1. Introduction
Globally, breast cancer (BC) stands out as the most diagnosed cancer and a leading contributor to cancer-related fatalities in women [
1]. BC presents itself with four molecular subtypes (luminal A, luminal B, HER2-enriched, and basal-like) based on a 50-gene expression signature (PAM50) [
2]. Besides this molecular classification, a more traditional classification based on the immunohistochemistry expression of key proteins such as estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2), and the proliferation marker Ki67, are commonly employed (reviewed in [
3]). BC cells can acquire invasiveness, leading to the formation of metastases, as they spread through the bloodstream or lymphatic system to various organs and sites in the body. Primary BC has been extensively studied and generally has a positive prognosis. In contrast, metastatic BC presents numerous challenges, including the presence of diverse cell types and resistance to treatment. These factors often contribute to the failure of therapeutic interventions [
4].
Therapy decisions usually depend on the features of primary tumors, as metastatic biopsies are relatively rare. Yet, primary tumors may not fully represent the heterogeneous characteristics of metastatic tumors. Frequently, these attributes demonstrate inconsistency in phenotypic markers [
5]. Consequently, therapy recommendations based on the characteristics of primary tumors alone might lead to poorer outcomes [
6,
7].
Malignant pleural effusion (MPE), which describes the presence of malignant cells in the pleural cavity, develops in 7% of BC patients [
8,
9]. The quality of life and prognosis of patients suffering from MPE are relatively poor and defined by chest pain and breathing difficulties [
10]. However, as MPE contains metastatic BC cells and can be obtained through a simple puncture, it offers more options for metastatic biopsies [
11]. Moreover, malignant cells in MPEs can serve as a source for the establishment of in vitro models that represent the characteristics of metastasized BC to improve therapy outcomes and approach precision oncology. The most common approaches include cell lines and patient-derived xenograft (PDX) models. While cell lines are widely used in BC research but are not capable of predicting drug response in patients, PDX models are invaluable for translational research but are expensive and limited in efficiency [
12].
Recently, patient-derived BC organoids have been shown to be valuable three-dimensional models for research and personalized oncology, as they represent the characteristics of their respective origin and can be used for long-term culturing [
13,
14]. Cultured in an extracellular matrix, organoids of different sources have already been used in various methods, such as high-throughput drug assays [
13,
15].
In the area of BC treatment, standard therapeutic approaches involve surgery, chemotherapy, radiotherapy, endocrine therapy, and targeted therapy [
16]. The latter approach includes immunotherapy, which has risen as a crucial element, presenting promising effectiveness and minimal safety concerns [
17]. The emergence of genetically modified T cell therapies, incorporating a chimeric antigen receptor (CAR) has achieved remarkable success in long-lasting clinical responses among individuals with hematologic malignancies [
18]. This sparked immense enthusiasm in the potential to address various types of cancer including BC. CAR-T cell therapy, a type of immunotherapy derived from adoptive T cell transfer, has been established to harness the patient’s own immune cells to fight cancer by triggering antigen-specific cytotoxic response [
19].
Conventional CAR constructs are limited in their ability to provide adjustable cytotoxicity and flexible selectivity against heterogenous tumors. These limitations result in potential risks such as uncontrolled CAR-T expansion, depletion of normal tissues expressing the target antigen, CAR-T exhaustion, and lack of activation toward antigen-negative tumor cells [
20,
21,
22].
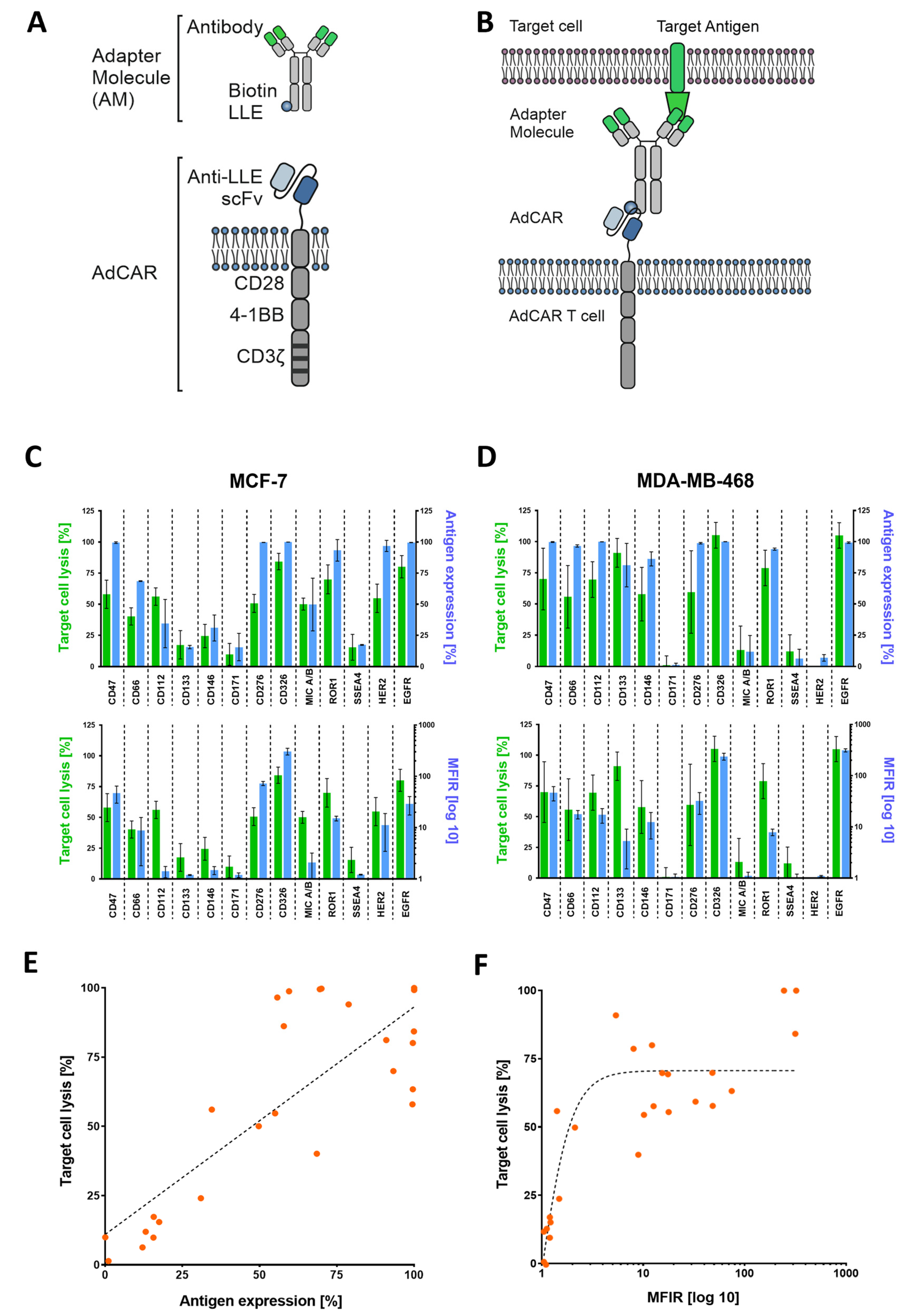
One sophisticated approach to circumvent these limitations is to separate target antigen recognition from CAR-T activation through the introduction of adapter molecules (AMs) and adapter CARs (AdCARs) [
23]. These AMs combine the functional components of an antigen-binding moiety and a CAR-binding moiety. Being able to conjugate clinically approved therapeutical antibodies, effectively turning them into AMs, enables flexible redirection of CAR-T cells toward different tumor-associated antigens (TAAs) and furthermore introduces a controllable on/off-switch.
In previous studies, we have demonstrated the remarkable effectiveness of AdCAR-T cells in vitro and in vivo by specifically targeting tumors associated with a broad spectrum of tumor-associated antigens (TAAs) across different cancer types [
23,
24]. This CAR technology provides the advantage of universal targeting of various cancer types and furthermore allows for the finely adjustable modulation of effector function and ultimately addresses the challenge of immune evasion due to antigen loss.
While CAR-T cell therapy has proven great achievements in hematologic malignancies, its effectiveness in battling solid tumors, including BC, still has room for improvement. In a previous study, we cultured tumor cells derived from MPE of advanced BC patients employing organoid technology and generated metastatic BC patient-derived organoids (MBC-PDOs). MBC-PDOs were then used as a platform to perform drug screenings applying various inhibitors [
14].
In this study, we cultured MBC-PDOs and analyzed their antigen expression patterns utilizing flow cytometry (FC). Our goal was to utilize MBC-PDOs as a screening platform to demonstrate feasibility of an AdCAR-T-based precision immunotherapy approach for flexible targeting of various tumor-associated target antigens. In the future, research on patient-derived models could have enormous potential for clinical applications, such as extending patients’ survival time and improving their quality of life.
2. Materials and Methods
2.1. Patient Cohort
All MBC-PDOs were previously established and characterized [
14]. The study was approved by the Ethics Committee of the Eberhard Karl University of Tübingen (ethical approval 288/2022BO2) and is compliant with all relevant ethical regulations regarding research involving human participants. For full patient characteristics see
Table S1.
2.2. Culturing 2D Cell Lines
BC cell lines MDA-MB-468 and MCF-7 were acquired from American Type Culture Collection (Manassas, VA, USA, HTB-22 and HTB-132). Cells were handled in DMEM-FBS (Dulbecco’s Modified Eagle Medium (41965-062), containing 10% FBS (10270-106), 1% Pen/Strep (15140-122) from Thermo Fisher Scientific, Waltham, MA, USA). Cells were recurrently checked for mycoplasma using a PCR Detection Kit (abm, Richmond, BC, Canada, G238).
2.3. Freezing and Thawing of CAR-T Cells
Cultured cells were centrifuged at 350× g for 5 min and resuspended in human serum albumin (HSA, CSL Behring GmbH, Marburg, Germany, 4356500002) containing 10% dimethyl sulfoxide (DMSO, PanReac Applichem, Darmstadt, Germany, A3672) at a cell density of 1–10 × 107 cells/mL; 1 mL aliquots were frozen overnight at −80 °C and subsequently transferred to liquid nitrogen storage. To thaw cells, frozen aliquots were rapidly thawed in a 37 °C water bath, diluted in pre-warmed TexMACS™ medium (Miltenyi Biotec, Bergisch Gladbach, Germany, 31870) without interleukins or antibiotics and centrifuged at 350× g for 5 min. Then, T cells were resuspended in TexMACS™ medium without supplementation at a density of 4 × 106 cells/mL and incubated for 4 h before adding IL-7/IL-15-supplemented (Miltenyi Biotec, Bergisch Gladbach, Germany, 130-095-367 and 130-093-955) TexMACSTM media to achieve a cell density of 1 × 106 cells/mL.
2.4. Isolation and Transduction of T Cells
Peripheral blood mononuclear cells (PBMCs) were isolated from freshly collected peripheral blood samples via Ficoll centrifugation (Biocoll®, BIO&SELL GmbH, Nürnberg, Germany, L 6115) from healthy voluntary donors at the University Children’s Hospital Tübingen. T cell isolation was performed using anti-CD4 and anti-CD8 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany, 130-045-101 and 130-045-201), subsequently activated with TransActTM (Miltenyi Biotec, Bergisch Gladbach, Germany, 130-128-758) and cultivated in TexMACSTM media supplemented with 10 ng/mL IL-7 and 5 ng/mL IL-15. After 36 h, activated T cells were transduced with AdCAR (LLE-CAR) lentivirus (provided by Miltenyi Biotec, Bergisch Gladbach, Germany) at a multiplicity of infection (MOI) of 3. Transduced T cells were maintained at 0.5–2 × 106 cells/mL in IL-7/IL-15-supplemented TexMACSTM media and furthermore monitored for lactate. CAR transduction efficiency was determined by flow cytometry at day 7 using AdCAR detection reagent (provided by Miltenyi Biotec, Bergisch Gladbach, Germany).
2.5. Adapter Molecule Conjugation
Biotin adapter conjugation was achieved at 30 °C for 1 h in DPBS buffer (Thermo Fisher Scientific, Waltham, MA, USA, 14190-094) using 5-fold molar excess of biotin-LC-LC-NHS (Thermo Fisher Scientific, Waltham, MA, USA, CAS-No. 89889-52-1). This was followed by separation of the antibody/label mix on a Sephadex G25 column (Cytiva Europe GmbH, Freiburg, Germany, 17085101). Protein holding fractions were collected, analyzed by absorption at 280 nm, and subsequently united. Successful conjugation was confirmed via flow cytometry on cell lines expressing the target.
2.6. Organoid Culture Setup
Cryopreserved MBC-PDOs from our previous study were used for the study [
14]. For the setup of organoid cultures, the required amount of cell suspension was mixed with basement membrane extract (BME; Cultrex Reduced Growth Factor Basement Membrane Extract, Type 2 Select, Bio-techne, Minneapolis, MN, USA, 3533-005-02) at a ratio of 30% cell suspension to 70% BME; 20 µL droplets were seeded on 48-well plates and placed upside down in an incubator (37 °C, 5% CO
2) to solidify for 30 min. BC culture medium (BCM;
Table S2, composition previously described [
13,
25]) was added to each well and renewed every 3–4 days. Cells were incubated at 37 °C and 5% CO
2 and pictures were taken frequently with EVOS M7000 microscope (Thermo Fisher Scientific, Waltham, MA, USA).
2.7. Passaging of MBC-PDOs
MBC-PDOs were passaged every 7 to 21 days, depend on organoid size and density. Organoids were recovered from the wells by resuspending the BME droplets in ice-cold DPBS containing 5 µM Y-27632 (DPBS/Y-27632). The organoid suspension was centrifuged at 500× g for 10 min and the supernatant was discarded. The BME-organoid pellet was dispersed with 1 mL of TrypLE™ Express Enzyme (1X; Thermo Fisher Scientific, Waltham, MA, USA, 12604013) at 37 °C in a water bath for 5 min. The suspension was then centrifuged (500× g for 10 min) and the supernatant was discarded. For further culture, the desired amount of cell pellet was resuspended in AdvDMEM+++ and mixed with BME at a ratio of 30% cell suspension to 70% BME and cultured as described above. To stock organoids, passaged cells were cryopreserved in Recovery™ Cell Culture Freezing Medium (Thermo Fisher Scientific, Waltham, MA, USA, 12648010) and stored in cryovials in liquid nitrogen.
2.8. Generation of Lentiviral Vector
Luciferase and GFP containing lentivirus were produced as previously described [
23]. After lipofection (Lipofectamine 3000, Thermo Fisher) of Lenti-XTM 293 T (Clontech/ TaKaRa Bio Company, San Jose, CA, USA, 631231) with second-generation packaging plasmid, VSV-G envelope plasmid and transfer plasmid, lentivirus containing supernatants were concentrated (Lenti-X concentrator, TaKaRa Bio Company, San Jose, CA, USA, 631231) and cryopreserved at −80 °C.
2.9. Viral Transduction and Sorting of MBC-PDOs
Third-generation-based lentiviral vector transfer plasmids containing luciferase and GFP were kindly provided by Irmela Jeremias, Helmholtz Center Munich, Munich, Germany (Irmela Jeremias, Helmholtz Center Munich, Germany, 12260). Lentiviral particles were produced as described above. All MBC-PDO lines were transduced at an MOI of 3. Subsequent transgene expression was analyzed by flow cytometry using the co-expressed fluorescent protein. Transduced cells were enriched by bulk fluorescence-activated cell sorting (FACS).
2.10. Flow Cytometry
Flow cytometry analysis was performed by staining 0.2 × 10
6 cells in fluorescence- activated cell sorting (FACS) tubes. The following antibodies by Miltenyi Biotec (Bergisch Gladbach, Germany) were used to determine antigen expression. Anti-CD47 (anti-human, Biotin, 130-101-342), anti-CD66 (anti-human, Biotin, 130-093-156), anti-CD112 (anti-human, Biotin, 130-109-000), anti-CD133 (anti-human, Biotin, 130-112-193), anti-CD146 (anti-human, Biotin, 130-092-850), anti-CD171 (anti-human, Biotin, 130-100-702), anti-CD276 (anti-human, Biotin, 130-118-579), anti-ROR1 (anti-human, Biotin, 130-118-018), anti-TROP2 (anti-human, Biotin, 130-115-096), anti-CD326 (EpCAM) (anti-human, Biotin, 130-111-114), HER2 (biotin-conjugated Trastuzumab, Kanjinti, 1144554A), and EGFR (biotin conjugated Cetuximab, Erbitux, G0157D) (
Table S3). CliniMACS
® buffer (Miltenyi Biotec, Bergisch Gladbach, Germany, 700-25) containing antibodies at an equimolar concentration of 20 µg/mL was added to each sample. The cells were furthermore stained with anti-Biotin (APC; Miltenyi Biotec, Bergisch Gladbach, Germany, 130-111-069) and analyzed via BD FACSCanto II (BD Biosciences, Franklin Lakes, NJ, USA).
2.11. Two-Dimensional Luciferase-Based Cytotoxicity Assay (LCA)
Tumor cells were plated in complete RPMI media (Thermo Fisher Scientific, Waltham, MA, USA, 31870) in white 96-well flat-bottomed plates (Greiner Holding, Kremsmünster, Austria, 655083) with 15,000 cells per well and were co-cultured with AdCAR-T cells at a 2:1 ratio. At timepoint 0 h, adapter molecules and synthetic D-luciferin (Sigma Aldrich, St. Louis, MO, USA, L9504) were added to each well at a final concentration of 10 ng/mL and 4 µg/mL, respectively. Bioluminescence was assessed after 24 h, 48 h, and 72 h using the Tecan SPARK microplate reader (Perkin Elmer, Waltham, MA, USA, 30086376) at 37 °C. Lysis was evaluated by the relative luminescence to tumor control wells without AdCAR-T cells.
2.12. Real-Time Impedance-Based Cytotoxicity Assay (ICA)
The impedance-based Real-Time Cytotoxicity Analyzer (RTCA) xCELLigence device (ACEA Biosciences Inc., Santa Clara, CA, USA, 106-0534) was used to assess label-free real-time cytotoxicity [
23]. MCF-7 and MD-MBA-468 cell lines were plated at 30,000 cells per well in RPMI 1640-based complete media in 96-well electronic microtiter plates E-Plate
® 96 (ACEA Biosciences Inc., Santa Clara, CA, USA, 2801035). After 24 h, effector cells were added according to indicated effector to target ratio. Therapeutic antibodies (AMs) were used at 10 ng/mL. Plates were incubated under 37 °C, 95% humidity, and 5% CO
2, and impedance was assessed every 15 min for 24 h.
2.13. Three-Dimensional Luciferase-Based Cytotoxicity Assay (LCA)
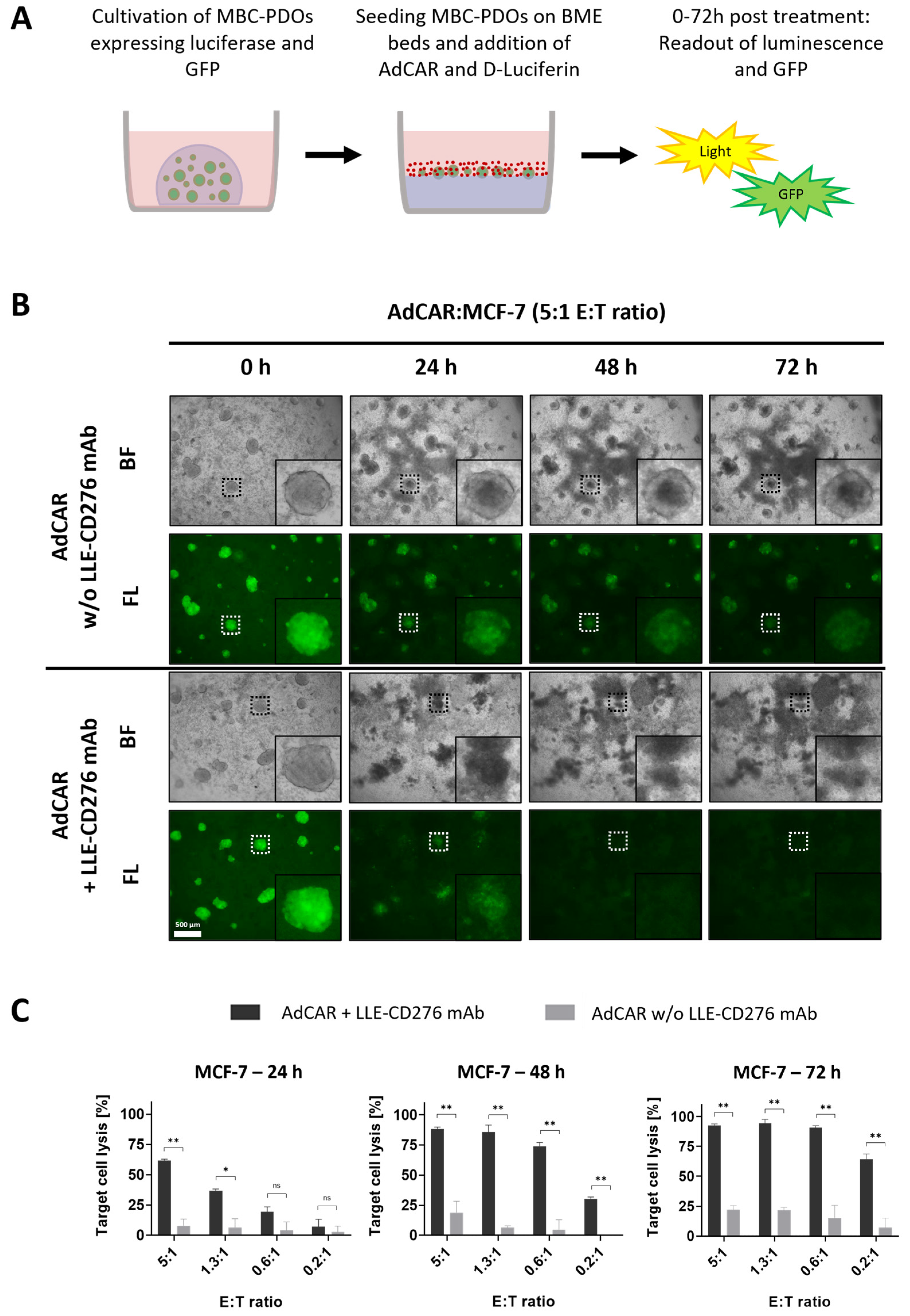
For the AdCAR-T cell treatment of MBC-PDOs and MCF-7, cells were cultured 7 to 21 days as described in
Section 2.7. The day before treatment, MBC-PDOs were recovered from the BME droplets by incubating the droplets in 1 mg/mL Dispase (Sigma-Aldrich, St. Louis, MO, USA, D4693) at 37 °C for 20 min. Droplet suspensions were diluted in 1% BSA and centrifuged at 250×
g for 10 min. The supernatant was removed, and organoid pellets were resuspended in the required amount of assay medium (BCM without Nicotinamide and Y-27632). Per well, 125 µL of organoid suspensions was seeded in 96-well plates (clear plates, 353072; white plates, 136102; both from Thermo Fisher Scientific, Waltham, MA, USA) coated with 40 µL BME-advDMEM (in a ratio of 50% BME and 50% AdvDMEM+++).
CAR-T cells were thawed the day before treatment and cultured in TexMacsTM medium lacking cytokines and including 10% FBS at 37 °C for 4 h. Cells were then diluted to a concentration of 1 × 106 cells/mL with TexMacsTM supplemented with IL-7 (10 ng/mL), IL-15 (5 ng/mL), penicillin (100 units/mL)/streptomycin (100 µg/mL), and cultured at 37 °C until the following day.
The following day, MBC-PDO cells were counted by the addition of TrypLE to one well per line. Organoids dispersed into single cells at 37 °C for 10 min and were pelleted and resuspended in 100 µL of AdvDMEM+++. MBC-PDO and CAR-T cells were counted using the Bio-Rad TC20™ Automated Cell Counter (Bio-Rad Laboratories, Inc., Hercules, CA, USA) according to manufacturer’s protocol.
Diluted in assay medium, 25 µL of CAR-T cells (various E:T ratios), 25 µL of biotinylated antibodies (80 ng/mL), and 25 µL D-Luciferin (2 ng/µL) were added to the wells, reaching a total volume of 200 µL. Wells of clear plates received assay medium instead of D-Luciferin; also, control wells received assay medium as a replacement for CAR-T cells and/or biotinylated antibodies. Readouts were performed after 24 h, 48 h, and 72 h by measuring the luminescence (white plates) with a Varioskan LUX (Thermo Fisher Scientific, Waltham, MA, USA) and capturing brightfield and fluorescence images (clear plates) of MCF-7 organoids and MBC-PDOs. Assays were carried out in multiple technical replicates, and results were normalized to untreated controls.
4. Discussion
Metastatic BC is associated with various challenges including cancer heterogeneity and treatment-resistant cells [
4]. As metastatic biopsies are relatively rare, most therapy decisions depend on the characteristics of primary tumors. However, primary tumors may not represent the heterogeneous features of metastatic tumors. Therefore, therapy decisions based solely on the characteristics of primary tumors can lead to a poor outcome [
6,
7].
BC exhibits significant heterogeneity and can be clinically categorized into various subtypes based on the presence or absence of hormone receptors (HRs) and the status of HER2 [
3]. Triple-negative BC, for one, which is defined by the absence of both HRs and HER2, is associated with a poorer prognosis, as endocrine therapy and HER2-targeted therapy are off the table [
3]. In this case, alternative target antigens, expressed on the tumor cell surface, need to be explored and clinically evaluated. For instance, CD276 has shown to be expressed in numerous solid tumors including BC [
26,
27]. In the present study, we provide a comprehensive analysis of target antigen expression patterns in BC cell lines and MBC-PDOs, demonstrating inter-individual heterogeneity. These results clearly demonstrate the need for patient-individualized treatment approaches.
The application of genetically engineered T cell treatments, which integrate a chimeric antigen receptor (CAR), has demonstrated impressive clinical responses in patients with hematological malignancies [
18]. Thus far, multiple obstacles have restricted successful translation of CAR-T therapies in solid tumors. Guiding CAR-T cells to reach and infiltrate the tumor poses a significant challenge, which is furthermore magnified by the immunosuppressive conditions found in the tumor microenvironment [
18]. Additionally, tumor cells have the capability to reduce antigen expression under the selective pressure exerted by CAR-T cells. Promiscuous antigen expression between tumor and physiological tissues can result in on-target off-tumor effects and associated life-threatening toxicities [
18]. This is particularly highlighted in the case study conducted by Morgan et al., where the application of HER2-targeted CAR-T cells triggered a cascading systemic inflammation, ultimately resulting in multi-organ failure that was attributed to CAR-T activation by a low-level expression of HER2 on lung epithelial cells [
28]. This emphasizes the necessity for advancements in the emerging field of CAR-T cell-based therapy in BC, where progress depends on discovering suitable TAAs and mechanisms for stringent control of CAR-T activity. This is especially pronounced in the context of triple-negative BC.
To date, numerous novel CAR-based target antigens have been evaluated against BC. The majority of these studies have been conducted in preclinical trials. Ongoing clinical trials with BC patients include the targeting of HER2, GD2, and EpCAM amongst others [
19]. In addition to targeting tumor cells, efforts have been directed toward the elimination of cells residing within the extracellular matrix [
29]. Combinatorial strategies with CAR-T cells and conventional BC therapy may lead to better efficacy, especially in terms of overcoming the suppressive tumor microenvironment. These approaches have yet to be assessed in clinical trials.
One elegant way to improve safety and flexibility of CAR-T cells in comparison to conventional CAR-T cell designs is to split antigen recognition from CAR-T cell activation. By introducing “adapter” molecules that bridge between TAA and CAR, this approach allows maximal control of CAR-T cell activity. Pioneered by the expression of an Fcγ receptor (CD16) [
30] or CD16-derived CAR construct [
31] in T cells to enable antibody-dependent cellular cytotoxicity (ADCC), multiple groups have re-invented the concept of “adapter”-mediated CAR-T activation [
32,
33,
34,
35]. We have recently reported on the development of the AdCAR platform [
23]. The AdCAR is directed against biotin in the context of a specific linker structure, referred to as linker–label–epitope (LLE). We did not see any interference with serum or protein-bound biotin. The LLE-tag can be chemically conjugated on any kind of binding molecule (e.g., mAbs, mAb fragments, natural or synthetic ligands), allowing highly flexible and convenient AM generation. This flexibility in AM generation provides an advantage over AMs that incorporate recombinant tags [
32,
33,
35], facilitating to build on clinically available mAbs. Moreover, we use the physiologically available vitamin biotin as a label. In contrast to other approaches using, for example, FITC, we expect less immunogenicity. Inherent to the design of all “adapter”-CAR systems is the beneficial safety profile, rendering these approaches a perfect fit for promiscuously expressed TAA. Moreover, AdCAR-T allows highly flexible and multiple targeting to prevent antigen escape and enable individualized targeting the regiment’s platform [
23]. First “adapter”-CAR systems have already entered the clinic (NCT04230265), targeting CD123 in adult AML, demonstrating complete remission in 2 out of 3 patients and underscoring the feasibility of “adapter”-CAR approaches [
36]. To explore this concept, we tested AdCAR-T cells with a variety of adapter molecules that target different antigens on MCF-7 and triple-negative MDA-MB-468 in 2D cultures (
Figure 1C–F). Most of the targets used in this study harbor clinical relevance [
19]. We observed a positive correlation between antigen expression and target cell lysis.
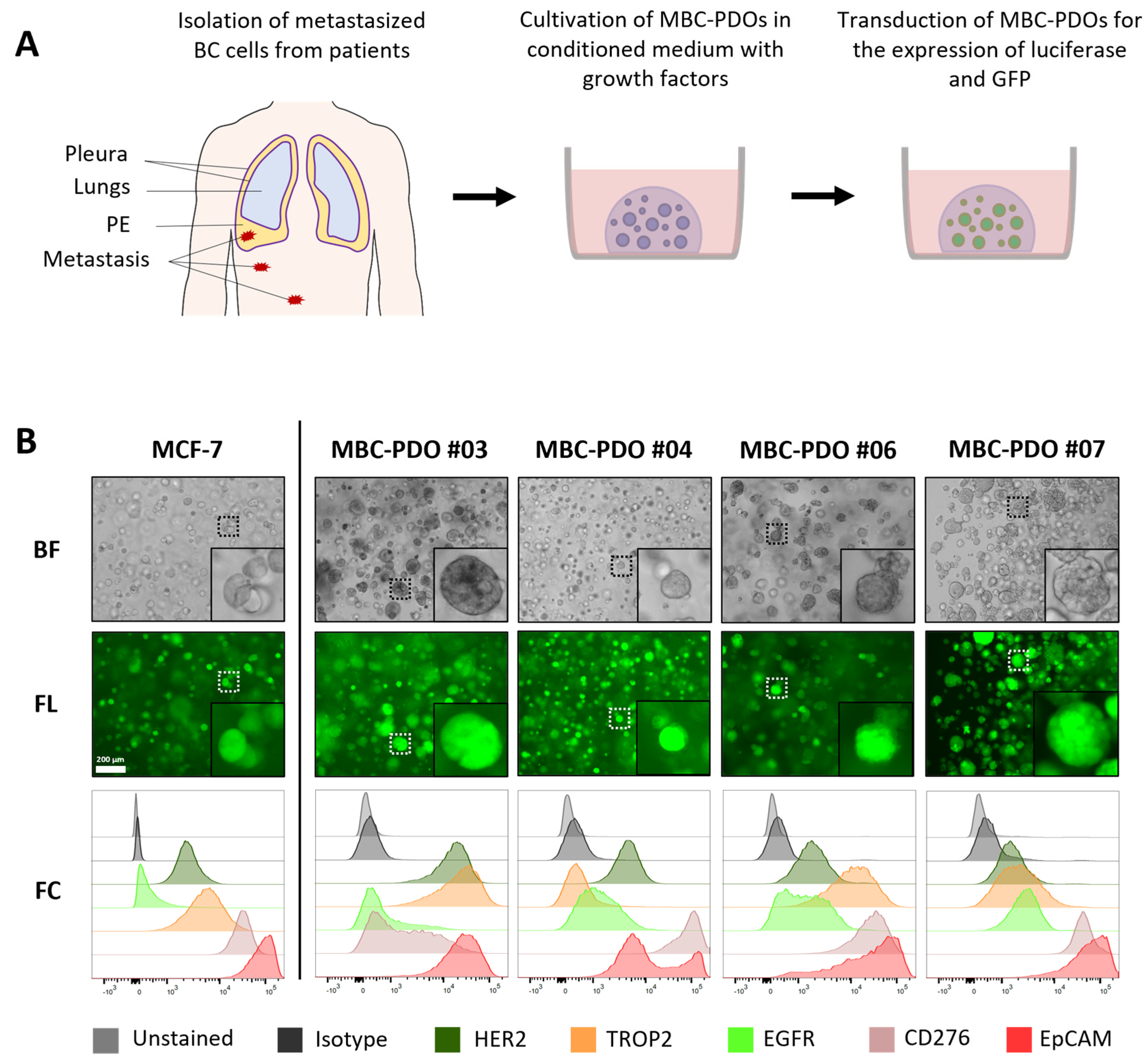
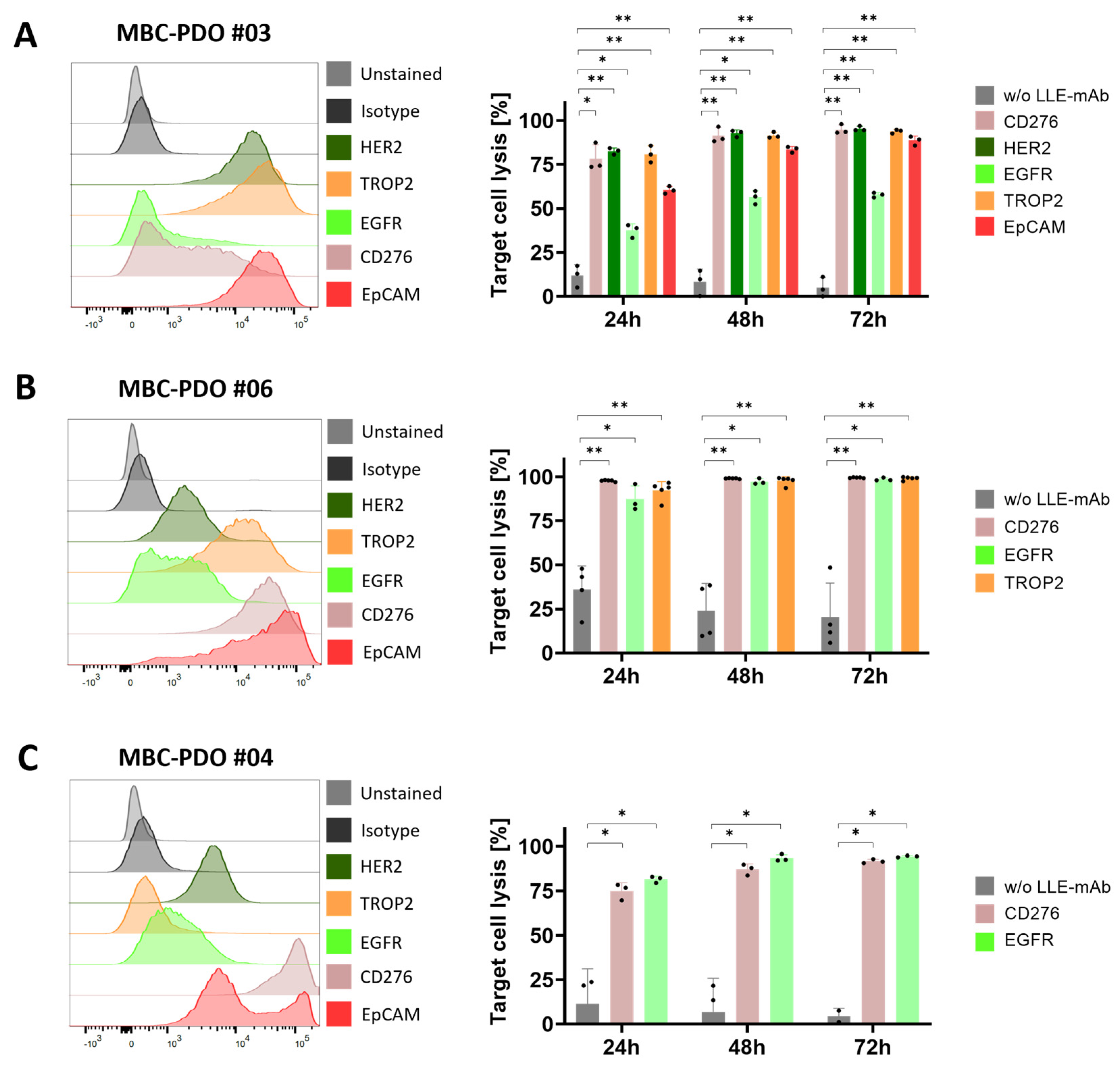
To demonstrate the feasibility of patient-individualized targeting and co-clinical functional validation, we utilized our MBC-PDOs’ platform [
14]. As shown before, MBC-PDOs preserve both receptor statuses and hotspot mutations across numerous passages. Therefore, MBC-PDOs provide a reliable in vitro platform for pre-clinical assessment of therapeutic effectiveness [
14]. For the first time, we applied this platform for systematic testing in the context of immunotherapy, particularly CAR-T cells. In accordance with individual antigen expression profiles assessed by flow cytometry (
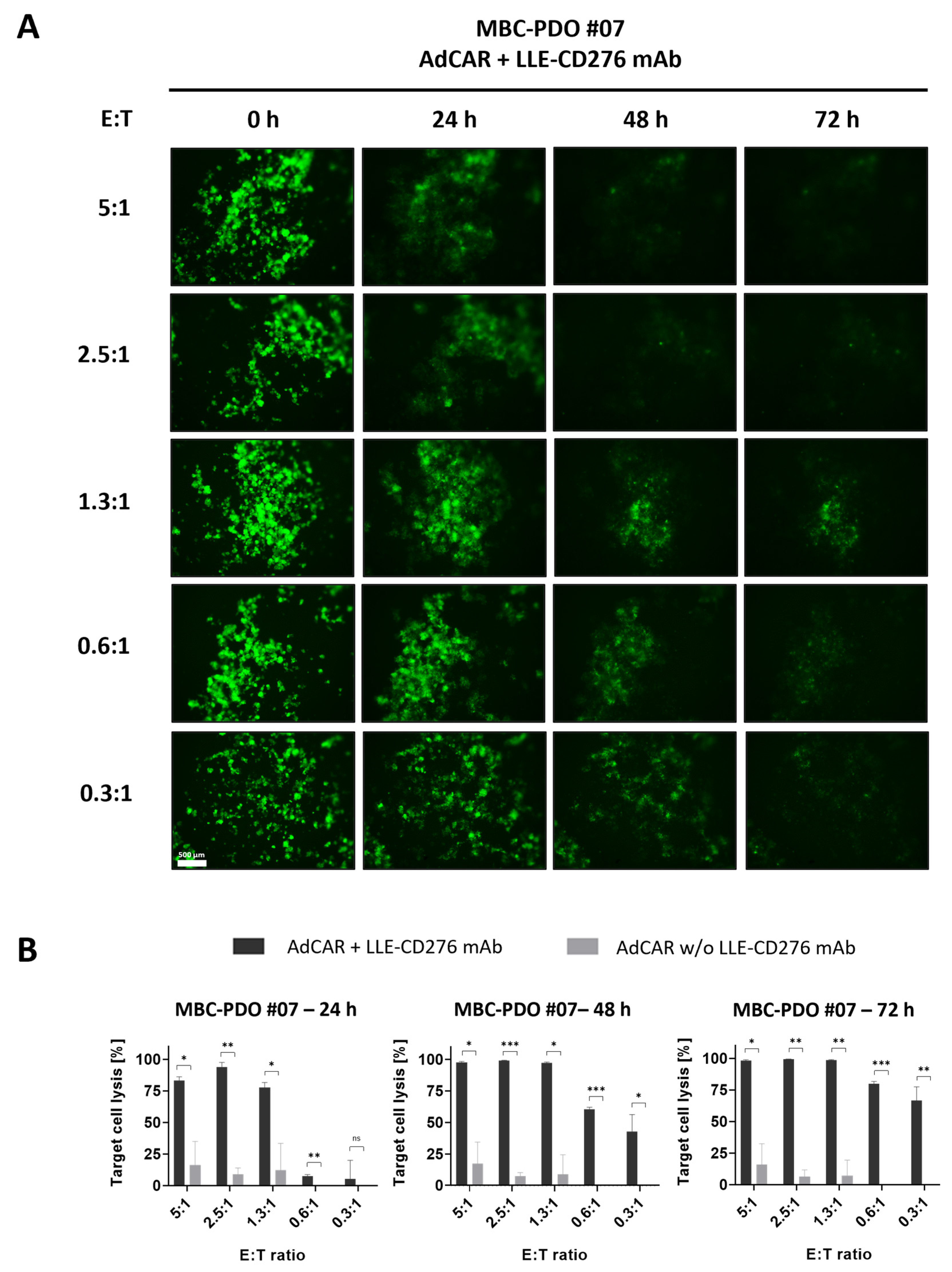
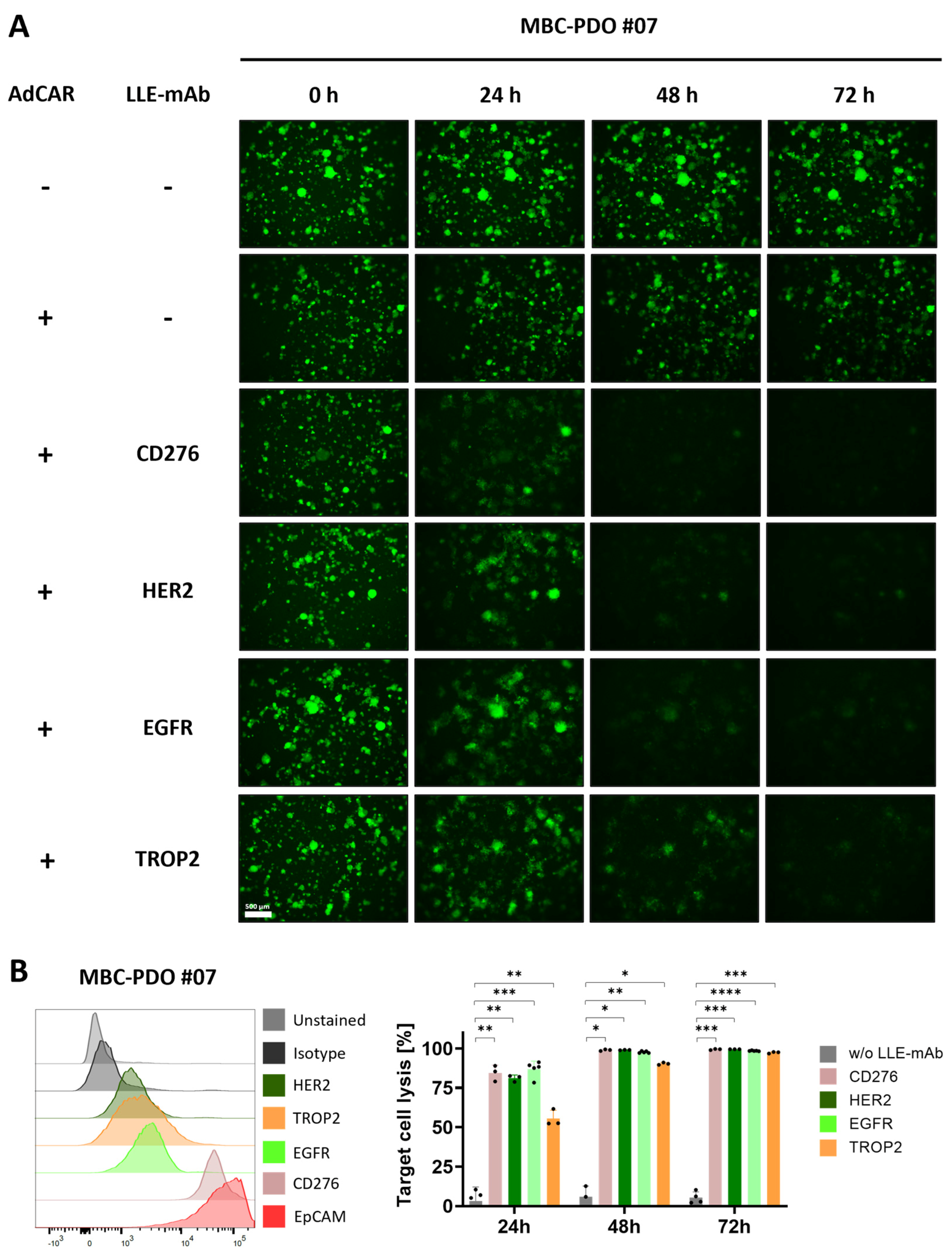
Figure 2B), AdCAR-T in combination with specific adapter molecules delivered potent lysis of MBC-PDOs. Specific lyses strongly correlated with antigen expression. For instance, MBC-PDO #03 demonstrated minor levels of EGFR and high levels of CD276, HER2, TROP2, and EpCAM expression (
Figure 6A). As a result, AdCAR-mediated cytolysis applying LLE-EGFR mAbs was low (38–58%), while the treatments with LLE-CD276, LLE-HER2, LLE-TROP2, and LLE-EpCAM led to complete target cell lysis. Future studies can include combinatorial approaches with multiple targets, as this was demonstrated to be superior compared to monotargeting [
24]. Whether targeting specific TAAs results in enhanced target cell lysis can be investigated in further studies While our approach is clearly functional and shows a correlation between antigen expression and target cell lysis in 2D culture (
Figure 1E,F), further investigation is needed within the 3D setting. Here, antigen staining alone does not seem to provide a clear prediction of AdCAR activity.
We, however, clearly demonstrate the feasibility of individually analyzing target antigen expression profiles and functionally validating patient-derived organoids’ efficacy of specific targeting. We further underscore the potential of AdCAR-T as an ideal tool for precision immunotherapy allowing individual selection target antigens, streamlining complex manufacturing and safety assessment of engineered cellular therapeutics. By building clinically tested and approved antibodies, this approach will facilitate clinical translation and accessibility of targeted cellular therapies to a very heterogeneous patient cohort.
Our MBC-PDO-based in vitro platform in combination with AdCAR-T cells pave the way for precision immunotherapy in solid tumor malignancies and can further be utilized in the assessment of existing and novel therapeutic approaches.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}