
Overlapping Stromal Alterations in Myeloid and Lymphoid Neoplasms

 , , , , ,
, , , , , 
Abstract
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
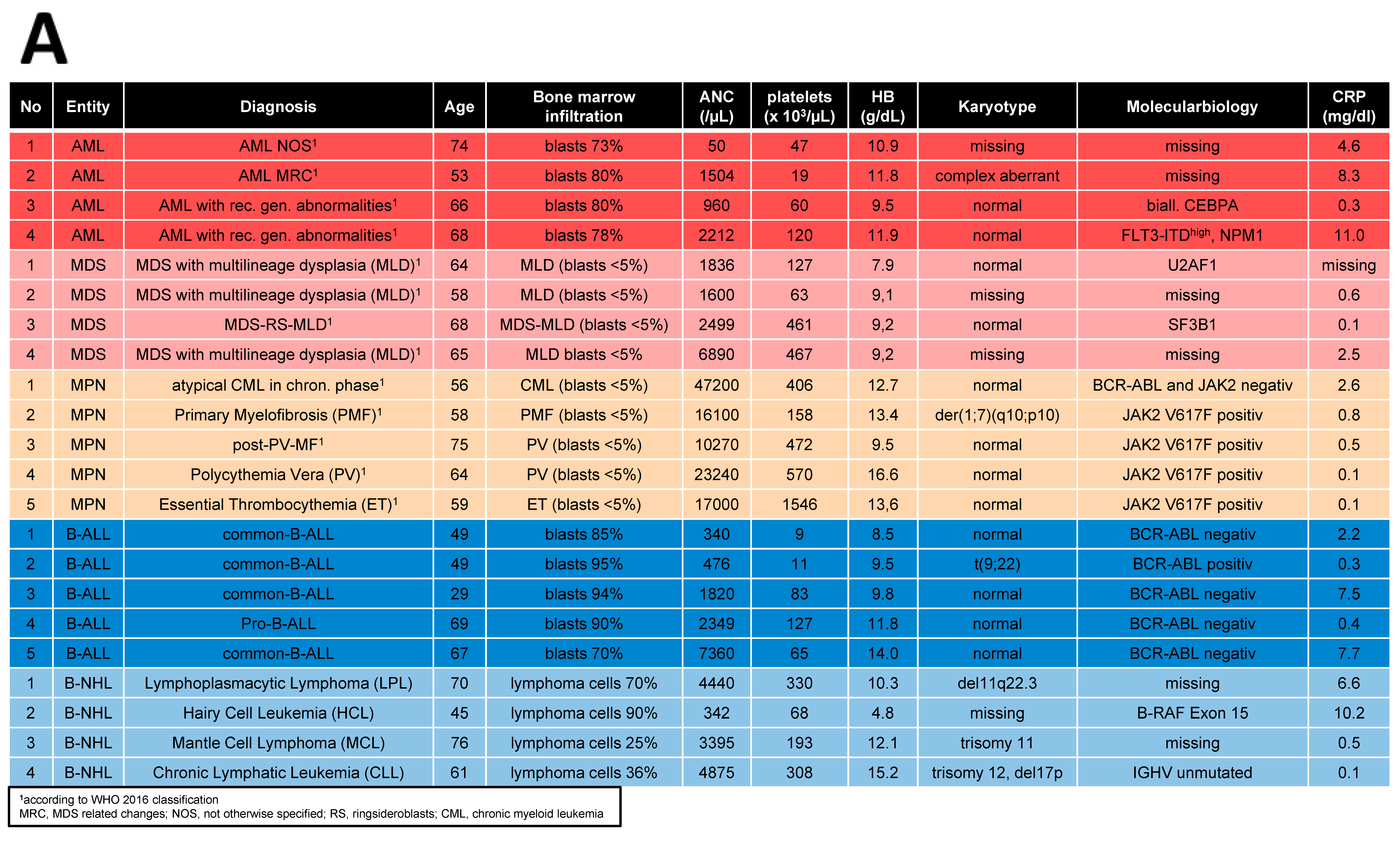
2.1. Patients, Healthy Controls, and Cell Preparation
2.2. Cell Culture Conditions and Reagents
2.3. Growth Properties and Cellular Senescence
2.4. Conditioned Media
2.5. Differentiation Properties
2.6. Hematopoietic Support—Long-Term Culture-Initiating Cells (LTC-IC Assay)
2.7. Co-Culture with Conditioned Media and Blocking of TGFB1
2.8. Quantitative Realtime-Polymerase Chain Reaction (qRT-PCR)
2.9. RNA-Sequencing
2.10. Bioinformatical Analysis
2.11. Data Access
2.12. Statistical Analysis
3. Results
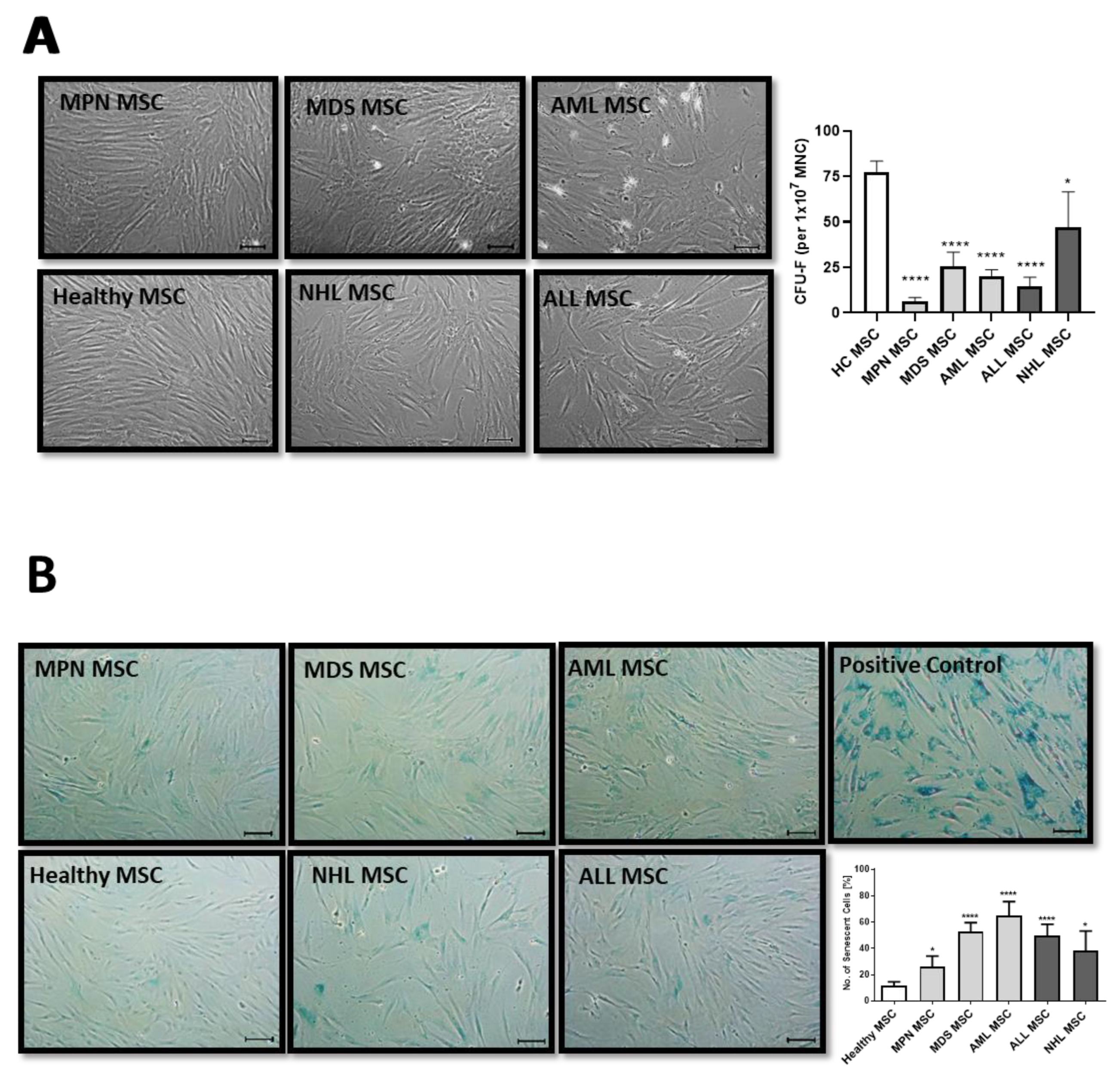
3.1. MSCs from Myeloid and Lymphoid Neoplasms Showed Similar Alterations in Growth Capacity and Cellular Senescence
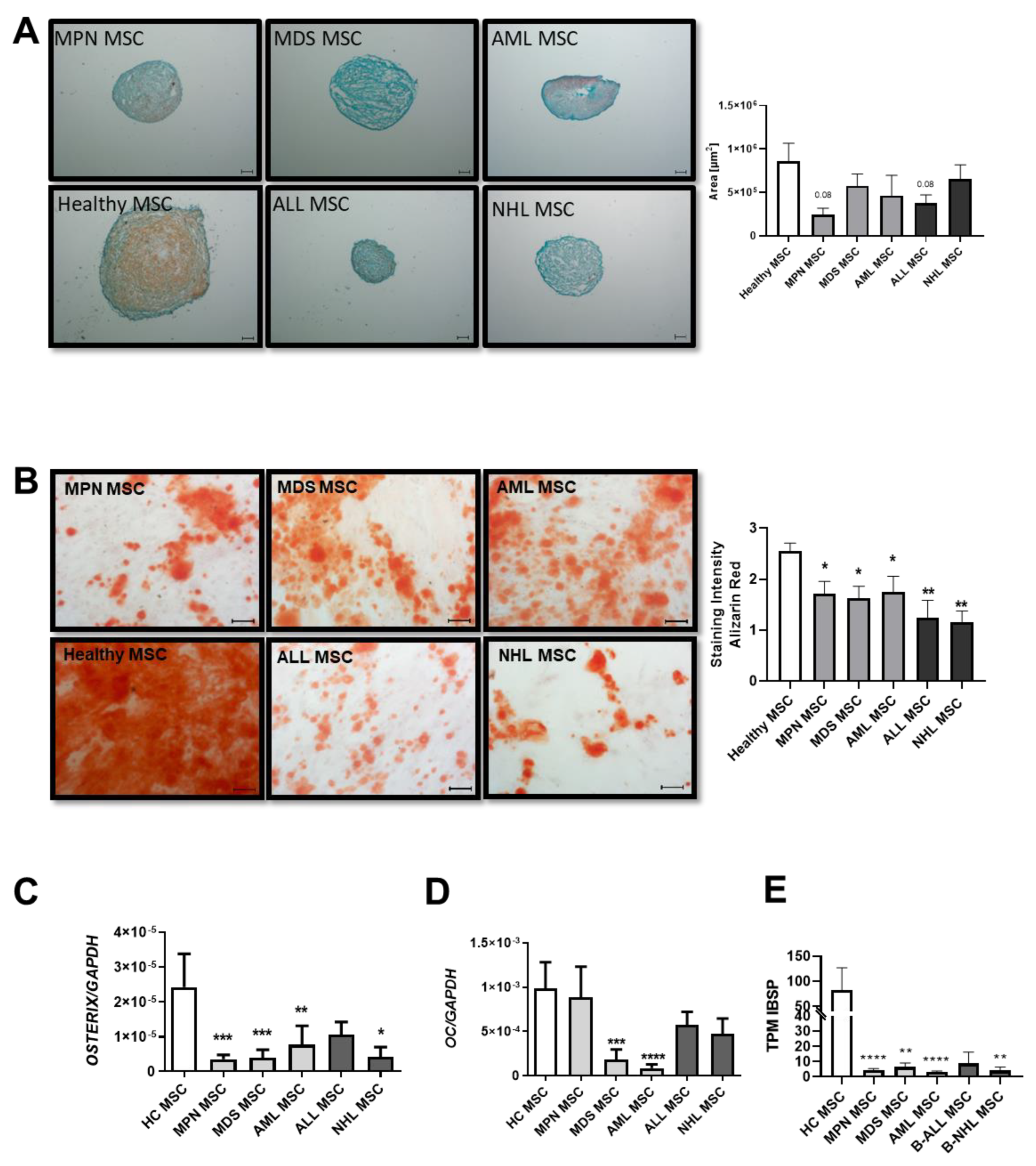
3.2. MSCs from Myeloid and Lymphoid Neoplasms Showed Similar Reduced Chondrogenic-Osteogenic Differentiation Capacity
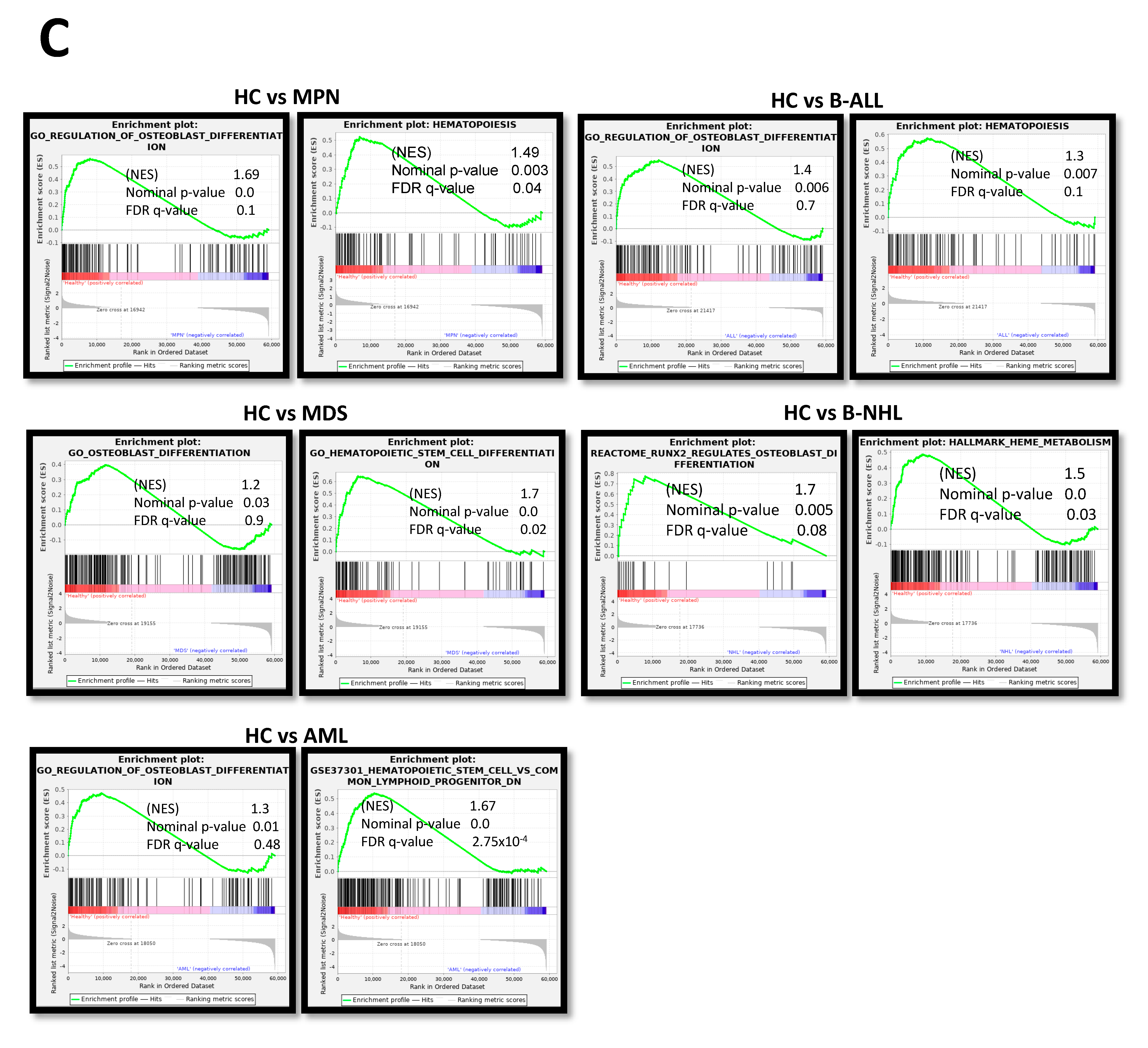
3.3. Insufficient Hematopoietic Support in Myeloid and Lymphoid Neoplasms
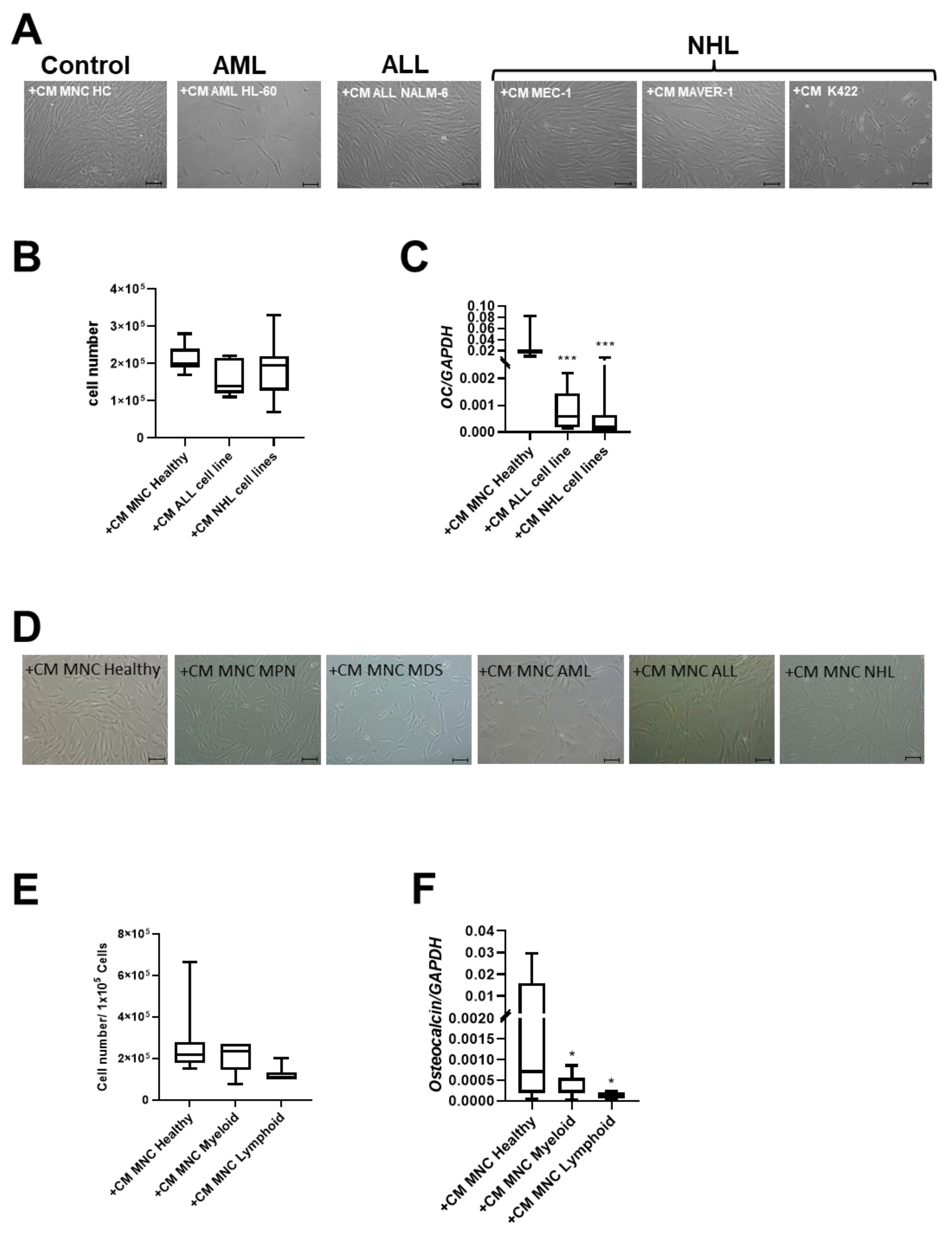
3.4. Patient-Derived and Cell Line-Derived Conditioned Media-Induced Deficits in MSCs
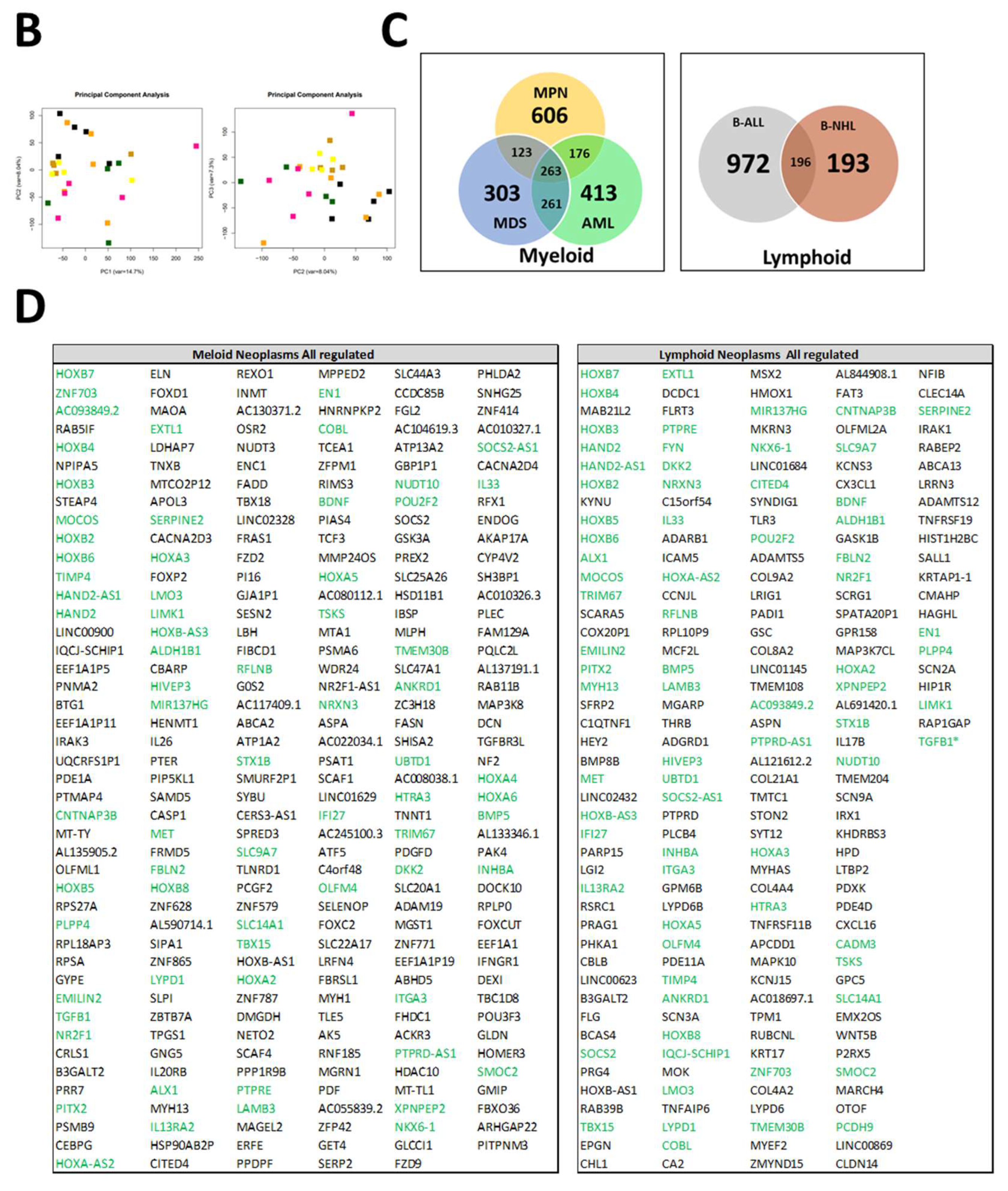
3.5. RNA Sequencing Distinguish MSCs from Myeloid and Lymphoid Neoplasms
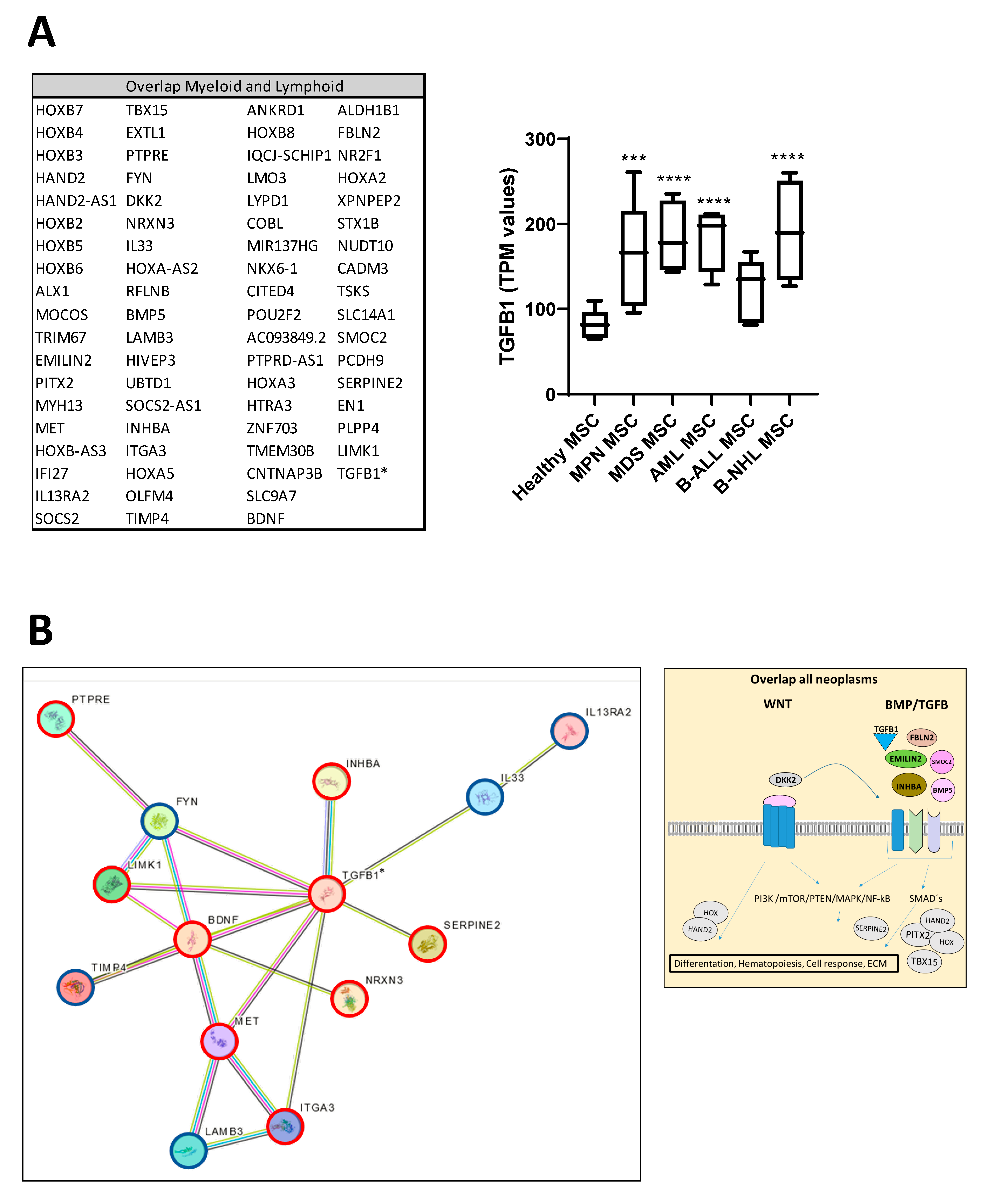
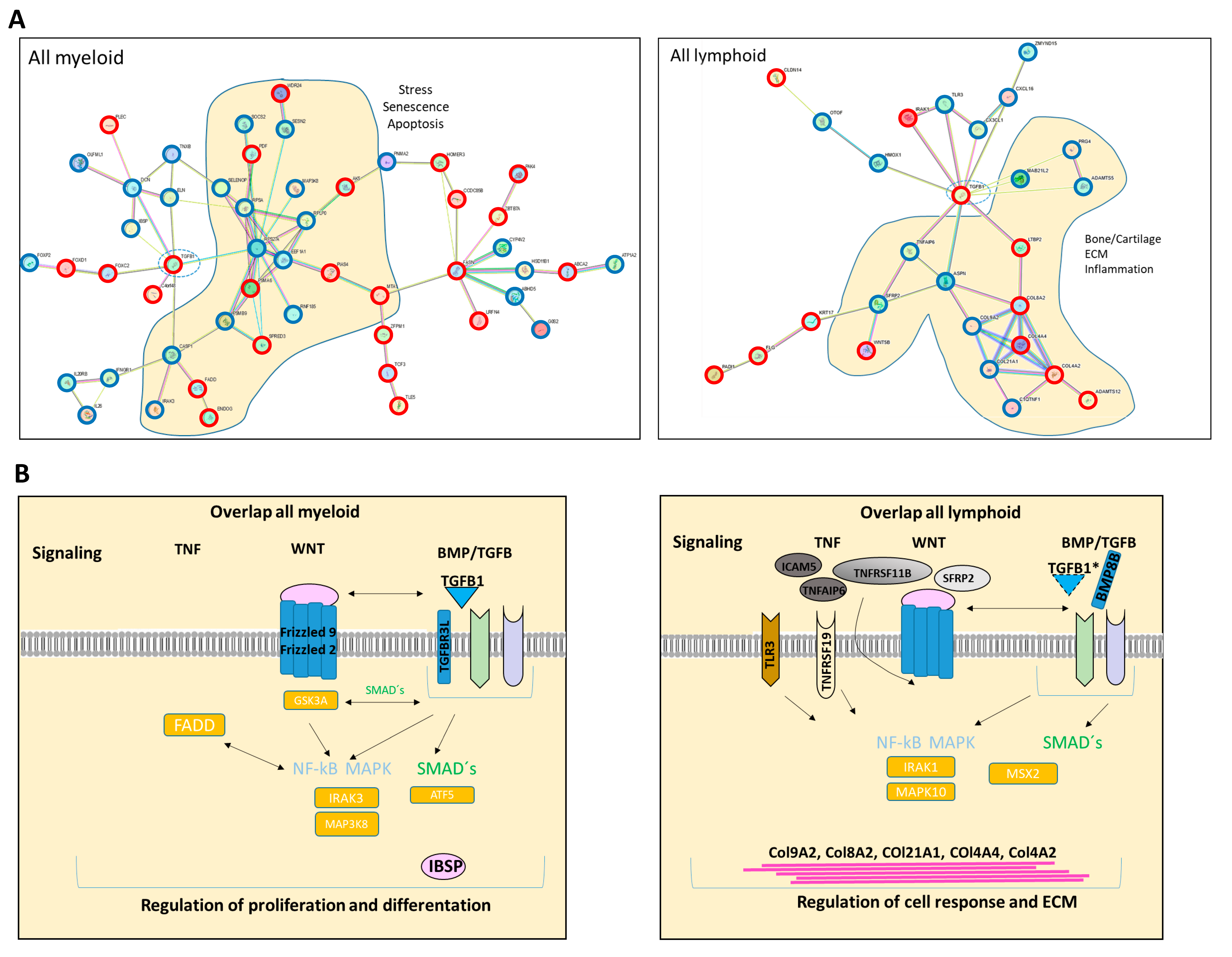
3.6. RNA Sequencing Revealed Overlapping Genetic Signatures Associated with Chondrogenic-Osteogenic Differentiation and Hematopoietic Supporting Capacity in MSCs from Myeloid and Lymphoid Neoplasms
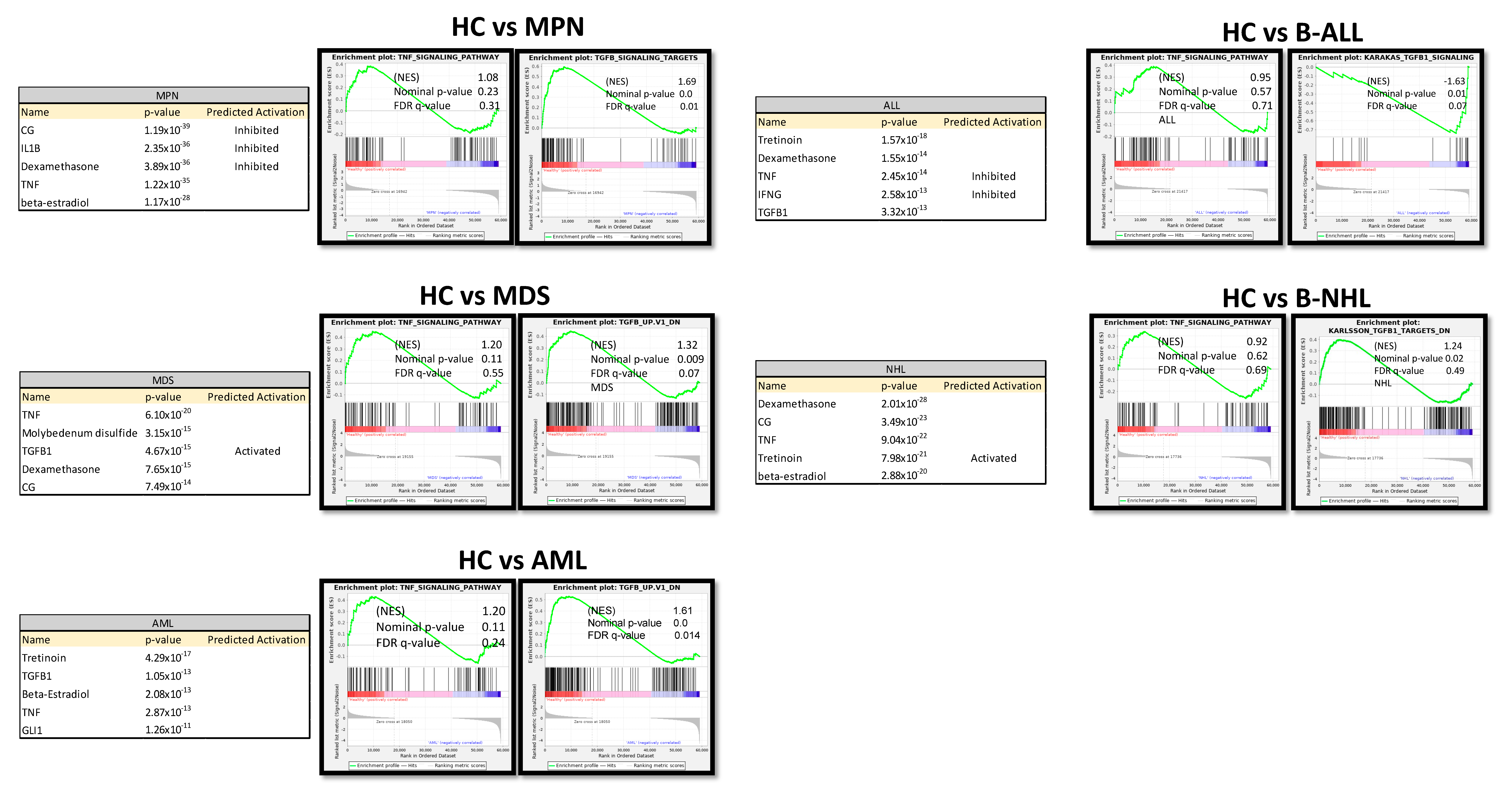
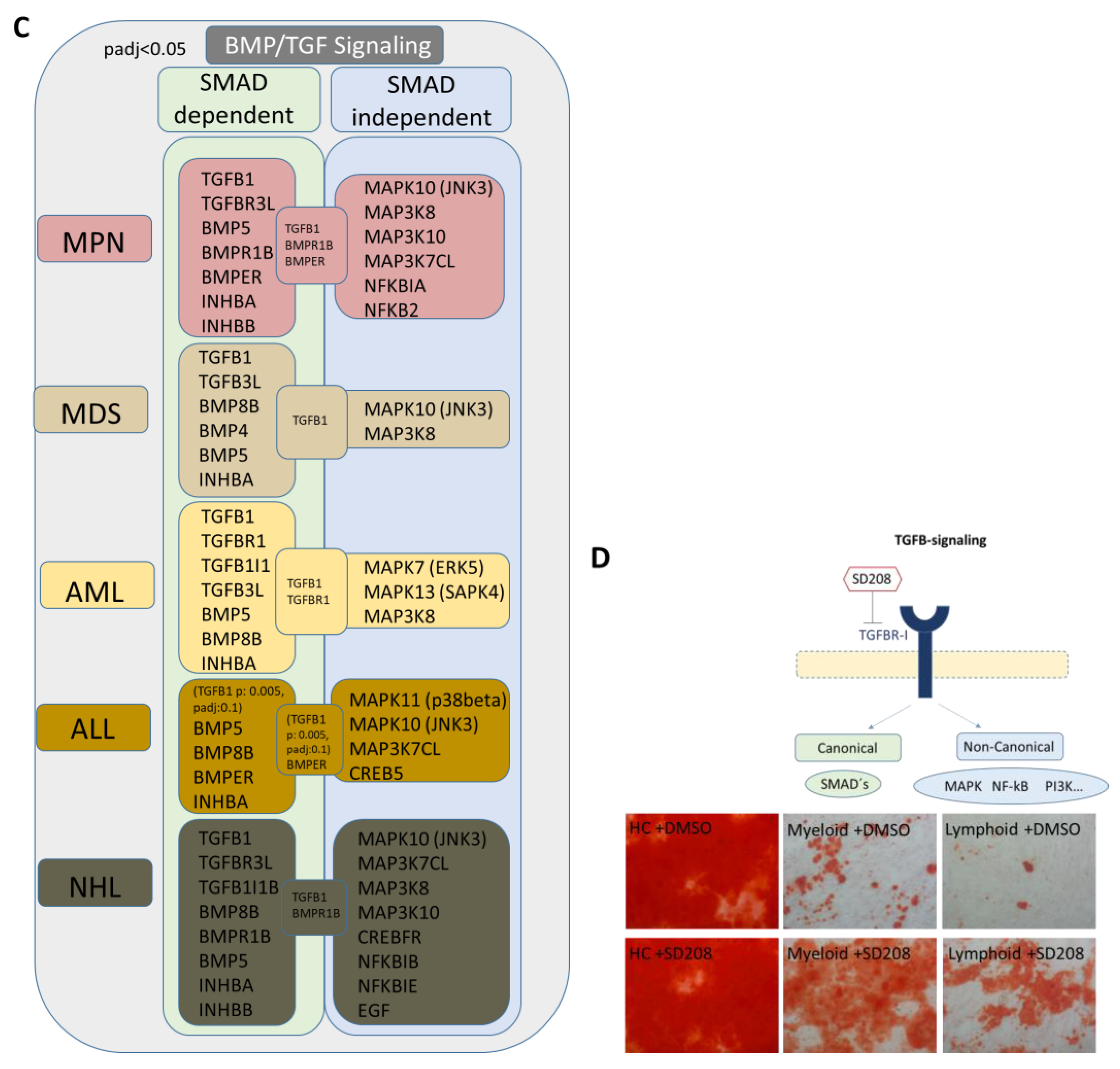
3.7. TGFB1 as Potential Overlapping Upstream Regulator
3.8. RNA Sequencing Revealed SMAD-Dependent and SMAD-Independent BMP/TGFB-Signaling in Myeloid and Lymphoid Neoplasms, Which Can Both Be Blocked as Potential Therapeutic Targets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Will, B.; Zhou, L.; Vogler, T.O.; Ben-Neriah, S.; Schinke, C.; Tamari, R.; Yu, Y.; Bhagat, T.D.; Bhattacharyya, S.; Barreyro, L.; et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood 2012, 120, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.J.; Tariman, J.D. Multiple Myeloma Genomics: A Systematic Review. Semin. Oncol. Nurs. 2017, 33, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Borkhardt, A.; Wuchter, C.; Viehmann, S.; Pils, S.; Teigler-Schlegel, A.; Stanulla, M.; Zimmermann, M.; Ludwig, W.D.; Janka-Schaub, G.; Schrappe, M.; et al. Infant acute lymphoblastic leukemia—Combined cytogenetic, immunophenotypical and molecular analy-sis of 77 cases. Leukemia 2002, 16, 1685–1690. [Google Scholar] [CrossRef] [PubMed]
- van Beers, E.J.; Muller, M.C.; Vlaar, A.P.; Spanjaard, L.; van den Bergh, W.M.; Group, H.-I.S. Haematological malignancy in the intensive care unit: Microbiology results and mortality. Eur. J. Haematol. 2016, 97, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Frattini, F.; Crestani, S.; Bonfanti, C. Bleeding complications in patients with hematologic malignancies. Semin. Thromb. Hemost. 2013, 39, 94–100. [Google Scholar] [PubMed]
- Jager, P.; Geyh, S.; Twarock, S.; Cadeddu, R.P.; Rabes, P.; Koch, A.; Maus, U.; Hesper, T.; Zilkens, C.; Rautenberg, C.; et al. Acute myeloid leukemia-induced functional inhibition of healthy CD34+ hematopoietic stem and progenitor cells. Stem Cells 2021, 39, 1270–1284. [Google Scholar] [CrossRef]
- Bruns, I.; Cadeddu, R.P.; Brueckmann, I.; Frobel, J.; Geyh, S.; Bust, S.; Fischer, J.C.; Roels, F.; Wilk, C.M.; Schildberg, F.A.; et al. Multiple myeloma-related deregulation of bone marrow-derived CD34(+) hematopoietic stem and progenitor cells. Blood 2012, 120, 2620–2630. [Google Scholar] [CrossRef]
- Geyh, S.; Oz, S.; Cadeddu, R.P.; Frobel, J.; Bruckner, B.; Kundgen, A.; Fenk, R.; Bruns, I.; Zilkens, C.; Hermsen, D.; et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia 2013, 27, 1841–1851. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brustle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegue, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Pozzi, S.; Patel, K.; Vaghela, N.; Fulciniti, M.T.; Veiby, P.; Hideshima, T.; Santo, L.; Cirstea, D.; Scadden, D.T.; et al. A novel role for CCL3 (MIP-1alpha) in myeloma-induced bone disease via osteocalcin downregulation and inhibition of osteoblast function. Leukemia 2011, 25, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Frisch, B.J.; Ashton, J.M.; Xing, L.; Becker, M.W.; Jordan, C.T.; Calvi, L.M. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood 2012, 119, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Geyh, S.; Rodriguez-Paredes, M.; Jager, P.; Khandanpour, C.; Cadeddu, R.P.; Gutekunst, J.; Wilk, C.M.; Fenk, R.; Zilkens, C.; Hermsen, D.; et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia 2016, 30, 683–691. [Google Scholar] [CrossRef]
- Geyh, S.; Rodriguez-Paredes, M.; Jager, P.; Koch, A.; Bormann, F.; Gutekunst, J.; Zilkens, C.; Germing, U.; Kobbe, G.; Lyko, F.; et al. Transforming growth factor beta1-mediated functional inhibition of mesenchymal stromal cells in myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2018, 103, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, R.A.; Wobus, M.; List, C.; Wehner, R.; Schonefeldt, C.; Brocard, B.; Mohr, B.; Rauner, M.; Schmitz, M.; Stiehler, M.; et al. Mesenchymal stromal cells from patients with myelodyplastic syndrome display distinct functional alterations that are modulated by lenalidomide. Haematologica 2013, 98, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.C.; Xu, Y.H.; Chen, H.X.; Wang, X.J. Acute Lymphoblastic Leukemia Cells Inhibit the Differentiation of Bone Mesenchymal Stem Cells into Osteoblasts In Vitro by Activating Notch Signaling. Stem Cells Int. 2015, 2015, 162410. [Google Scholar] [CrossRef]
- Bogun, L.; Koch, A.; Scherer, B.; Fenk, R.; Maus, U.; Bormann, F.; Köhrer, K.; Petzsch, P.; Wachtmeister, T.; Zukovs, R.; et al. Stromal alterations in patients with MGUS, smoldering myeloma and multiple myeloma. Blood Adv. 2024. [Google Scholar] [CrossRef]
- Frith, J.; Genever, P. Transcriptional control of mesenchymal stem cell differentiation. Transfus. Med. Hemother. 2008, 35, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Gage. The canonical Wnt signaling antagonist DKK2 is an essential effecotr of PITX2 function during normal eye development. Dev. Biol. 2008, 317, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K. The T-box transcription factor Tbx15 is required for skeletal development. Mech. Dev. 2005, 122, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Vanegas, N.P.; Ruiz-Aparicio, P.F.; Uribe, G.I.; Linares-Ballesteros, A.; Vernot, J.P. Leukemia-Induced Cellular Senescence and Stemness Alterations in Mesenchymal Stem Cells Are Reversible upon Withdrawal of B-Cell Acute Lymphoblastic Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 8166. [Google Scholar] [CrossRef] [PubMed]
- Balandran, J.C.; Davila-Velderrain, J.; Sandoval-Cabrera, A.; Zamora-Herrera, G.; Teran-Cerqueda, V.; Garcia-Stivalet, L.A.; Limon-Flores, J.A.; Armenta-Castro, E.; Rodriguez-Martinez, A.; Leon-Chavez, B.A.; et al. Patient-Derived Bone Marrow Spheroids Reveal Leukemia-Initiating Cells Supported by Mesenchymal Hypoxic Niches in Pediatric B-ALL. Front. Immunol. 2021, 12, 746492. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, S.; Mihara, K.; Downing, J.R.; Pui, C.H.; Campana, D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J. Clin. Investig. 2007, 117, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.J.; Goodell, L.; Glod, J.; Gelinas, C.; Rabson, A.B.; Strair, R.K. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of B-cell activating factor and activation of the canonical and non-canonical nuclear factor kappaB pathways. Haematologica 2012, 97, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Maeda, S.; Aburatani, H.; Kitamura, K.; Miyoshi, H.; Miyazono, K.; Imamura, T. Pitx2 prevents osteoblastic transdifferentiation of myoblasts by bone morphogenetic proteins. J. Biol. Chem. 2008, 283, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Maguer-Satta, V. Targeting BMP signaling in the bone marrow microenvironment of myeloid leukemia. Biochem. Soc. Trans. 2020, 48, 411–418. [Google Scholar] [CrossRef]
- Ame-Thomas, P.; Maby-El Hajjami, H.; Monvoisin, C.; Jean, R.; Monnier, D.; Caulet-Maugendre, S.; Guillaudeux, T.; Lamy, T.; Fest, T.; Tarte, K. Human mesenchymal stem cells isolated from bone marrow and lymphoid organs support tumor B-cell growth: Role of stromal cells in follicular lymphoma pathogenesis. Blood 2007, 109, 693–702. [Google Scholar] [CrossRef]
- Purroy, N. Co-culture of primary CLL cells with bone marrow mesenchymal cells, CD40 ligand and CpG ODN promotes proliferation of chemoresistant CLL cells phenotypically comparable to those proliferating in vivo. Oncotarget 2015, 6, 7632–7643. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; De Veirman, K.; De Becker, A.; Vanderkerken, K.; Van Riet, I. Mesenchymal stem cells in multiple myeloma: A therapeutical tool or target? Leukemia 2018, 32, 1500–1514. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Deng, C.; Li, Y.P. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-beta signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 2022, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Oetjen, K.A.; Wang, T.; Xu, H.; Abou-Ezzi, G.; Krambs, J.R.; Uttarwar, S.; Duncavage, E.J.; Link, D.C. TGF-beta signaling in myeloproliferative neoplasms contributes to myelofibrosis without disrupting the hematopoietic niche. J. Clin. Investig. 2022, 132, e154092. [Google Scholar] [CrossRef]
- Timmins, M.A.; Ringshausen, I. Transforming Growth Factor-Beta Orchestrates Tumour and Bystander Cells in B-Cell Non-Hodgkin Lymphoma. Cancers 2022, 14, 1772. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | % | |
|---|---|---|
| Patients No. | 70 | |
| Sex | ||
| Male | 42 | 60 |
| Female | 28 | 40 |
| AML No. | 25 | |
| Median age, years (Range) | 61 | (25–74) |
| Diagnosis WHO 2016 1 | ||
| Median BM infiltration, % (Range) | 69 | (35–95) |
| Median ANC, ×103/µL (Range) | 16.50 | (0.00–21.00) |
| Median platelets, ×103/µL (Range) | 60 | (6–279) |
| Median HB, g/dL (Range) | 9.4 | (4.2–11.9) |
| MDS No. | 16 | |
| Median age, years (Range) | 68 | (47–81) |
| Diagnosis WHO 2016 2 | ||
| Median BM infiltration, % (Range) | Dysplasia 2, blasts > 5% in 6 | |
| Median ANC, ×103/µL (Range) | 1.76 | (0.20–6.89) |
| Median platelets, ×103/µL (Range) | 69 | (27–467) |
| Median HB, g/dL (Range) | 9.2 | 6.6–11.2 |
| MPN No. | 11 | |
| Median age, years (Range) | 60 | (22–75) |
| Diagnosis WHO 2016 3 | ||
| Median BM infiltration, % (Range) | Proliferation, blasts < 5% | |
| Median ANC, ×103/µL (Range) | 16.10 | (5.40–47.20) |
| Median platelets, ×103/µL (Range) | 534 | (54–1564) |
| Median HB, g/dL (Range) | 13.6 | (9.3–16.6) |
| ALL No. | 9 | |
| Median age, years (Range) | 47 | (22–69) |
| Diagnosis 4 | ||
| Median BM infiltration, % (Range) | 85 | (40–95) |
| Median ANC, ×103/µL (Range) | 2.35 | (0.34–15.20) |
| Median platelets, ×103/µL (Range) | 53 | (9–138) |
| Median HB, g/dL (Range) | 11.8 | (8.5–16.7) |
| NHL No. | 9 | |
| Median age, years (Range) | 63 | (38–76) |
| Diagnosis 5 | ||
| Median BM infiltration, % (Range) | 25 | (10–90) |
| Median ANC, ×103/µL (Range) | 3.40 | (0.33–8.90) |
| Median platelets, ×103/µL (Range) | 193 | (68–345) |
| Median HB, g/dL (Range) | 12.5 | (4.8–15.2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogun, L.; Koch, A.; Scherer, B.; Germing, U.; Fenk, R.; Maus, U.; Bormann, F.; Köhrer, K.; Petzsch, P.; Wachtmeister, T.; et al. Overlapping Stromal Alterations in Myeloid and Lymphoid Neoplasms. Cancers 2024, 16, 2071. https://doi.org/10.3390/cancers16112071
Bogun L, Koch A, Scherer B, Germing U, Fenk R, Maus U, Bormann F, Köhrer K, Petzsch P, Wachtmeister T, et al. Overlapping Stromal Alterations in Myeloid and Lymphoid Neoplasms. Cancers. 2024; 16(11):2071. https://doi.org/10.3390/cancers16112071
Chicago/Turabian StyleBogun, Lucienne, Annemarie Koch, Bo Scherer, Ulrich Germing, Roland Fenk, Uwe Maus, Felix Bormann, Karl Köhrer, Patrick Petzsch, Thorsten Wachtmeister, and et al. 2024. "Overlapping Stromal Alterations in Myeloid and Lymphoid Neoplasms" Cancers 16, no. 11: 2071. https://doi.org/10.3390/cancers16112071
APA StyleBogun, L., Koch, A., Scherer, B., Germing, U., Fenk, R., Maus, U., Bormann, F., Köhrer, K., Petzsch, P., Wachtmeister, T., Kobbe, G., Dietrich, S., Haas, R., Schroeder, T., Geyh, S., & Jäger, P. (2024). Overlapping Stromal Alterations in Myeloid and Lymphoid Neoplasms. Cancers, 16(11), 2071. https://doi.org/10.3390/cancers16112071