
Genome-Wide DNA Methylation Profiling as a Prognostic Marker in Pituitary Adenomas—A Pilot Study
 , , ,
, , ,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
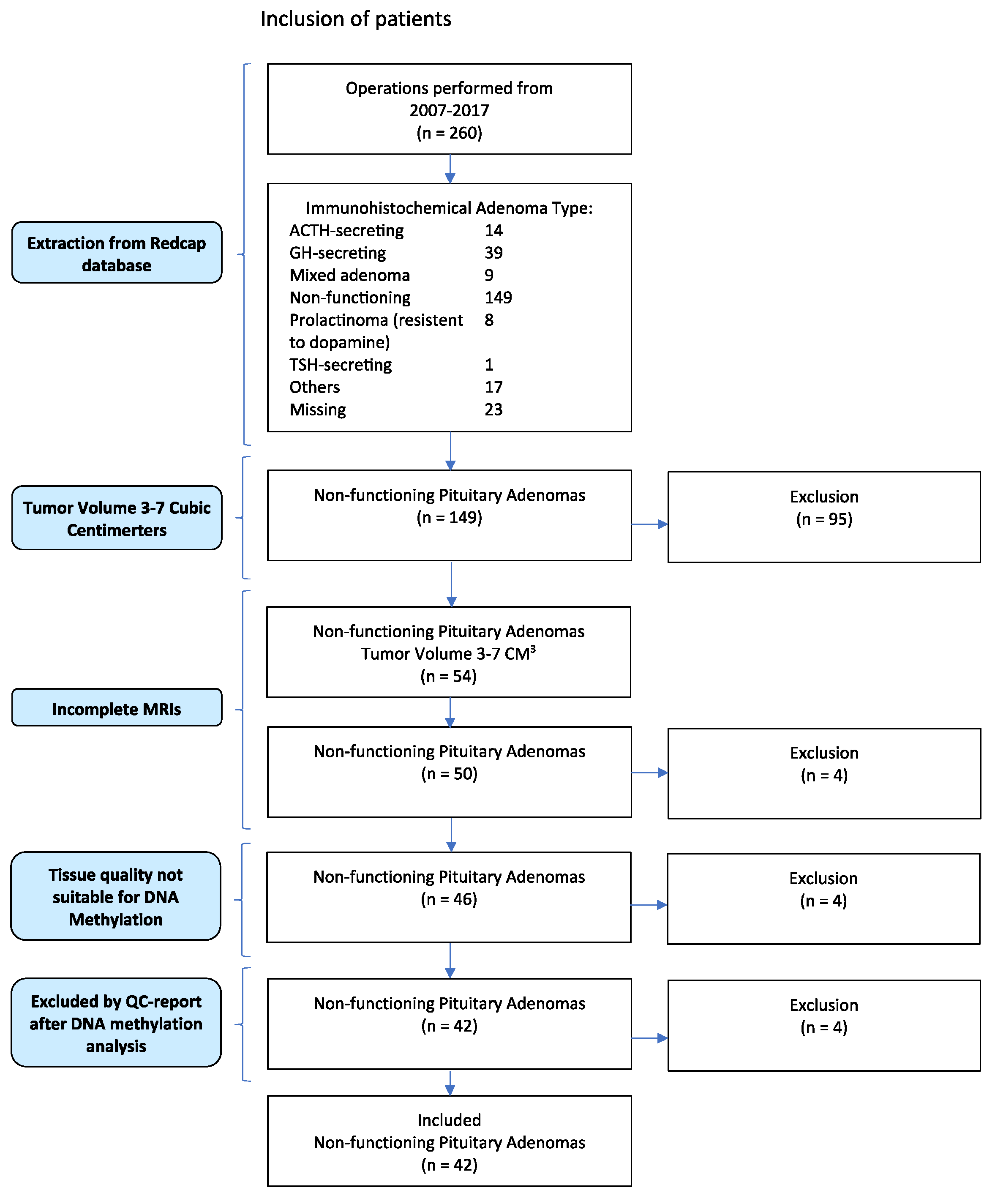
2.1. Patient Inclusion
2.2. Radiological Classification
2.3. DNA Methylation
2.4. Statistical Analysis
2.5. Immunohistochemistry
3. Results
3.1. WHO Classification from 2017
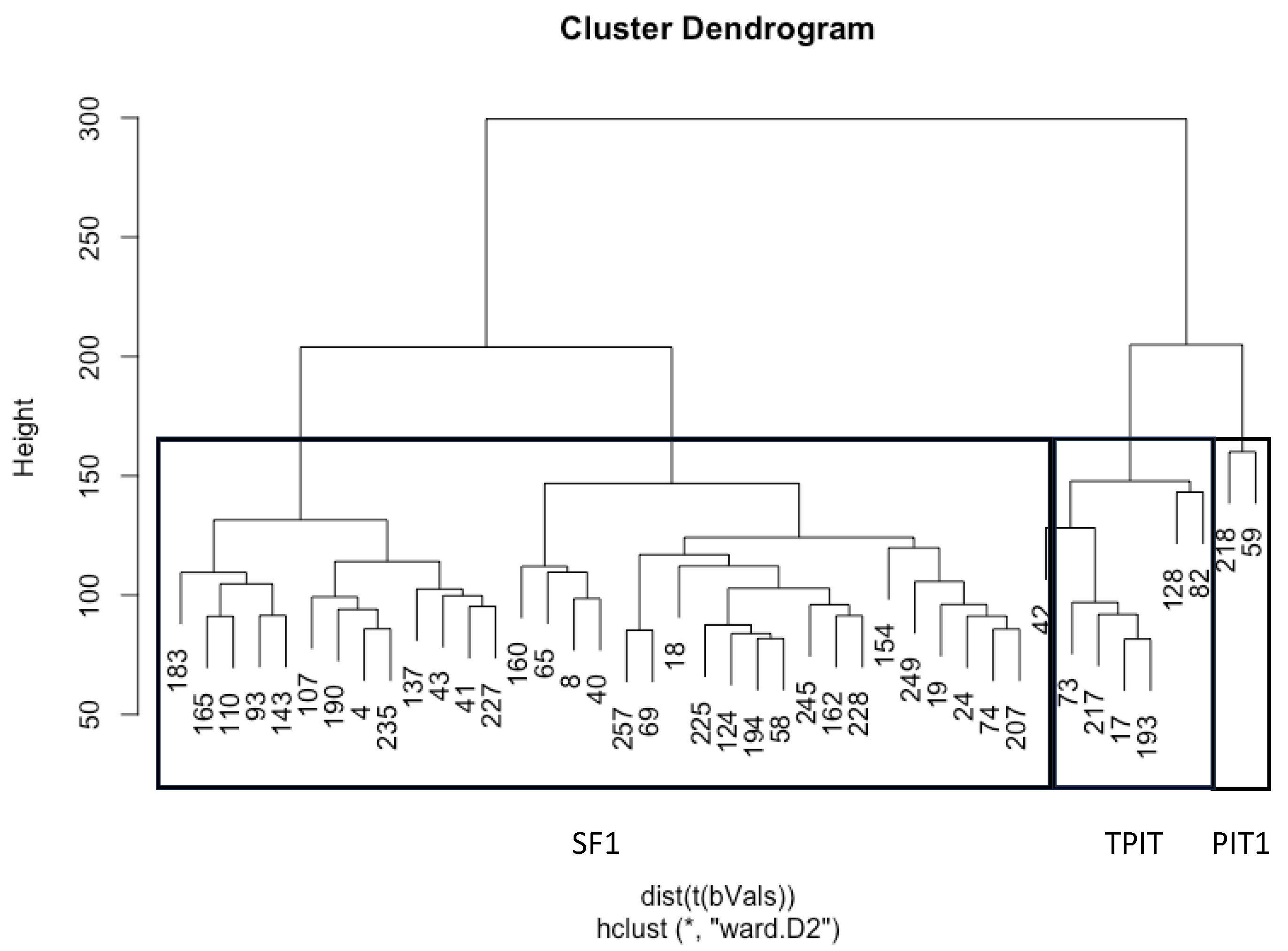
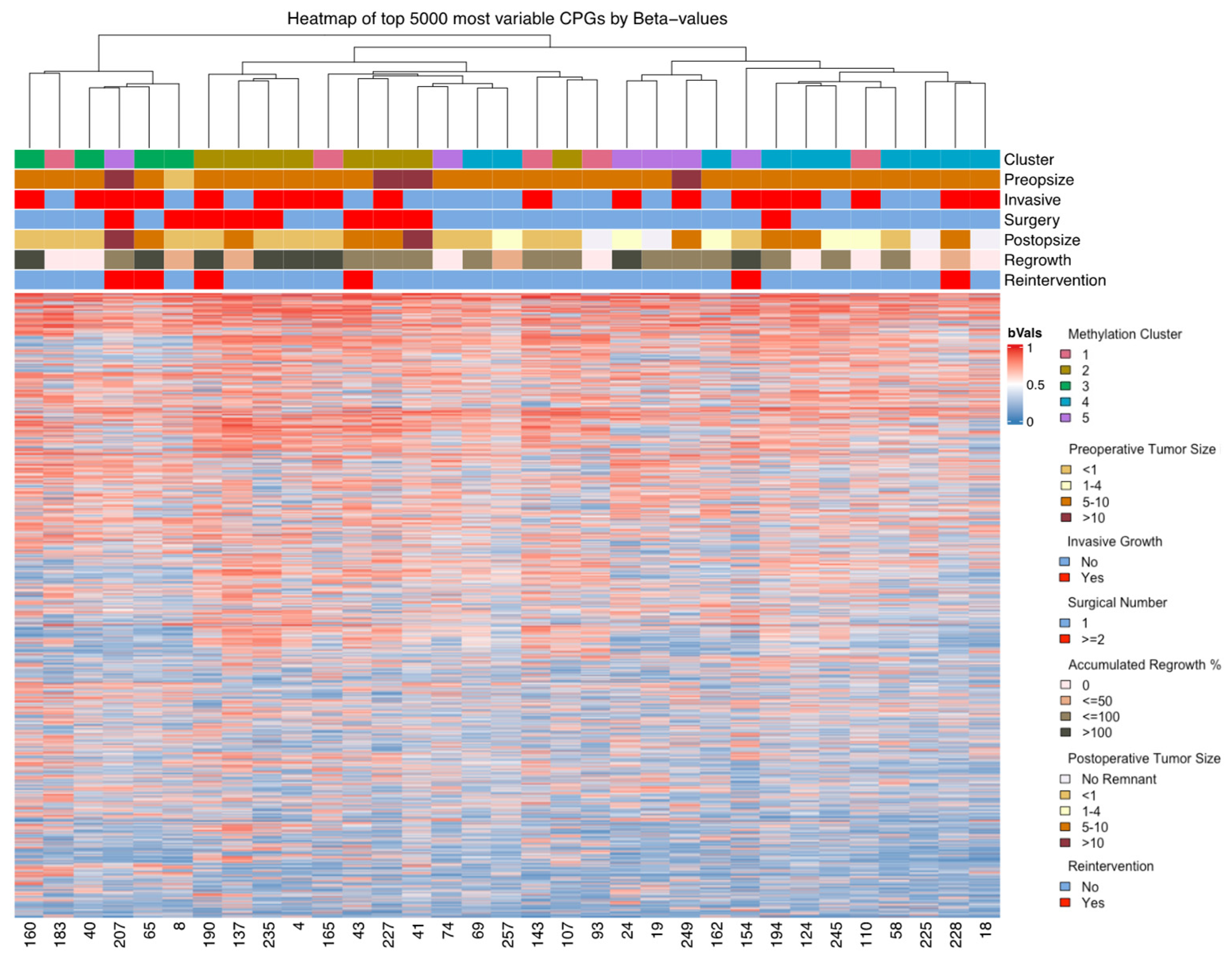
3.2. DNA Methylation
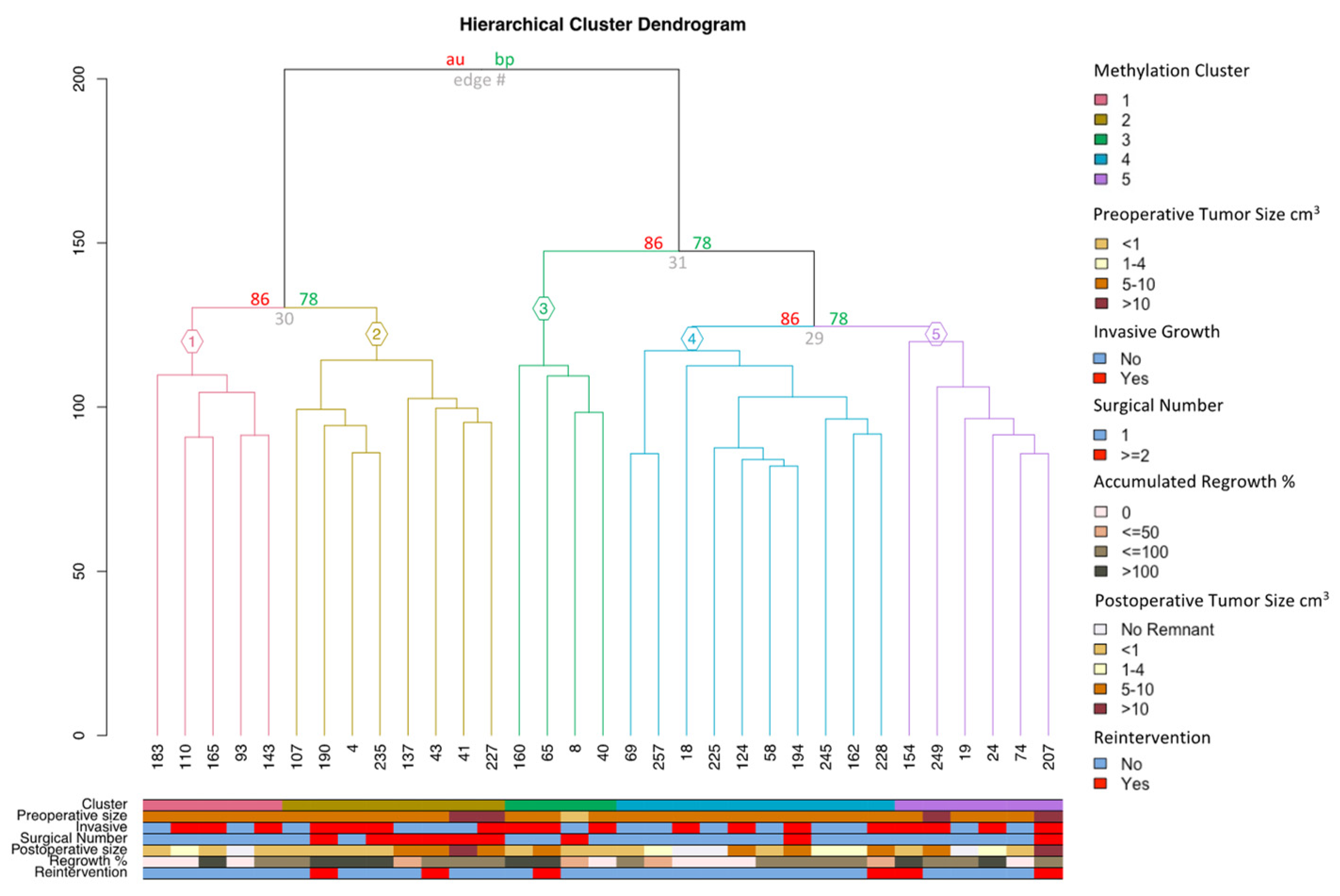
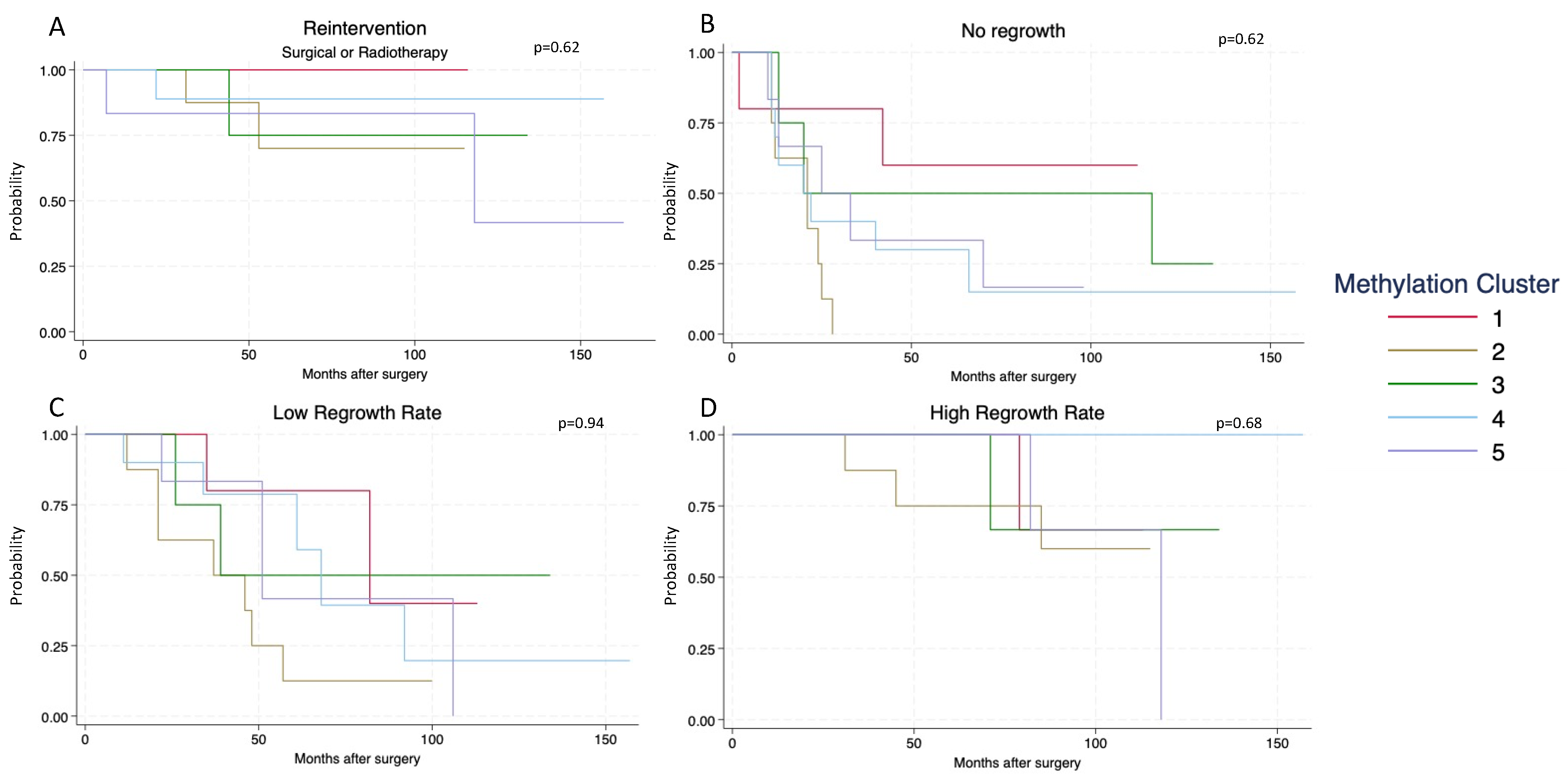
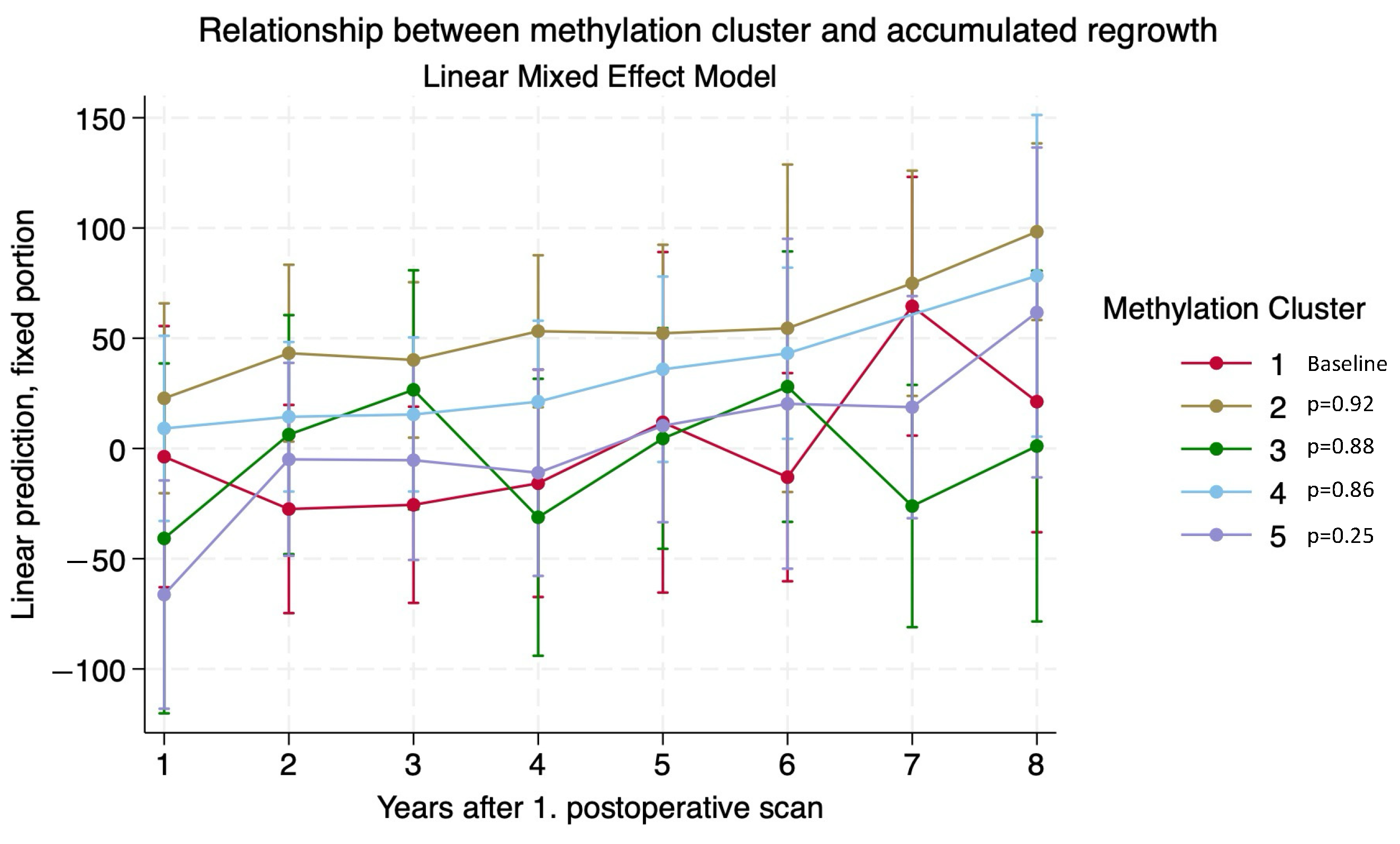
3.3. Adenoma Regrowth Potential and DNA Methylation
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molitch, M.E. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol. Metab. Clin. N. Am. 2008, 37, 151–171. [Google Scholar] [CrossRef]
- Mete, O.; Lopes, M.B. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2017, 28, 228–243. [Google Scholar] [CrossRef]
- Burcea, I.F.; Nastase, V.N.; Poiana, C. Pituitary transcription factors in the immunohistochemical and molecular diagnosis of pituitary tumours—A systematic review. Endokrynol. Pol. 2021, 72, 53–63. [Google Scholar] [CrossRef]
- Taniguchi-Ponciano, K.; Andonegui-Elguera, S.; Pena-Martinez, E.; Silva-Roman, G.; Vela-Patino, S.; Gomez-Apo, E.; Chavez-Macias, L.; Vargas-Ortega, G.; Espinosa-de-Los-Monteros, L.; Gonzalez-Virla, B.; et al. Transcriptome and methylome analysis reveals three cellular origins of pituitary tumors. Sci. Rep. 2020, 10, 19373. [Google Scholar] [CrossRef]
- Nishioka, H.; Inoshita, N.; Mete, O.; Asa, S.L.; Hayashi, K.; Takeshita, A.; Fukuhara, N.; Yamaguchi-Okada, M.; Takeuchi, Y.; Yamada, S. The Complementary Role of Transcription Factors in the Accurate Diagnosis of Clinically Nonfunctioning Pituitary Adenomas. Endocr. Pathol. 2015, 26, 349–355. [Google Scholar] [CrossRef]
- Manojlovic-Gacic, E.; Engstrom, B.E.; Casar-Borota, O. Histopathological classification of non-functioning pituitary neuroendocrine tumors. Pituitary 2018, 21, 119–129. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell. 2008, 30, 755–766. [Google Scholar] [CrossRef]
- Campagna, M.P.; Xavier, A.; Lechner-Scott, J.; Maltby, V.; Scott, R.J.; Butzkueven, H.; Jokubaitis, V.G.; Lea, R.A. Epigenome-wide association studies: Current knowledge, strategies and recommendations. Clin. Epigenet. 2021, 13, 214. [Google Scholar] [CrossRef]
- Schroder, C.; Leitao, E.; Wallner, S.; Schmitz, G.; Klein-Hitpass, L.; Sinha, A.; Jockel, K.H.; Heilmann-Heimbach, S.; Hoffmann, P.; Nothen, M.M.; et al. Regions of common inter-individual DNA methylation differences in human monocytes: Genetic basis and potential function. Epigenet. Chromatin 2017, 10, 37. [Google Scholar] [CrossRef]
- Yong, W.S.; Hsu, F.M.; Chen, P.Y. Profiling genome-wide DNA methylation. Epigenet. Chromatin 2016, 9, 26. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Silva-Júnior, R.; Bueno, A.C.; Martins, C.S.; Coelli-Lacchini, F.; Ozaki, J.G.O.; Almeida, E.S.D.C.; Marrero-Gutiérrez, J.; Santos, A.C.D.; Garcia-Peral, C.; Machado, H.R.; et al. Integrating Methylome and Transcriptome Signatures Expands the Molecular Classification of the Pituitary Tumors. J. Clin. Endocrinol. Metab. 2023, 108, 1452–1463. [Google Scholar] [CrossRef]
- Mosella, M.S.; Sabedot, T.S.; Silva, T.C.; Malta, T.M.; Dezem, F.S.; Asmaro, K.P.; Wells, M.; Mukherjee, A.; Poisson, L.M.; Snyder, J.; et al. DNA methylation-based signatures classify sporadic pituitary tumors according to clinicopathological features. Neuro Oncol. 2021, 23, 1292–1303. [Google Scholar] [CrossRef]
- Ling, C.; Pease, M.; Shi, L.; Punj, V.; Shiroishi, M.S.; Commins, D.; Weisenberger, D.J.; Wang, K.; Zada, G. A pilot genome-scale profiling of DNA methylation in sporadic pituitary macroadenomas: Association with tumor invasion and histopathological subtype. PLoS ONE 2014, 9, e96178. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Fita, K.D.; Rotermund, R.; Piffko, A.; Schmid, S.; Capper, D.; Buslei, R.; Buchfelder, M.; Burkhardt, T.; Matschke, J.; et al. Genome-wide DNA methylation profiles distinguish silent from non-silent ACTH adenomas. Acta Neuropathol. 2020, 140, 95–97. [Google Scholar] [CrossRef]
- Cheng, S.; Li, C.; Xie, W.; Miao, Y.; Guo, J.; Wang, J.; Zhang, Y. Integrated analysis of DNA methylation and mRNA expression profiles to identify key genes involved in the regrowth of clinically non-functioning pituitary adenoma. Aging 2020, 12, 2408–2427. [Google Scholar] [CrossRef]
- Garcia-Martinez, A.; Sottile, J.; Sanchez-Tejada, L.; Fajardo, C.; Camara, R.; Lamas, C.; Barbera, V.M.; Pico, A. DNA Methylation of Tumor Suppressor Genes in Pituitary Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2019, 104, 1272–1282. [Google Scholar] [CrossRef]
- Zhang, C.; Qian, Y.; Qiao, Y.; Li, Y.; Wang, W.; Li, J.; Deng, X. Analysis of whole genome-wide microRNA transcriptome profiling in invasive pituitary adenomas and non-invasive pituitary adenomas. Chin. Neurosurg. J. 2019, 5, 27. [Google Scholar] [CrossRef]
- Hauser, B.M.; Lau, A.; Gupta, S.; Bi, W.L.; Dunn, I.F. The Epigenomics of Pituitary Adenoma. Front. Endocrinol. 2019, 10, 290. [Google Scholar] [CrossRef]
- Hallén, T.; Johannsson, G.; Dahlén, R.; Glad, C.A.M.; Örndal, C.; Engvall, A.; Carén, H.; Skoglund, T.; Olsson, D.S. Genome-wide DNA Methylation Differences in Nonfunctioning Pituitary Adenomas with and Without Postsurgical Progression. J. Clin. Endocrinol. Metab. 2022, 107, 2318–2328. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Wan, X.; Xu, Y.; Chen, J.; Shu, K.; Lei, T. Prognostic Factors for Recurrence in Pituitary Adenomas: Recent Progress and Future Directions. Diagnostics 2022, 12, 977. [Google Scholar] [CrossRef]
- Caulley, L.; Whelan, J.; Khoury, M.; Mavedatnia, D.; Sahlollbey, N.; Amrani, L.; Eid, A.; Doyle, M.A.; Malcolm, J.; Alkherayf, F.; et al. Post-operative surveillance for somatotroph, lactotroph and non-functional pituitary adenomas after curative resection: A systematic review. Pituitary 2023, 26, 73–93. [Google Scholar] [CrossRef]
- Moller, M.W.; Andersen, M.S.; Pedersen, C.B.; Kristensen, B.W.; Poulsen, F.R. Intraoperative low field MRI in transsphenoidal pituitary surgery. Endocr. Connect. 2018, 7, 897–906. [Google Scholar] [CrossRef]
- Moller, M.W.; Andersen, M.S.; Glintborg, D.; Pedersen, C.B.; Halle, B.; Kristensen, B.W.; Poulsen, F.R. Endoscopic vs. microscopic transsphenoidal pituitary surgery: A single centre study. Sci. Rep. 2020, 10, 21942. [Google Scholar] [CrossRef]
- Hardy, J. Excision of pituitary adenomas by trans-sphenoidal approach. Union Med. Can. 1962, 91, 933–945. [Google Scholar]
- Micko, A.S.; Wohrer, A.; Wolfsberger, S.; Knosp, E. Invasion of the cavernous sinus space in pituitary adenomas: Endoscopic verification and its correlation with an MRI-based classification. J. Neurosurg. 2015, 122, 803–811. [Google Scholar] [CrossRef]
- Araujo-Castro, M.; Acitores Cancela, A.; Vior, C.; Pascual-Corrales, E.; Rodriguez Berrocal, V. Radiological Knosp, Revised-Knosp, and Hardy-Wilson Classifications for the Prediction of Surgical Outcomes in the Endoscopic Endonasal Surgery of Pituitary Adenomas: Study of 228 Cases. Front. Oncol. 2021, 11, 807040. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef]
- Asa, S.L.; Mete, O.; Perry, A.; Osamura, R.Y. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2022, 33, 6–26. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, F.; Morandi, L.; Zoli, M.; Mazzatenta, D.; Righi, A.; Evangelisti, S.; Ambrosi, F.; Tonon, C.; Giannini, C.; Lloyd, R.V.; et al. Epigenomic and somatic mutations of pituitary tumors with clinical and pathological correlations in 111 patients. Clin. Endocrinol. 2022, 97, 763–772. [Google Scholar] [CrossRef]
- Giuffrida, G.; D’Argenio, V.; Ferrau, F.; Lasorsa, V.A.; Polito, F.; Aliquo, F.; Ragonese, M.; Cotta, O.R.; Alessi, Y.; Oteri, R.; et al. Methylome Analysis in Nonfunctioning and GH-Secreting Pituitary Adenomas. Front. Endocrinol. 2022, 13, 841118. [Google Scholar] [CrossRef]
- Spada, A.; Mantovani, G.; Lania, A.G.; Treppiedi, D.; Mangili, F.; Catalano, R.; Carosi, G.; Sala, E.; Peverelli, E. Pituitary Tumors: Genetic and Molecular Factors Underlying Pathogenesis and Clinical Behavior. Neuroendocrinology 2022, 112, 15–33. [Google Scholar] [CrossRef]
- Serioli, S.; Agostini, L.; Pietrantoni, A.; Valeri, F.; Costanza, F.; Chiloiro, S.; Buffoli, B.; Piazza, A.; Poliani, P.L.; Peris-Celda, M.; et al. Aggressive PitNETs and Potential Target Therapies: A Systematic Review of Molecular and Genetic Pathways. Int. J. Mol. Sci. 2023, 24, 15719. [Google Scholar] [CrossRef]
- Toader, C.; Dobrin, N.; Tataru, C.I.; Covache-Busuioc, R.A.; Bratu, B.G.; Glavan, L.A.; Costin, H.P.; Corlatescu, A.D.; Dumitrascu, D.I.; Ciurea, A.V. From Genes to Therapy: Pituitary Adenomas in the Era of Precision Medicine. Biomedicines 2023, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jeon, H.M.; Batara, D.C.; Lee, S.; Lee, S.J.; Yin, J.; Park, S.I.; Park, M.; Seo, J.B.; Hwang, J.; et al. CREB5 promotes the proliferation and self-renewal ability of glioma stem cells. Cell Death Discov. 2024, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Seward, C.H.; Song, Y.; Inamdar, M.; Leddy, A.M.; Zhang, H.; Yoo, J.; Kao, W.C.; Pawlowski, H.; Stubbs, L.J. Galnt17 loss-of-function leads to developmental delay and abnormal coordination, activity, and social interactions with cerebellar vermis pathology. Dev. Biol. 2022, 490, 155–171. [Google Scholar] [CrossRef]
- Dong, Z.; Yeo, K.S.; Lopez, G.; Zhang, C.; Dankert Eggum, E.N.; Rokita, J.L.; Ung, C.Y.; Levee, T.M.; Her, Z.P.; Howe, C.J.; et al. GAS7 Deficiency Promotes Metastasis in MYCN-Driven Neuroblastoma. Cancer Res. 2021, 81, 2995–3007. [Google Scholar] [CrossRef]
- Letellier, E.; Haan, S. SOCS2: Physiological and pathological functions. Front. Biosci. 2016, 8, 189–204. [Google Scholar]
- Lan, L.; Cao, H.; Chi, W.; Meng, W.; Zhao, L.; Cui, W.; Wang, B. Aberrant DNA hypermethylation-silenced LINC00886 gene accelerates malignant progression of laryngeal carcinoma. Pathol. Res. Pract. 2020, 216, 152877. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Luo, Y.; Xu, W.; Han, L.; Liu, W.; Liao, T.; Yang, Y.; Wang, Y. LINC00886 Negatively Regulates Malignancy in Anaplastic Thyroid Cancer. Endocrinology 2023, 164, bqac204. [Google Scholar] [CrossRef] [PubMed]
- Herrgott, G.A.; Asmaro, K.P.; Wells, M.; Sabedot, T.S.; Malta, T.M.; Mosella, M.S.; Nelson, K.; Scarpace, L.; Barnholtz-Sloan, J.S.; Sloan, A.E.; et al. Detection of tumor-specific DNA methylation markers in the blood of patients with pituitary neuroendocrine tumors. Neuro Oncol. 2022, 24, 1126–1139. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | Methylation Clusters of SF1-Lineage NFPAs | p-Value | |||||
|---|---|---|---|---|---|---|---|
| All | 1 | 2 | 3 | 4 | 5 | ||
| Total (%) | 42 (100) | 5 (15.6) | 8 (24.2) | 4 (12.5) | 10 (31.3) | 6 (18.8) | |
| Age (median) (min–max) | 61 (26–87) | 63 (43–77) | 59 (53–68) | 62.5 (50–80) | 53 (26–87) | 66 (49–76) | p = 0.83 |
| Invasive growth | 18 (42.9) | 3 | 4 | 3 | 4 | 4 | |
| Regrowth Rate No Low Medium High | NA | 3 0 1 1 | 0 1 4 3 | 1 1 1 1 | 2 3 5 0 | 1 1 2 2 | |
| Radiotherapy | 2 (4.8) | 0 | 1 | 0 | 0 | 1 | |
| Re-operations | 6 (14.3) | 0 | 2 | 1 | 1 | 2 | |
| Mean preoperative tumor volume (cm3) (95%CI) | 7.9 (4.9–10.9) | 6.06 (5.6–6.5) | 6.2 (4.6–7.9) | 5.9 (2.9–8.8) | 6.1 (5.1–7.1) | 9.1 (4.8–13.2) | p = 0.38 |
| Mean postoperative tumor volume (cm3) (95%CI) | 4.4 (1.2–7.6) | 1.2 (0.4–2.1) | 3.5 (1.6–5.3) | 3.6 (1.1–6.0) | 1.9 (0.8–3.1) | 4.8 (–0.3–10.0) | p = 0.26 |
| Median follow-up time (months) (min–max) | 75 (12–167) | 82 (66–130) | 88 (45–159) | 96 (58–147) | 60 (12–157) | 86.5 (46–167) | p = 0.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Møller, M.W.; Andersen, M.S.; Halle, B.; Pedersen, C.B.; Boldt, H.B.; Tan, Q.; Jurmeister, P.S.; Herrgott, G.A.; Castro, A.V.; Petersen, J.K.; et al. Genome-Wide DNA Methylation Profiling as a Prognostic Marker in Pituitary Adenomas—A Pilot Study. Cancers 2024, 16, 2210. https://doi.org/10.3390/cancers16122210
Møller MW, Andersen MS, Halle B, Pedersen CB, Boldt HB, Tan Q, Jurmeister PS, Herrgott GA, Castro AV, Petersen JK, et al. Genome-Wide DNA Methylation Profiling as a Prognostic Marker in Pituitary Adenomas—A Pilot Study. Cancers. 2024; 16(12):2210. https://doi.org/10.3390/cancers16122210
Chicago/Turabian StyleMøller, Morten Winkler, Marianne Skovsager Andersen, Bo Halle, Christian Bonde Pedersen, Henning Bünsow Boldt, Qihua Tan, Philipp Sebastian Jurmeister, Grayson A. Herrgott, Ana Valeria Castro, Jeanette K. Petersen, and et al. 2024. "Genome-Wide DNA Methylation Profiling as a Prognostic Marker in Pituitary Adenomas—A Pilot Study" Cancers 16, no. 12: 2210. https://doi.org/10.3390/cancers16122210
APA StyleMøller, M. W., Andersen, M. S., Halle, B., Pedersen, C. B., Boldt, H. B., Tan, Q., Jurmeister, P. S., Herrgott, G. A., Castro, A. V., Petersen, J. K., & Poulsen, F. R. (2024). Genome-Wide DNA Methylation Profiling as a Prognostic Marker in Pituitary Adenomas—A Pilot Study. Cancers, 16(12), 2210. https://doi.org/10.3390/cancers16122210