S.L.; Karjalainen, K.; Ruedl, C. A discrete subset of monocyte-derived cells among typical conventional type 2 dendritic cells can efficiently cross-present. Cell Rep. 2017, 21, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.M.D.; Decker, A.H.; Florez-Grau, G.; Bakdash, G.; Roring, R.J.; Stelloo, S.; Vermeulen, M.; Piet, B.; Aarntzen, E.; Verdoes, M.; et al. Inhibition of CSF-1r and IL-6r prevents conversion of cDC2s into immune incompetent tumor-induced DC3s boosting DC-driven therapy potential. Cell Rep. Med. 2024, 5, 101386. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, C.A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; van den Hoogen, L.L.; Leong, J.Y.; Lee, B.; et al. Single-cell analysis of human mononuclear phagocytes reveals subset-defining markers and identifies circulating inflammatory dendritic cells. Immunity 2019, 51, 573–589.e578. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.L.; Batista, N.V.; Wang, K.C.; Zhou, A.C.; Watts, T.H. Gitrl on inflammatory antigen presenting cells in the lung parenchyma provides signal 4 for t-cell accumulation and tissue-resident memory t-cell formation. Mucosal Immunol. 2019, 12, 363–377. [Google Scholar] [CrossRef]
- Girard, M.; Law, J.C.; Edilova, M.I.; Watts, T.H. Type I interferons drive the maturation of human DC3s with a distinct costimulatory profile characterized by high gitrl. Sci. Immunol. 2020, 5, eabe0347. [Google Scholar] [CrossRef]
- Marciscano, A.E.; Anandasabapathy, N. The role of dendritic cells in cancer and anti-tumor immunity. Semin. Immunol. 2021, 52, 101481. [Google Scholar] [CrossRef]
- Smalley, I.; Chen, Z.H.; Phadke, M.; Li, J.N.; Yu, X.Q.; Wyatt, C.; Evernden, B.; Messina, J.L.; Sarnaik, A.; Sondak, V.K.; et al. Single-cell characterization of the immune microenvironment of melanoma brain and leptomeningeal metastases. Clin. Cancer Res. 2021, 27, 4109–4125. [Google Scholar] [CrossRef] [PubMed]
- Kusmartsev, S.; Gabrilovich, D.I. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Metastasis Rev. 2006, 25, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Di Blasio, S.; van Wigcheren, G.F.; Becker, A.; van Duffelen, A.; Gorris, M.; Verrijp, K.; Stefanini, I.; Bakker, G.J.; Bloemendal, M.; Halilovic, A.; et al. The tumour microenvironment shapes dendritic cell plasticity in a human organotypic melanoma culture. Nat. Commun. 2020, 11, 2749. [Google Scholar] [CrossRef] [PubMed]
- Subtil, B.; van der Hoorn, I.A.E.; Cuenca-Escalona, J.; Becker, A.M.D.; Alvarez-Begue, M.; Iyer, K.K.; Janssen, J.; van Oorschot, T.; Poel, D.; Gorris, M.A.J.; et al. cDC2 plasticity and acquisition of a DC3-like phenotype mediated by IL-6 and PGE2 in a patient-derived colorectal cancer organoids model. Eur. J. Immunol. 2024, 54, e2350891. [Google Scholar] [CrossRef] [PubMed]
- Kvedaraite, E.; Ginhoux, F. Human dendritic cells in cancer. Sci. Immunol. 2022, 7, eabm9409. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.S.; Zhou, X.; Yan, W.B.; Li, Y.J.; Du, C.R.; Wang, X.S.; Shen, C.Y.; Wang, Q.F.; Ying, H.M.; Lu, X.G.; et al. Dissecting the heterogeneity of the microenvironment in primary and recurrent nasopharyngeal carcinomas using single-cell rna sequencing. Oncoimmunology 2022, 11, 2026583. [Google Scholar] [CrossRef]
- Liu, Y.; He, S.; Wang, X.L.; Peng, W.; Chen, Q.Y.; Chi, D.M.; Chen, J.R.; Han, B.W.; Lin, G.W.; Li, Y.Q.; et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat. Commun. 2021, 12, 741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Peng, L.; Luo, Y.; Zhang, S.; Pu, Y.; Chen, Y.; Guo, W.; Yao, J.; Shao, M.; Fan, W.; et al. Dissecting esophageal squamous-cell carcinoma ecosystem by single-cell transcriptomic analysis. Nat. Commun. 2021, 12, 5291. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Acharya, N.; Subramanian, A.; Purohit, V.; Tabaka, M.; Hou, Y.; He, D.; Dixon, K.O.; Lambden, C.; Xia, J.; et al. TIM-3 adapter protein bat3 acts as an endogenous regulator of tolerogenic dendritic cell function. Sci. Immunol. 2022, 7, eabm0631. [Google Scholar] [CrossRef]
- Harding, J.J.; Moreno, V.; Bang, Y.J.; Hong, M.H.; Patnaik, A.; Trigo, J.; Szpurka, A.M.; Yamamoto, N.; Doi, T.; Fu, S.; et al. Blocking TIM-3 in treatment-refractory advanced solid tumors: A phase ia/b study of LY3321367 with or without an anti-PD-L1 antibody. Clin. Cancer Res. 2021, 27, 2168–2178. [Google Scholar] [CrossRef]
- Chevolet, I.; Speeckaert, R.; Schreuer, M.; Neyns, B.; Krysko, O.; Bachert, C.; Van Gele, M.; van Geel, N.; Brochez, L. Clinical significance of plasmacytoid dendritic cells and myeloid-derived suppressor cells in melanoma. J. Transl. Med. 2015, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.O.; Schmidt, H.; Moller, H.J.; Donskov, F.; Hoyer, M.; Sjoegren, P.; Christensen, I.J.; Steiniche, T. Intratumoral neutrophils and plasmacytoid dendritic cells indicate poor prognosis and are associated with pSTAT3 expression in AJCC stage I/II melanoma. Cancer 2012, 118, 2476–2485. [Google Scholar] [CrossRef] [PubMed]
- Aspord, C.; Leccia, M.T.; Charles, J.; Plumas, J. Plasmacytoid dendritic cells support melanoma progression by promoting Th2 and regulatory immunity through OX40L and ICOSL. Cancer Immunol. Res. 2013, 1, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Li, X.; Porter, J.L.; Ostrodi, D.H.; Yang, B.; Li, J.; Wang, Y.; Zhang, J.; Bai, L.; Jiao, S. Level of plasmacytoid dendritic cells is increased in non-small cell lung carcinoma. Tumour Biol. 2014, 35, 2247–2252. [Google Scholar] [CrossRef]
- Laheurte, C.; Seffar, E.; Gravelin, E.; Lecuelle, J.; Renaudin, A.; Boullerot, L.; Malfroy, M.; Marguier, A.; Lecoester, B.; Gaugler, B.; et al. Interplay between plasmacytoid dendritic cells and tumor-specific t cells in peripheral blood influences long-term survival in non-small cell lung carcinoma. Cancer Immunol. Immunother. 2023, 72, 579–589. [Google Scholar] [CrossRef]
- Ghirelli, C.; Reyal, F.; Jeanmougin, M.; Zollinger, R.; Sirven, P.; Michea, P.; Caux, C.; Bendriss-Vermare, N.; Donnadieu, M.H.; Caly, M.; et al. Breast cancer cell-derived gm-csf licenses regulatory Th2 induction by plasmacytoid predendritic cells in aggressive disease subtypes. Cancer Res. 2015, 75, 2775–2787. [Google Scholar] [CrossRef]
- Bosteels, C.; Neyt, K.; Vanheerswynghels, M.; van Helden, M.J.; Sichien, D.; Debeuf, N.; De Prijck, S.; Bosteels, V.; Vandamme, N.; Martens, L.; et al. Inflammatory type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity 2020, 52, 1039–1056.e1039. [Google Scholar] [CrossRef]
- Gavish, A.; Tyler, M.; Greenwald, A.C.; Hoefflin, R.; Simkin, D.; Tschernichovsky, R.; Galili Darnell, N.; Somech, E.; Barbolin, C.; Antman, T.; et al. Hallmarks of transcriptional intratumour heterogeneity across a thousand tumours. Nature 2023, 618, 598–606. [Google Scholar] [CrossRef]
- Kilmister, E.J.; Koh, S.P.; Weth, F.R.; Gray, C.; Tan, S.T. Cancer metastasis and treatment resistance: Mechanistic insights and therapeutic targeting of cancer stem cells and the tumor microenvironment. Biomedicines 2022, 10, 2988. [Google Scholar] [CrossRef]
- O’Sullivan, E.; Keogh, A.; Henderson, B.; Finn, S.P.; Gray, S.G.; Gately, K. Treatment strategies for KRAS-mutated non-small-cell lung cancer. Cancers 2023, 15, 1635. [Google Scholar] [CrossRef]
- Longaray, J.B.; Dias, C.K.; Scholl, J.N.; Battastini, A.M.O.; Figueiro, F. Investigation of co-treatment multi-targeting approaches in breast cancer cell lines. Eur. J. Pharmacol. 2024, 966, 176328. [Google Scholar] [CrossRef]
- Shen, H.; Yang, E.S.; Conry, M.; Fiveash, J.; Contreras, C.; Bonner, J.A.; Shi, L.Z. Predictive biomarkers for immune checkpoint blockade and opportunities for combination therapies. Genes Dis. 2019, 6, 232–246. [Google Scholar] [CrossRef]
- Zhou, X.; Ni, Y.; Liang, X.; Lin, Y.; An, B.; He, X.; Zhao, X. Mechanisms of tumor resistance to immune checkpoint blockade and combination strategies to overcome resistance. Front. Immunol. 2022, 13, 915094. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzoccoli, L.; Liu, B. Dendritic Cells in Shaping Anti-Tumor T Cell Response. Cancers 2024, 16, 2211. https://doi.org/10.3390/cancers16122211
Mazzoccoli L, Liu B. Dendritic Cells in Shaping Anti-Tumor T Cell Response. Cancers. 2024; 16(12):2211. https://doi.org/10.3390/cancers16122211
Chicago/Turabian StyleMazzoccoli, Luciano, and Bei Liu. 2024. "Dendritic Cells in Shaping Anti-Tumor T Cell Response" Cancers 16, no. 12: 2211. https://doi.org/10.3390/cancers16122211
APA StyleMazzoccoli, L., & Liu, B. (2024). Dendritic Cells in Shaping Anti-Tumor T Cell Response. Cancers, 16(12), 2211. https://doi.org/10.3390/cancers16122211