Simple Summary
Glucose metabolism was the first metabolic pathway to be discovered and serves a central role across all life by generating energy. Cancer cells hijack glycolysis to promote growth and metastasis. Understanding the molecular basis of glycolytic rewiring in cancers could illuminate new treatment approaches.
Abstract
Glycolysis is the central metabolic pathway across all kingdoms of life. Intensive research efforts have been devoted to understanding the tightly orchestrated processes of converting glucose into energy in health and disease. Our review highlights the advances in knowledge of how metabolic and gene networks are integrated through the precise spatiotemporal compartmentalization of rate-limiting enzymes. We provide an overview of technically innovative approaches that have been applied to study phosphofructokinase-1 (PFK1), which represents the fate-determining step of oxidative glucose metabolism. Specifically, we discuss fast-acting chemical biology and optogenetic tools that have delineated new links between metabolite fluxes and transcriptional reprogramming, which operate together to enact tissue-specific processes. Finally, we discuss how recent paradigm-shifting insights into the fundamental basis of glycolytic regulatory control have shed light on the mechanisms of tumorigenesis and could provide insight into new therapeutic vulnerabilities in cancer.
1. Introduction
Glycolysis, originally known as the Embden–Meyerhof–Parnas pathway, is a fundamental metabolic process across all life. The net gain of glycolysis generates two ATPs and pyruvates, which subsequently leads to 36 ATPs from oxidative phosphorylation (Figure 1). Intermediates throughout the pathway also generate precursors for serine (one carbon metabolism), sugars (protein glycosylation), and nucleotide synthesis (pentose phosphate pathway) []. Glucose enters the cell through transporters at the plasma membrane and becomes rapidly phosphorylated to trap glucose inside []. There are 10 core enzymes in the glycolytic pathway that are found throughout the cytoplasm []. A wide range of cellular mechanisms have evolved in order to regulate glycolytic flux. For instance, glycolytic enzymes can be controlled on the level of protein expression, post-translational modification codes, and through precise spatiotemporal compartmentation []. The present review seeks to provide an overview of recent discoveries in this field of research. As a lens for discussing these emerging concepts, we focus on phosphofructokinase-1 (PFK1), the gatekeeper of glycolysis []. We highlight the emerging methodologies that have uncovered novel spatiotemporal control mechanisms such as optogenetics, a genetic approach that uses light to noninvasively control proteins rapidly and reversibly with precise spatial resolution [,,]. Moreover, we discuss the implications of PFK1 that becomes dysregulated in cancer and how this new breadth of knowledge could offer vulnerabilities to be exploited for therapeutics.
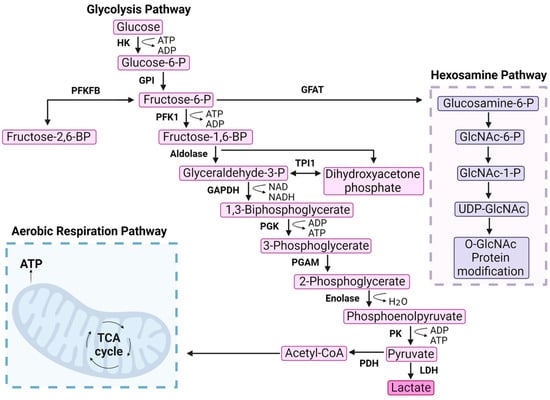
Figure 1.
Phosphofructokinase-1 is the gatekeeper of glycolysis. Glycolysis is a multistep pathway that converts glucose into usable energy for the cell. Glucose enters the cell through transporters and is phosphorylated by HK. The first committed irreversible step of glycolysis is catalyzed by PFK1 that phosphorylates fructose-6-phosphate (F6P) to generate fructose-1,6-bisphosphate (FBP). Several side products are produced from intermediates throughout the pathway, as highlighted by F6P entry into the hexosamine pathway via glutamine fructose-6-phosphate amidotransferase (GFAT). F6P can also be converted into fructose-2,6-bisphosphate. Aerobic respiration is included on the bottom left as the entry point of acetyl-CoA. Hexokinase (HK), phosphoglucoisomerase (GPI), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB), phosphofructokinase-1 (PFK1), aldolase, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK), phosphoglycerate mutase (PGAM), enolase, pyruvate kinase (PK), lactate dehydrogenase (LDH), pyruvate dehydrogenase (PDH), glutamine fructose-6-phosphate amidotransferase (GFAT), triosephosphate isomerase 1 (TPI1).
2. PFK Protein Structure and Composition
2.1. PFK1 Isoforms
PFK1 is localized in the cytosol and consists of three domains with approximately 780 amino acids (85 kDa) (Figure 2). PFK1 catalyzes an irreversible reaction that commits glucose to glycolytic breakdown by converting fructose-6-phosphate (F6P) into fructose-1,6-bisphosphate (FBP) []. Vertebrates have three isoforms of PFK1 that share a 95% sequence homology and were named based on the tissue of their discovery, including PFK-M (muscle), PFK-L (liver), and PFK-P (platelet) []. Most human tissues have all three isoforms, albeit with different levels of expression. PFK-P is the most highly expressed form across all tissue types []. Distinct enzymatic regulation has been reported for each PFK1 isoform, despite their similar amino acid composition.
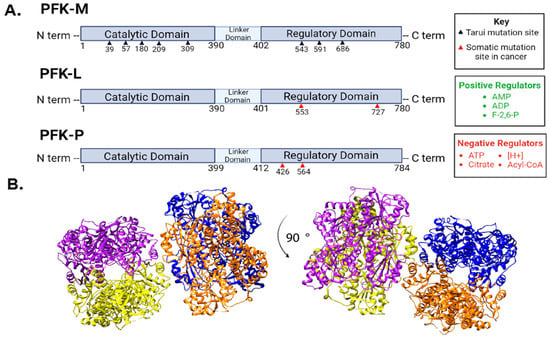
Figure 2.
Phosphofructokinase-1 isoforms and regulation. (A) There are three PFK1 isoforms that each have three distinct domains. PFK-M and PFK-L are 780 amino acids long, while PFK-P is 784 amino acids long. PFK-M is the only isoform to contain mutation sites involved in Tarui disease, denoted by black triangles below PFK-M. Red arrows denote reported cancer-related mutations in PFK-L and PFK-P discussed in the text. All three isoforms respond to the listed regulators, although some isoforms are more sensitive to certain regulators than others. [H+] indicates an increase in protons or acidity as a PFK1 negative regulator. Fructose-2,6-bisphosphate (F-2,6-BP). (B) Representative images of PFK1 structure from different angles (90-degree rotation) taken from protein database entry 4XYK with subunits color-coded using UCSF Chimera Software. Each of the four PFK1subunits are depicted in a different color (blue, orange, yellow, and purple).
2.2. PFK1 Expression, Activity and Genetic Mutations
Metabolic reprogramming is a classic hallmark of cancer [,]. Most notably, cancerous cells exhibit an increased rate of glycolysis independent of the amount of oxygen present. This phenomenon, known as aerobic glycolysis or the Warburg effect, is thought to create a metabolic environment that supports high levels of cell proliferation through the accumulation of nutrients and free energy []. It is not surprising that PFK1 expression is also increased in many cancers, seeing as it is one of the key enzymes that determines glycolytic rate. It has been previously shown that increasing PFK1 expression promotes aerobic glycolysis, directly connecting PFK1 expression with the metabolic phenotype seen in cancer []. PFK1 expression increases in breast cancer, lung cancer, brain cancer, bladder cancer, and colon cancer [,,,,]. This increase in PFK1 expression is not observed across all three isoforms equally, with L or P being the predominant isoforms in cancer []. The reason for this biased isoform expression in cancer remains unknown. Overexpression of TAp73 protein, which is an isoform of the tumor suppressor p73, is often overexpressed in tumors and increases the expression of PFK-L to promote glycolysis []. Additionally, the activation of the hypoxia-inducible factor pathway, which commonly promotes tumor growth, has been tied to increased PFK1 activity through allosteric activation, typically by fructose-2,6-bisphosphate (F-2,6-BP) [,,,].
Genetic mutations that alter glycolytic activity have been identified in PFK-P and PFK-L in several types of cancers [,]. The expression of PFK-P cancer mutants N426S and D564N in rat-derived breast cancer cells demonstrate an increase in PFK1 activity and lactic acid production, which is suggestive of aerobic glycolysis (Table 1) []. PFK-L cancer mutation in aspartate (D553N) correlates with decreased glycolysis in breast cancer, suggesting an intentional metabolic redirection to the pentose phosphate pathway []. Additionally, a K727A mutation of PFK-L led to constitutive activity that increased glycolysis and invasiveness in ovarian cancer (Table 1) []. In contrast, genetic mutations in PFK-M were reported in the 1960s to cause glycogen storage disease type VII, known as Tarui disease. Tarui disease is a rare autosomal recessive disorder that is characterized by PFK-M deficiency. A lack of the PFK1 enzyme results in an inability to break down glucose and leads to skeletal muscle deterioration and indirect kidney damage []. Together, the altered functions of glucose metabolism that result from genetic mutations in PFK1 highlight the fundamental importance of maintaining a tight regulation of PFK1 activities.

Table 1.
Phosphofructokinase-1 mutations in cancer impact enzymatic activity. The mutations displayed are not comprehensive but reflect those discussed in the text.
2.3. PFKFB Isoforms
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB) plays an essential role in coordinating glycolytic and gluconeogenic fluctuations in metabolism []. PFKFB was identified after the initial discovery of F-2,6-BP as an allosteric regulator of PFK1 [,]. There are four isoforms of PFKFB (PFKFB1, PFKFB2, PFKFB3, and PFKFB4) that serve as bifunctional homodimers responsible for catalyzing the creation and degradation of F-2,6-BP (Figure 1). PFKFB proteins have two domains that act as independent enzymes: the N terminal domain serves as a kinase, known as phosphofructokinase-2 (PFK2), while the C terminal domain serves as a phosphatase named fructo-2,6-bisphosphatase (FBPase2) [,]. The two differing domain activities are regulated primarily by phosphorylation, where the addition of a phosphate simultaneously inhibits kinase activity and promotes phosphatase activity, while dephosphorylation has the opposite effect []. PFKFB isoforms have some degree of tissue specificity: PFKFB1 is highly expressed in skeletal and cardiac muscle as well as the liver while PFKFB2 is primarily found in cardiac muscle. PFKFB3 is expressed in most organs, while PFKFB4 is mainly expressed in the testes [,,]. PFKFB isoforms only have partial tissue specificity and can still be found to be co-expressed in various tissues []. PFKFB isoforms are also differentiated by their kinase to phosphatase activity ratios, where certain isoforms more often catalyze one reaction over the other [,].
2.4. PFKFB Expression and Activity
PFKFB is an essential determinant in the regulation of carbohydrate metabolism by controlling the concentration of F-2,6-BP in cells, which is the most potent activator of PFK1 activity. Therefore, its altered expression in certain cancers can lead to aberrant glucose metabolism. Certain cancers exhibit a biased isoform expression of PFKFB similarly to PFK1. PFKFB1 has not been reported to have obvious anomalous expression in cancer [,,]. Meanwhile, PFKFB2 has been shown to be overexpressed in pancreas, lung, and prostate cancer [,,]. PFKFB2 has also been reported to have an opposite expression pattern in colorectal cancer, where decreased PFKFB2 was correlated with poor prognosis in patients []. PFKFB3 and PFKFB4 are the most overexpressed and active isoforms in a plethora of cancers; these two isoforms are responsible for the common upregulation of F-2,6-BP seen in cancer cells [,,,,,]. PFKFB3 has the highest kinase to bisphosphatase activity ratio among all the isoforms, producing the most F-2,6-BP and triggering increased glycolytic flux [,,]. Alternatively, PFKFB4 has been suggested to indirectly shunt glucose into the pentose phosphate pathway, which provides the cell with a means to combat reactive oxygen species (ROS) [,]. The biased expression of both PFKFB3 and PFKFB4 could allow for metabolic fine tuning that accommodates rapid growth during tumorigenesis.
3. PFK1 Regulation
3.1. Metabolites
PFK1 plays a unique role in glycolysis on multiple levels. First, the conversion of F6P to FBP represents the slowest enzymatic step in the glycolytic pathway. On the atomic level, PFK1 has two states of the quaternary structure known as the low-activity T state and the high-activity R state []. An inactive dimeric PFK1 has also been reported []. PFK1 is allosterically regulated by metabolites []. ATP, citrate, and phosphoenolpyruvate (PEP) stabilize the T state to decrease catalytic activity while AMP and FBP stabilize the R state to increase PFK1 activity (Figure 2). On a cellular level, this tuning of PFK1 operates as a way to modulate glycolytic flow based on the metabolic demands of a specific cell state. For instance, when energy levels are sufficient, high levels of ATP, citrate, and PEP are produced from the TCA cycle and glycolysis, respectively. Once energy levels drop, increased levels of AMP trigger PFK1 to become more active so as to provide glucose and energy from glycolysis []. In the early 1980s, F-2,6-BP was identified as a novel allosteric regulator of PFK1 []. Now known as PFK1′s most potent activator, F-2,6-BP increases PFK1 activity while relieving ATP inhibition, which increases glucose uptake and metabolic flux [,]. It is interesting to note that the muscle isoform, PFK-M, has the highest binding affinities for both F6P (K0.5F6P 147 µM) and ATP (K0.5ATP 152 µM) while PFK-P has the lowest binding affinities (K0.5ATP 276 µM and K0.5F6P 1333 µM). In contrast, PFK-L demonstrates high ATP binding affinity (K0.5ATP 160 µM) but low F6P binding affinity (K0.5F6P 1360 µM) [,]. These in vitro biochemical measurements highlight the distinct properties of each PFK1 isoform and suggest that the expression of each PFK1 paralog may contribute to fulfilling the unique metabolic demands across differing cell types and tissues.
PFK1 expression has been tied to cancer in numerous ways, the most obvious being to promote glycolysis and cancerous cell growth. One possibility for this discrepancy in isoform expression may be due to isoforms’ reactivity to allosteric regulation. Each isoform has different affinities for distinct allosteric regulators, with PFK-L and PFK-P being more responsive to allosteric activators than PFK-M []. It has also been suggested that PFK-M possesses a stable state of activity in order to maintain basal metabolic control compared with the other two isoforms. Another possibility may be due to differences in spatiotemporal regulation where isoform region-specific clustering into cellular domains could also serve to increase glycolysis. Considering that each isoform has different sensitivity levels to activity triggers, cancer cells may be preferentially expressing isoforms that respond the most efficiently to unique metabolic cues within the tumor microenvironment.
3.2. Post-Translational Modifications
Post-translational modifications (PTMs) serve to both regulate PFK1 activity and dictate localization in the cell, simultaneously in some cases. In 1975, it was first reported that PFK1 is phosphorylated in liver extracts []. Since this discovery, phosphorylation has been shown to promote the localization of PFK1 onto actin structures, which in turn increases enzyme activity []. Additionally, Akt phosphorylation at residue S386 has been shown to stabilize protein levels by blocking subsequent ubiquitination and proteasomal degradation []. PFK1 acetylation also drives the formation of multienzyme complexes that contribute to glucose metabolism (Table 2) [].
Table 2.
Summary of phosphofructokinase-1 post-translational modifications (PTM) and their effect on enzyme activity.
Several PFK1 PTMs that occur in physiologic conditions are hijacked to promote cancer cell metabolism. In ovarian cancer, S-nitrosylation has also been reported to promote PFK1 activity by creating resistance to negative feedback regulators []. Hypoxia leads to PFK1 serine 529 modification with O-GlcNAcylation, which inhibits enzymatic activity and redirects glucose flux into the pentose phosphate pathway to promote growth in multiple solid tumor cancer cell lines, including breast, prostate, liver, colon, and cervical cells (Table 2) []. Blocking the O-GlcNAcylation of PFK1 reduced cancer cell proliferation in vitro and decreased tumorigenesis in vivo []. In these cases, cancer cell growth was enabled by PFK1 inhibition by shunting metabolites into the pentose phosphate pathway to generate nucleic acids for rapid proliferation while also contributing to the prevention of oxidative stress in cancer microenvironments [,] (Figure 1).
A biased expression of PFK1 may allow the cell to have better control over PFK1 activity and cellular growth []. A recent report by Rossi et al. highlighted that interactions between PFK-P and phosphoglycerate dehydrogenase (PHGDH) were critical for regulating the delicate balance between glycolysis and the sialic acid pathway. By inhibiting PFK1 activity, its substrate F6P was more readily available to be shunted into the hexosamine pathway, which allowed for cellular glycosylation that aided in metastasis. Once the cancerous cells mobilized, PFK1 activity resumed to provide nutrients and energy to allow the cell to grow. By controlling PFK1 activity, cancer cells can better adapt to both mobilization and growth in the body, two aspects that are crucial to the spread of cancer.
4. PFK1 Spatiotemporal Regulation
The biochemical regulation of glycolytic enzymes has been the focus of intensive research investigations. Precise biochemical reactions of PFK1 are determined by subcellular localization in the cytosol, along the plasma membrane, and even at the mitochondrial interface []. Localized expression of PFK1 and FBP has also been seen during certain stages of embryogenesis. In murine embryos, localization during chorioallantoic branching was correlated with a decrease in glycolysis while localization during organogenesis was tied to an increase in glycolysis at the site of higher expression [,,]. This research suggests that spatiotemporal regulation is a requirement for precise metabolic control during crucial moments of growth and maturation. Localized expression of glycolytic enzymes has also been seen in other species during development, such as in avian and amphibian embryos [,]. Recent research has also begun to show that subcellular localization of both PFK1 and PFKFB in cancer can serve as a predictor for cancer recurrence []. These emerging studies highlight the need for understanding spatiotemporal regulation and its significance in development and disease. Such an architectural remodeling of PFK1 throughout the cell is rapid, dynamic, and highly reversible, which suggests that such mechanisms may have evolved as additional approaches to tune the production of metabolites depending on the tissue-specific demands.
4.1. Plasma Membrane
The plasma membrane is a hotspot of metabolic activity being the interface between intracellular and extracellular matrices. Glucose enters the cell at the plasma membrane through glucose transporters []. As the entry point for glucose into the cell, activated PFK1 at the plasma membrane ensures efficient and rapid energy production. Caveolae are plasma membrane invaginations that are driven by the activity of caveolin proteins []. Additionally, caveolin can also serve scaffolding functions for metabolic pathways such as glycolysis. Indeed, PFK-M is known to associate with multiple isoforms of caveolin, and PFK-M recruitment to caveolae depends on the specific Cav-3 isoform expression level. It has been reported that human mutations in Cav-3 reduce PFK-M expression and membrane recruitment in skeletal muscle tissues [,]. High extracellular glucose concentrations also promote the recruitment and targeting of PFK-M into caveolin-enriched caveolae domains, which suggests that PFK-M is recruited to promote glycolysis at the plasma membrane surface []. In line with this, PFK1 activators stabilize Cav-2/PFK-M complexes [].
4.2. Cytoskeleton
Cytoskeletal dynamics enable cellular adhesion, migration, proliferation, and homeostatic maintenance. Beyond these classic roles, cytoskeletal remodeling also coordinates cellular metabolism and metabolic enzyme activities. Actin filaments (F-actin) are linear polymers of globular actin []. In the case of PFK1, it has been reported for over 30 years that PFK-M interacts with actin filaments to increase PFK1 enzymatic activities []. This interaction is amplified by PFK1 phosphorylation, which promotes its affinity for actin []. Additionally, actin binds at the adenosine triphosphate (ATP) activation site on PFK1, where it acts as an allosteric activity upregulator []. Global glycolytic stimulators such as insulin have also been shown to drive PFK-M associations with actin []. Recently, the PFK-P isoform has also been reported to associate with F-actin []. Decreases in the product of PFK1, FBP, coupled with the dissociation of PFK-P from actin further suggests that PFK-P interactions with actin increase activity, as similarly seen with PFK-M []. More specifically, F-actin bundling and colocalization with PFK-P have been correlated with increased glycolysis, which indicates that PFK-P may also sense and respond to cellular mechanical cues from the cell environment []. In contrast, PFK1 localization with the microtubule component tubulin was found to decrease enzyme activity []. This is due to the preferential binding of PFK1 dimers, which are an inactive form of PFK1 []. Similar to F-actin, PFK1 is associated with microtubule bundling and has even been shown to drive microtubule bundling through the formation of cross-bridges [,,]. While both F-actin and microtubules are essential cytoskeletal constituents of cellular architectures, these studies further highlight key functions in metabolic regulation through PFK1 and glycolysis.
4.3. Cytosolic Phase Condensates
Phase condensates are membrane-less structural assemblies of proteins that form upon changes in solubility states. A recent report systematically interrogating the localization of PFK1 in vivo was performed using a Caenorhabditis elegans model []. Dynamic remodeling of PFK1 occurred in response to hypoxia. Diffuse PFK1 localized within the cytosol during normoxic conditions and was rapidly redirected into phase-separated condensates during hypoxic energy stress conditions. The authors propose that the liquid-like properties and spheroid shapes of phase condensates enable rapid internal molecular reorganization to promote PFK1 signaling dynamics. PFK1 condensates were formed through a heterologous self-association cryptochrome 2 domain and also recruited the downstream glycolytic enzyme, aldolase. Notably, PFK1 condensates were not correlated with stress granules. PFK1 in Caenorhabditis elegans genetically maps to the conserved region of the F674 residue, which is critical for tetramer formation and PFK multivalent interactions []. Together, this study in a living organism showcases how the glycolytic PFK1 enzyme can dynamically form condensates in vivo. Whether PFK1 metabolic regulation via sub-compartments occurs across mammalian systems has yet to be examined.
4.4. Cytosolic Filaments
PFK-L polymerizes into filaments throughout the cytoplasm []. Transmission negative-stain electron microscopy revealed that filaments were an average of six tetramers long (~lengths of 65.4 nm), where tetramers are composed of two structurally distinct dimers. The unusual helical symmetry of PFK-L conformations is critical to enable the last subunit to be able to bind another subunit in a linear manner, or alternatively, to introduce a kink into the filament. These dynamic structural conformations occurred between the F6P binding pocket and the C-terminus. A chimera of the C-terminal regulatory domain of PFK-L fused to the catalytic region of PFK-P led to key insights into the isoform-selective properties driving filament formation. Indeed, this chimera delineated that the PFK-L C-terminus was responsible for determining filament assembly as it was sufficient to drive filament formation. The remaining questions involve how the enzymatic activity of PFK-L is changed upon entry into filaments and how PFK-L removal from PFK-P and PFK-M cytosolic complexes impacts glycolysis. Given that both F6P and citrate promote PFK-L filament assembly, it is tempting to speculate that this dynamic remodeling process may regulate glycolytic flux. On a larger scale, authors note that filament localization appeared as punctate in cells with sizes that could indicate a potential phase transition of PFK-L into P granules, stress granules, or aggresomes. Further investigations into this question could provide essential molecular insights into a major question in metabolism regarding how the precise location of enzyme activities directly impacts the levels of downstream metabolites.
4.5. Glucosome
The term metabolon was first termed by Paul Srere as a supramolecular complex of sequential metabolic enzymes and cellular structural elements []. A type of metabolon, the glucosome, is defined as a multienzyme assembly that is formed by metabolic enzymes during glucose metabolism. Glucosomes are spatiotemporally controlled subcellular domains that regulate glucose flux in the cell where both glycolytic and gluconeogenic enzymes have been found inside. The importance of this structure is further highlighted by the presence of specific enzymes that catalyze rate-limiting reactions [,]. PFK-L has been previously reported to be a driver of glucosome formation, specifically through acetyl modifications on lysine residues []. PFK-L-induced glucosomes also include downstream rate-limiting enzymes such as PKM2, FBPase, and PEPCK1 (Figure 3).
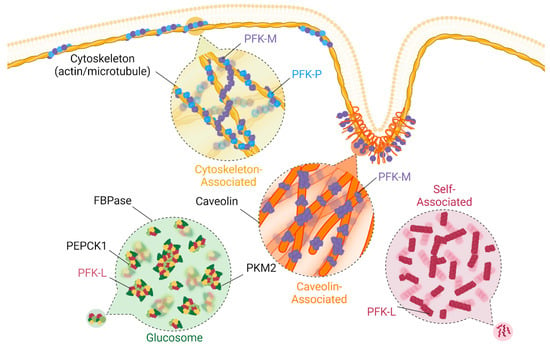
Figure 3.
Spatiotemporal regulation of phosphofructokinase-1. PFK1 can be spatiotemporally regulated in a variety of ways, which seem to vary based on isoform type. Isoform PFK-L can self-associate into filaments in the cytosol in response to both positive and negative regulators, as well as drive glucosome formation with a variety of other metabolic enzymes. PFK-M and PFK-P are known to associate with actin, with PFK-M creating bridges between different actin filaments and between microtubules. PFK-M can associate with caveolin as well.
The glucosome is multi-enzymatic and regulates enzymatic activities of multiple rate-limiting metabolic enzymes. While the exact function of the glucosome is currently under investigation, it has been suggested that assembly serves to quickly sequester enzymes from the cytosol and halt glucose metabolism. Given this, glucosome formation could downregulate metabolic pathways as membrane-less assemblies allow for quicker and more transient protein sequestration. Furthermore, the absence of membranes suggest that glucosome formation could provide a cost-effective mechanism to regulate cellular contents. Finally, compartmentalization of PFK1 along with other glycolytic enzymes into glucosomes could maximize metabolic rate, as having these enzymes in proximity shuttles the product of one reaction directly into the next reaction without the interference of product diffusion in the cytosol. Increasing protein proximity has been shown to increase positive cooperative effects []. While it was recently discovered that glucosomes are regulated by cell cycle-associated signaling pathways, this regulation is size-specific and only applies to smaller-sized glucosomes [,]. Thus, the full extent of underlying mechanisms that control the formation and dissociation of such glycolytic clusters still remains unknown [].
Although the glucosome remains to be a relatively new concept in the global understanding of cellular metabolic regulation, the discovery of this signaling hub provides insights into how rate-limiting enzymes are finely tuned on a rapid time scale. Future work aimed at untangling this regulatory pathway across cell types and tissues is warranted. Filling these gaps in knowledge could have broad implications for understanding cancer cell metabolism and for designing interventions that target the required mechanisms that execute these precise metabolic shifts in glycolysis.
4.6. Mechanosensation
Recent work underscored a fundamental mechanosensation mechanism through PFK1 and the regulation of downstream glycolytic activity. This key discovery was uncovered by the simple observation that F6P levels were elevated on soft substrate and coincided with reduced levels of downstream metabolites in the glycolytic pathway []. This led authors to explore whether protein levels of glycolytic enzymes contribute to such metabolite fluxes. Indeed, PFK1 levels were specifically reduced in conditions when F6P became elevated, while all other glycolytic enzymes remained unchanged. Importantly, authors further demonstrated that stiff substrates increased PFK1 interactions with actin bundles that segregated PFK1 from degradation enzyme TRIM21 and from the proteasomal ubiquitin machinery. This further highlights the importance of protein degradation pathways in the tuning metabolism, where previous studies have largely focused on protein expression. Furthermore, downregulation of several E3 ubiquitin ligases such as TRIM21 have been reported in cancer and could highlight how the loss of such proteins could offer an additional approach through which cancer increases glycolysis. While this report focused on human lung tumors, the stiffness of substrates represents a broad phenomenon across multiple types of cancers and could represent a more global view on mechanisms influencing glycolysis during tumorigenesis.
5. Future Perspectives
Metabolism changes as a cancer develops from a small, premalignant lesion to an aggressive primary tumor and then metastasizes. Overall, PFK1 activity is closely tied to cancer, particularly cancers of the female reproductive system [,,,,,,,,]. Additionally, the downregulation of PFK1 in cancer has also shown to be advantageous for creating more metabolite availability for other biosynthetic pathways [,]. Knowledge of how PFK1 is fundamentally regulated and how this regulation goes awry in cancers will provide insight into understanding context-dependent metabolic fluidity [,]. Both the up- and downregulation of PFK1 can promote cancer cell growth; insights on these alternate pathways and appreciating their context-dependent use will elucidate novel mechanisms and targets for cancer treatment.
For instance, optogenetics provide exquisite spatial resolution and fast reversibility to acutely and dynamically dissect a metabolic pathway action [,,]. Fluorescence single-cell microscopy has also been emphasized as a classic and indispensable tool capable of monitoring subcellular protein localization with great target specificity []. Overall, the enzymatic activity of PFK1 is tightly orchestrated across cell types and tissues through multiple spatiotemporal mechanisms. While many spatiotemporally controlled processes are dedicated to tuning PFK1 activity, how these regulatory principles contribute to metabolic stages and tumor types are still beginning to unfold. PFK1 is an irreversible, essential component of glycolysis, which suggests that interventions aimed at PFK1 could offer an important vulnerability for disrupting glycolytic-driven cancer cell metabolism. Furthermore, all three mammalian PFK1 isoforms generate FBP; however, the unique highly specific regulation of each isoform suggests that the moonlighting functions of PFK1 may also contribute to processes in cancer that have yet to be uncovered. As organisms became more complex during evolution, cells may have devised subcellular localization as a dynamic mechanism to provide tighter regulation on cellular processes. Thus, the cell may have evolved such approaches in order to compensate for the irreversibility of PFK1 enzymatic activity.
6. Conclusions
In conclusion, this review highlights the numerous ways that PFK1 is spatiotemporally regulated. We draw attention to the importance of this regulation and isoform discrimination for possible therapeutic targets in cancer. We also elucidate new methodologies for further studying and understanding PFK1 compartmentalization. Both PFK1 and PFKFB are tightly regulated in cells to control glycolytic flux, and aberrant enzyme regulation in cancer leads to the dysregulation of glycolysis. Both increases and decreases in glucose metabolism can promote cancer cell growth and proliferation, depending on cell context. Spatiotemporal regulation is also emerging as an important type of enzyme regulation that can have profound effects on cell metabolism and cell growth. While the connection between spatiotemporal regulation and aberrant metabolic regulation in cancer is not clear, investigating the relationship between the two topics can uncover novel regulatory mechanisms in cancer metabolism that can provide new therapeutic targets for clinical treatment.
Author Contributions
Conceptualization, M.C. and L.V.A.; methodology, M.C. and L.V.A.; writing—original draft preparation, M.C. and L.V.A.; writing—review and editing, M.C. and L.V.A.; funding acquisition, L.V.A. All authors have read and agreed to the published version of the manuscript.
Funding
Ono Pharmaceuticals, University of California Irvine Startup; Chao Research Cancer Center CRCC Startup Funds.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank all members of the Albrecht lab and researchers of cell metabolism that inspire and fuel the fields of cell biology and cancer research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef]
- Tanner, L.B.; Goglia, A.G.; Wei, M.H.; Sehgal, T.; Parsons, L.R.; Park, J.O.; White, E.; Toettcher, J.E.; Rabinowitz, J.D. Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst. 2018, 7, 49–62. [Google Scholar] [CrossRef]
- Voronkova, M.A.; Hansen, H.L.; Cooper, M.P.; Miller, J.; Sukumar, N.; Geldenhuys, W.J.; Robart, A.R.; Webb, B.A. Cancer-Associated Somatic Mutations in Human Phosphofructokinase-1 Reveal a Critical Electrostatic Interaction for Allosteric Regulation of Enzyme Activity. Biochem. J. 2023, 480, 1411–1427. [Google Scholar] [CrossRef]
- Guglielmi, G.; Falk, H.J.; Renzis, S.D. Optogenetic Control of Protein Function: From Intracellular Processes to Tissue Morphogenesis. Trends Cell Biol. 2016, 26, 864–874. [Google Scholar] [CrossRef]
- Karunarathne, W.K.A.; O’Neill, P.R.; Gautam, N. Subcellular Optogenetics—Controlling Signaling and Single-Cell Behavior. J. Cell Sci. 2015, 128, 15–25. [Google Scholar] [CrossRef]
- Toettcher, J.E.; Voigt, C.A.; Weiner, O.D.; Lim, W.A. The Promise of Optogenetics in Cell Biology: Interrogating Molecular Circuits in Space and Time. Nat. Methods 2011, 8, 35–38. [Google Scholar] [CrossRef]
- Fernandes, P.M.; Kinkead, J.; McNae, I.; Michels, P.A.M.; Walkinshaw, M.D. Biochemical and Transcript Level Differences between the Three Human Phosphofructokinases Show Optimisation of Each Isoform for Specific Metabolic Niches. Biochem. J. 2020, 477, 4425–4441. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer Metabolic Reprogramming: Importance, Main Features, and Potentials for Precise Targeted Anti-Cancer Therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Li, L.; Li, L.; Li, W.; Chen, T.; Zou, B.; Zhao, L.; Wang, H.; Wang, X.; Xu, L.; Liu, X.; et al. TAp73-Induced Phosphofructokinase-1 Transcription Promotes the Warburg Effect and Enhances Cell Proliferation. Nat. Commun. 2018, 9, 4683. [Google Scholar] [CrossRef]
- Lim, J.S.; Shi, Y.; Park, S.H.; Jeon, S.M.; Zhang, C.; Park, Y.-Y.; Liu, R.; Li, J.; Cho, W.-S.; Du, L.; et al. Mutual Regulation between Phosphofructokinase 1 Platelet Isoform and VEGF Promotes Glioblastoma Tumor Growth. Cell Death Dis. 2022, 13, 1002. [Google Scholar] [CrossRef]
- Zancan, P.; Sola-Penna, M.; Furtado, C.M.; Da Silva, D. Differential Expression of Phosphofructokinase-1 Isoforms Correlates with the Glycolytic Efficiency of Breast Cancer Cells. Mol. Genet. Metab. 2010, 100, 372–378. [Google Scholar] [CrossRef]
- Sun, C.; Xiong, D.; Yan, Y.; Geng, J.; Liu, M.; Yao, X. Genetic Alteration in Phosphofructokinase Family Promotes Growth of Muscle-Invasive Bladder Cancer. Int. J. Biol. Markers 2016, 31, 286–293. [Google Scholar] [CrossRef]
- Shen, J.; Jin, Z.; Lv, H.; Jin, K.; Jonas, K.; Zhu, C.; Chen, B. PFKP Is Highly Expressed in Lung Cancer and Regulates Glucose Metabolism. Cell Oncol. 2020, 43, 617–629. [Google Scholar] [CrossRef]
- Lu, T.-J.; Yang, Y.-F.; Cheng, C.-F.; Tu, Y.-T.; Chen, Y.-R.; Lee, M.-C.; Tsai, K.-W. Phosphofructokinase Platelet Overexpression Accelerated Colorectal Cancer Cell Growth and Motility. J. Cancer 2023, 14, 943–951. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The Role of Hypoxia-Inducible Factors in Tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef]
- Golinska, M.; Troy, H.; Chung, Y.-L.; McSheehy, P.M.; Mayr, M.; Yin, X.; Ly, L.; Williams, K.J.; Airley, R.E.; Harris, A.L.; et al. Adaptation to HIF-1 Deficiency by Upregulation of the AMP/ATP Ratio and Phosphofructokinase Activation in Hepatomas. BMC Cancer 2011, 11, 198. [Google Scholar] [CrossRef]
- Bartrons, R.; Caro, J. Hypoxia, Glucose Metabolism and the Warburg’s Effect. J. Bioenerg. Biomembr. 2007, 39, 223–229. [Google Scholar] [CrossRef]
- Mor, I.; Cheung, E.C.; Vousden, K.H. Control of Glycolysis through Regulation of PFK1: Old Friends and Recent Additions. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 211–216. [Google Scholar] [CrossRef]
- Webb, B.A.; Forouhar, F.; Szu, F.-E.; Seetharaman, J.; Tong, L.; Barber, D.L. Structures of Human Phosphofructokinase-1 and Atomic Basis of Cancer-Associated Mutations. Nature 2015, 523, 111–114. [Google Scholar] [CrossRef]
- Chen, S.; Wu, Y.; Gao, Y.; Wu, C.; Wang, Y.; Hou, C.; Ren, M.; Zhang, S.; Zhu, Q.; Zhang, J.; et al. Allosterically Inhibited PFKL via Prostaglandin E2 Withholds Glucose Metabolism and Ovarian Cancer Invasiveness. Cell Rep. 2023, 42, 113246. [Google Scholar] [CrossRef]
- Tarui, S.; Giichi, O.; Ikura, Y.; Tanaka, T.; Suda, M.; Nishikawa, M. Phosphofructokinase Deficiency in Skeletal Muscle. A New Type Glycogenosis. Biochem. Biophys. Res. Commun. 1965, 19, 517–523. [Google Scholar] [CrossRef]
- Okar, D.A.; Lange, A.J.; Manzano, À.; Navarro-Sabatè, A.; Riera, L.; Bartrons, R. PFK-2/FBPase-2: Maker and Breaker of the Essential Biofactor Fructose-2,6-Bisphosphate. Trends Biochem. Sci. 2001, 26, 30–35. [Google Scholar] [CrossRef]
- Van Schaftingen, E.; Hue, L.; Hers, H.G. Fructose 2,6-Bisphosphate, the Probably Structure of the Glucose- and Glucagon-Sensitive Stimulator of Phosphofructokinase. Biochem. J. 1980, 192, 897–901. [Google Scholar] [CrossRef]
- Pilkis, S.J.; Claus, T.H.; Kurland, I.J.; Lange, A.J. 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase: A Metabolic Signaling Enzyme. Annu. Rev. Biochem. 1995, 64, 799–835. [Google Scholar] [CrossRef]
- El-Maghrabi, M.R.; Pilkis, S.J. Rat Liver 6-Phosphofructo 2-Kinase/Fructose 2,6-Bisphosphatase: A Review of Relationships between the Two Activities of the Enzyme. J. Cell. Biochem. 1984, 26, 1–17. [Google Scholar] [CrossRef]
- Hu, K.-F.; Shu, C.-W.; Lee, C.-H.; Tseng, C.-J.; Chou, Y.-H.; Liu, P.-F. Comparative Clinical Significance and Biological Roles of PFKFB Family Members in Oral Squamous Cell Carcinoma. Cancer Cell Int. 2023, 23, 257. [Google Scholar] [CrossRef]
- Kotowski, K.; Rosik, J.; Machaj, F.; Supplitt, S.; Wiczew, D.; Jabłońska, K.; Wiechec, E.; Ghavami, S.; Dzięgiel, P. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers 2021, 13, 909. [Google Scholar] [CrossRef]
- Minchenko, O.; Opentanova, I.; Caro, J. Hypoxic Regulation of the 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase Gene Family (PFKFB-1–4) Expression in Vivo. FEBS Lett. 2003, 554, 264–270. [Google Scholar] [CrossRef]
- Yi, M.; Ban, Y.; Tan, Y.; Xiong, W.; Li, G.; Xiang, B. 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3 and 4: A Pair of Valves for Fine-Tuning of Glucose Metabolism in Human Cancer. Mol. Metab. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Ros, S.; Schulze, A. Balancing Glycolytic Flux: The Role of 6-Phosphofructo-2-Kinase/Fructose 2,6-Bisphosphatases in Cancer Metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef]
- Bartrons, R.; Simon-Molas, H.; Rodríguez-García, A.; Castaño, E.; Navarro-Sabaté, À.; Manzano, A.; Martinez-Outschoorn, U.E. Fructose 2,6-Bisphosphate in Cancer Cell Metabolism. Front. Oncol. 2018, 8, 331. [Google Scholar] [CrossRef]
- Ozcan, S.C.; Sarioglu, A.; Altunok, T.H.; Akkoc, A.; Guzel, S.; Guler, S.; Imbert-Fernandez, Y.; Muchut, R.J.; Iglesias, A.A.; Gurpinar, Y.; et al. PFKFB2 Regulates Glycolysis and Proliferation in Pancreatic Cancer Cells. Mol. Cell Biochem. 2020, 470, 115–129. [Google Scholar] [CrossRef]
- Sha, L.; Lv, Z.; Liu, Y.; Zhang, Y.; Sui, X.; Wang, T.; Zhang, H. Shikonin Inhibits the Warburg Effect, Cell Proliferation, Invasion and Migration by Downregulating PFKFB2 Expression in Lung Cancer. Mol. Med. Rep. 2021, 24, 560. [Google Scholar] [CrossRef]
- Moon, J.-S.; Jin, W.-J.; Kwak, J.-H.; Kim, H.-J.; Yun, M.-J.; KIM, J.-W.; Park, S.W.; Kim, K.-S. Androgen Stimulates Glycolysis for de Novo Lipid Synthesis by Increasing the Activities of Hexokinase 2 and 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase 2 in Prostate Cancer Cells. Biochem. J. 2010, 433, 225–233. [Google Scholar] [CrossRef]
- Liu, F.; Wei, X.; Chen, Z.; Chen, Y.; Hu, P.; Jin, Y. PFKFB2 Is a Favorable Prognostic Biomarker for Colorectal Cancer by Suppressing Metastasis and Tumor Glycolysis. J. Cancer Res. Clin. Oncol. 2023, 149, 10737–10752. [Google Scholar] [CrossRef]
- Da, Q.; Huang, L.; Huang, C.; Chen, Z.; Jiang, Z.; Huang, F.; Shen, T.; Sun, L.; Yan, Z.; Ye, X.; et al. Glycolytic Regulatory Enzyme PFKFB3 as a Prognostic and Tumor Microenvironment Biomarker in Human Cancers. Aging 2023, 15, 4533–4559. [Google Scholar] [CrossRef]
- Trojan, S.E.; Markiewicz, M.J.; Leśkiewicz, K.; Kocemba-Pilarczyk, K.A. The Influence of PFK-II Overexpression on Neuroblastoma Patients’ Survival May Be Dependent on the Particular Isoenzyme Expressed, PFKFB3 or PFKFB4. Cancer Cell Int. 2019, 19, 292. [Google Scholar] [CrossRef]
- Kessler, R.; Fleischer, M.; Springsguth, C.; Bigl, M.; Warnke, J.-P.; Eschrich, K. Prognostic Value of PFKFB3 to PFKFB4 mRNA Ratio in Patients with Primary Glioblastoma (IDH-Wildtype). J. Neuropathol. Exp. Neurol. 2019, 78, 865–870. [Google Scholar] [CrossRef]
- Sakakibara, R.; Kato, M.; Okamura, N.; Nakagawa, T.; Komada, Y.; Tominaga, N.; Shimojo, M.; Fukasawa, M. Characterization of a Human Placental Fructose-6-Phosphate, 2-Kinase/Fructose- 2,6—Bisphosphatase1. J. Biochem. 1997, 122, 122–128. [Google Scholar] [CrossRef]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in Cancer. Signal Transduct. Target. Ther. 2017, 2, 17044. [Google Scholar] [CrossRef]
- Minchenko, O.H.; Tsuchihara, K.; Minchenko, D.O.; Bikfalvi, A.; Esumi, H. Mechanisms of Regulation of PFKFB Expression in Pancreatic and Gastric Cancer Cells. World J. Gastroenterol. 2014, 20, 13705–13717. [Google Scholar] [CrossRef]
- Le Bras, G.; Auzat, I.; Garel, J.R. Tetramer-Dimer Equilibrium of Phosphofructokinase and Formation of Hybrid Tetramers. Biochemistry 1995, 34, 13203–13210. [Google Scholar] [CrossRef]
- Schöneberg, T.; Kloos, M.; Brüser, A.; Kirchberger, J.; Sträter, N. Structure and Allosteric Regulation of Eukaryotic 6-Phosphofructokinases. Biol. Chem. 2013, 394, 977–993. [Google Scholar] [CrossRef]
- Van Schaftingen, E.; Jett, M.F.; Hue, L.; Hers, H.G. Control of Liver 6-Phosphofructokinase by Fructose 2,6-Bisphosphate and Other Effectors. Proc. Natl. Acad. Sci. USA 1981, 78, 3483–3486. [Google Scholar] [CrossRef]
- Clem, B.F.; O’Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A., II; Klarer, A.C.; Redman, R.; Miller, D.M.; Trent, J.O.; et al. Targeting 6-Phosphofructo-2-Kinase (PFKFB3) as a Therapeutic Strategy against Cancer. Mol. Cancer Ther. 2013, 12, 1461–1470. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Marín-Hernández, A.; Gallardo-Pérez, J.C.; Quezada, H.; Encalada, R.; Rodríguez-Enríquez, S.; Saavedra, E. Phosphofructokinase Type 1 Kinetics, Isoform Expression, and Gene Polymorphisms in Cancer Cells. J. Cell. Biochem. 2012, 113, 1692–1703. [Google Scholar] [CrossRef]
- Brand, I.A.; Söling, H.D. Activation and Inactivation of Rat Liver Phosphofructokinase by Phosphorylation—Dephosphorylation. FEBS Lett. 1975, 57, 163–168. [Google Scholar] [CrossRef]
- Roberts, S.J.; Somero, G.N. Binding of Phosphofructokinase to Filamentous Actin. Biochemistry 1987, 26, 3437–3442. [Google Scholar] [CrossRef]
- Lee, J.-H.; Liu, R.; Li, J.; Zhang, C.; Wang, Y.; Cai, Q.; Qian, X.; Xia, Y.; Zheng, Y.; Piao, Y.; et al. Stabilization of Phosphofructokinase 1 Platelet Isoform by AKT Promotes Tumorigenesis. Nat. Commun. 2017, 8, 949. [Google Scholar] [CrossRef]
- Kohnhorst, C.L.; Kyoung, M.; Jeon, M.; Schmitt, D.L.; Kennedy, E.L.; Ramirez, J.; Bracey, S.M.; Luu, B.T.; Russell, S.J.; An, S. Identification of a Multienzyme Complex for Glucose Metabolism in Living Cells. J. Biol. Chem. 2017, 292, 9191–9203. [Google Scholar] [CrossRef]
- Gao, W.; Huang, M.; Chen, X.; Chen, J.; Zou, Z.; Li, L.; Ji, K.; Nie, Z.; Yang, B.; Wei, Z.; et al. The Role of S-Nitrosylation of PFKM in Regulation of Glycolysis in Ovarian Cancer Cells. Cell Death Dis. 2021, 12, 408. [Google Scholar] [CrossRef]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A.; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. PFK1 Glycosylation Is a Key Regulator of Cancer Cell Growth and Central Metabolic Pathways. Science 2012, 337, 975–980. [Google Scholar] [CrossRef]
- Tong, X.; Zhao, F.; Thompson, C.B. The Molecular Determinants of de Novo Nucleotide Biosynthesis in Cancer Cells. Curr. Opin. Genet. Dev. 2009, 19, 32–37. [Google Scholar] [CrossRef]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef]
- Craven, P.A.; Basford, R.E. ADP-Induced Binding of Phosphofructokinase to the Brain Mitochondrial Membrane. Biochim. Biophys. Acta General. Subj. 1974, 354, 49–56. [Google Scholar] [CrossRef]
- Miyazawa, H.; Yamaguchi, Y.; Sugiura, Y.; Honda, K.; Kondo, K.; Matsuda, F.; Yamamoto, T.; Suematsu, M.; Miura, M. Rewiring of Embryonic Glucose Metabolism via Suppression of PFK-1 and Aldolase during Mouse Chorioallantoic Branching. Development 2017, 144, 63–73. [Google Scholar] [CrossRef]
- Bulusu, V.; Prior, N.; Snaebjornsson, M.T.; Kuehne, A.; Sonnen, K.F.; Kress, J.; Stein, F.; Schultz, C.; Sauer, U.; Aulehla, A. Spatiotemporal Analysis of a Glycolytic Activity Gradient Linked to Mouse Embryo Mesoderm Development. Dev. Cell 2017, 40, 331–341.e4. [Google Scholar] [CrossRef]
- Miyazawa, H.; Snaebjornsson, M.T.; Prior, N.; Kafkia, E.; Hammarén, H.M.; Tsuchida-Straeten, N.; Patil, K.R.; Beck, M.; Aulehla, A. Glycolytic Flux-Signaling Controls Mouse Embryo Mesoderm Development. eLife 2022, 11, e83299. [Google Scholar] [CrossRef]
- Fernandes-Silva, H.; Alves, M.G.; Araújo-Silva, H.; Silva, A.M.; Correia-Pinto, J.; Oliveira, P.F.; Moura, R.S. Lung Branching Morphogenesis Is Accompanied by Temporal Metabolic Changes towards a Glycolytic Preference. Cell Biosci. 2021, 11, 134. [Google Scholar] [CrossRef]
- Pegoraro, C.; Maczkowiak, F.; Monsoro-Burq, A.H. Pfkfb (6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase) Isoforms Display a Tissue-Specific and Dynamic Expression during Xenopus Laevis Development. Gene Expr. Patterns 2013, 13, 203–211. [Google Scholar] [CrossRef]
- Cheung, R.A.; Kraft, A.M.; Petty, H.R. Relocation of Phosphofructokinases within Epithelial Cells Is a Novel Event Preceding Breast Cancer Recurrence That Accurately Predicts Patient Outcomes. Am. J. Physiol.-Cell Physiol. 2021, 321, C654–C670. [Google Scholar] [CrossRef]
- Pragallapati, S.; Manyam, R. Glucose Transporter 1 in Health and Disease. J. Oral. Maxillofac. Pathol. 2019, 23, 443–449. [Google Scholar] [CrossRef]
- Razani, B.; Lisanti, M.P. Caveolin-Deficient Mice: Insights into Caveolar Function Human Disease. J. Clin. Invest. 2001, 108, 1553–1561. [Google Scholar] [CrossRef]
- Vallejo, J.; Hardin, C.D. Metabolic Organization in Vascular Smooth Muscle: Distribution and Localization of Caveolin-1 and Phosphofructokinase. Am. J. Physiol.-Cell Physiol. 2004, 286, C43–C54. [Google Scholar] [CrossRef]
- Sotgia, F.; Bonuccelli, G.; Minetti, C.; Woodman, S.E.; Capozza, F.; Kemp, R.G.; Scherer, P.E.; Lisanti, M.P. Phosphofructokinase Muscle-Specific Isoform Requires Caveolin-3 Expression for Plasma Membrane Recruitment and Caveolar Targeting. Am. J. Pathol. 2003, 163, 2619–2634. [Google Scholar] [CrossRef]
- Scherer, P.E.; Lisanti, M.P. Association of Phosphofructokinase-M with Caveolin-3 in Differentiated Skeletal Myotubes: Dynamic Regulation by Extracellular Glucose and Intracellular Metabolites. J. Biol. Chem. 1997, 272, 20698–20705. [Google Scholar] [CrossRef]
- Dominguez, R.; Holmes, K.C. Actin Structure and Function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef]
- Liou, R.-S.; Anderson, S. Activation of Rabbit Muscle Phosphofructokinase by F-Actin and Reconstituted Thin Filaments. Biochemistry 1980, 19, 2684–2688. [Google Scholar] [CrossRef]
- Roberts, S.J.; Somero, G.N. Properties of the Interaction between Phosphofructokinase and Actin. Arch. Biochem. Biophys. 1989, 269, 284–294. [Google Scholar] [CrossRef]
- Silva, A.P.P.; Alves, G.G.; Araújo, A.H.B.; Sola-Penna, M. Effects of Insulin and Actin on Phosphofructokinase Activity and Cellular Distribution in Skeletal Muscle. An. Acad. Bras. Ciênc. 2004, 76, 541–548. [Google Scholar] [CrossRef]
- Rossi, M.; Altea-Manzano, P.; Demicco, M.; Doglioni, G.; Bornes, L.; Fukano, M.; Vandekeere, A.; Cuadros, A.M.; Fernández-García, J.; Riera-Domingo, C.; et al. PHGDH Heterogeneity Potentiates Cancer Cell Dissemination and Metastasis. Nature 2022, 605, 747–753. [Google Scholar] [CrossRef]
- Glass-Marmor, L.; Beitner, R. Taxol (Paclitaxel) Induces a Detachment of Phosphofructokinase from Cytoskeleton of Melanoma Cells and Decreases the Levels of Glucose 1,6-Bisphosphate, Fructose 1,6-Bisphosphate and ATP. Eur. J. Pharmacol. 1999, 370, 195–199. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical Regulation of Glycolysis via Cytoskeleton Architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Lehotzky, A.; Telegdi, M.; Liliom, K.; Ovádi, J. Interaction of Phosphofructokinase with Tubulin and Microtubules. Quantitative evaluation of the mutual effects. J. Biol. Chem. 1993, 268, 10888–10894. [Google Scholar] [CrossRef]
- Vértessy, B.G.; Orosz, F.; Kovács, J.; Ovádi, J. Alternative Binding of Two Sequential Glycolytic Enzymes to Microtubules: Molecular Studies in the Phosphofructokinase/Aldolase/Microtubule System. J. Biol. Chem. 1997, 272, 25542–25546. [Google Scholar] [CrossRef]
- Lehotzky, A.; Palfia, Z.; Kovacs, J.; Molnar, A.; Ovadi, J. Ligand-Modulated Cross-Bridging of Microtubules by Phosphofructokinase. Biochem. Biophys. Res. Commun. 1994, 204, 585–591. [Google Scholar] [CrossRef]
- Vértessy, B.G.; Kovács, J.; Löw, P.; Lehotzky, A.; Molnár, A.; Orosz, F.; Ovádi, J. Characterization of Microtubule−Phosphofructokinase Complex: Specific Effects of MgATP and Vinblastine. Biochemistry 1997, 36, 2051–2062. [Google Scholar] [CrossRef]
- Jang, S.; Xuan, Z.; Lagoy, R.C.; Jawerth, L.M.; Gonzalez, I.J.; Singh, M.; Prashad, S.; Kim, H.S.; Patel, A.; Albrecht, D.R.; et al. Phosphofructokinase Relocalizes into Subcellular Compartments with Liquid-like Properties in Vivo. Biophys. J. 2021, 120, 1170–1186. [Google Scholar] [CrossRef]
- Webb, B.A.; Dosey, A.M.; Wittmann, T.; Kollman, J.M.; Barber, D.L. The Glycolytic Enzyme Phosphofructokinase-1 Assembles into Filaments. J. Cell Biol. 2017, 216, 2305–2313. [Google Scholar] [CrossRef]
- Zhang, Y.; Fernie, A.R. Metabolons, Enzyme–Enzyme Assemblies That Mediate Substrate Channeling, and Their Roles in Plant Metabolism. Plant Commun. 2021, 2, 100081. [Google Scholar] [CrossRef]
- Kurganov, B.I.; Sugrobova, N.P.; Mil’man, L.S. Supramolecular Organization of Glycolytic Enzymes. J. Theor. Biol. 1985, 116, 509–526. [Google Scholar] [CrossRef]
- Menard, L.; Maughan, D.; Vigoreaux, J. The Structural and Functional Coordination of Glycolytic Enzymes in Muscle: Evidence of a Metabolon? Biology 2014, 3, 623–644. [Google Scholar] [CrossRef]
- Jaeger, M.G.; Winter, G.E. Fast-Acting Chemical Tools to Delineate Causality in Transcriptional Control. Mol. Cell 2021, 81, 1617–1630. [Google Scholar] [CrossRef]
- Schmitt, D.L.; Dranchak, P.; Parajuli, P.; Blivis, D.; Voss, T.; Kohnhorst, C.L.; Kyoung, M.; Inglese, J.; An, S. High-Throughput Screening Identifies Cell Cycle-Associated Signaling Cascades That Regulate a Multienzyme Glucosome Assembly in Human Cells. PLoS ONE 2023, 18, e0289707. [Google Scholar] [CrossRef]
- Jeon, M.; Schmitt, D.L.; Kyoung, M.; An, S. Size-Specific Modulation of a Multienzyme Glucosome Assembly during the Cell Cycle. ACS Bio Med Chem Au 2023, 3, 461–470. [Google Scholar] [CrossRef]
- Schmitt, D.L.; An, S. Spatial Organization of Metabolic Enzyme Complexes in Cells. Biochemistry 2017, 56, 3184–3196. [Google Scholar] [CrossRef]
- Coelho, R.G.; Calaça, I.C.; Celestrini, D.M.; Correia-Carneiro, A.H.P.; Costa, M.M.; Zancan, P.; Sola-Penna, M. Hexokinase and Phosphofructokinase Activity and Intracellular Distribution Correlate with Aggressiveness and Invasiveness of Human Breast Carcinoma. Oncotarget 2015, 6, 29375–29387. [Google Scholar] [CrossRef]
- Moon, J.-S.; Kim, H.E.; Koh, E.; Park, S.H.; Jin, W.-J.; Park, B.-W.; Park, S.W.; Kim, K.-S. Krüppel-like Factor 4 (KLF4) Activates the Transcription of the Gene for the Platelet Isoform of Phosphofructokinase (PFKP) in Breast Cancer. J. Biol. Chem. 2011, 286, 23808–23816. [Google Scholar] [CrossRef]
- Yeerken, D.; Hong, R.; Wang, Y.; Gong, Y.; Liu, R.; Yang, D.; Li, J.; Fan, J.; Chen, J.; Zhang, W.; et al. PFKP Is Transcriptionally Repressed by BRCA1/ZBRK1 and Predicts Prognosis in Breast Cancer. PLoS ONE 2020, 15, e0233750. [Google Scholar] [CrossRef]
- Inaishi, T.; Shibata, M.; Ichikawa, T.; Kanda, M.; Hayashi, M.; Soeda, I.; Takeuchi, D.; Takano, Y.; Tsunoda, N.; Kodera, Y.; et al. Platelet Isoform of Phosphofructokinase Accelerates Malignant Features in Breast Cancer. Oncol. Rep. 2022, 47, 9. [Google Scholar] [CrossRef]
- Umar, S.M.; Kashyap, A.; Kahol, S.; Mathur, S.R.; Gogia, A.; Deo, S.V.S.; Prasad, C.P. Prognostic and Therapeutic Relevance of Phosphofructokinase Platelet-Type (PFKP) in Breast Cancer. Exp. Cell Res. 2020, 396, 112282. [Google Scholar] [CrossRef]
- El-Bacha, T.; de Freitas, M.S.; Sola-Penna, M. Cellular Distribution of Phosphofructokinase Activity and Implications to Metabolic Regulation in Human Breast Cancer. Mol. Genet. Metab. 2003, 79, 294–299. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, Y.; Cai, Y.; Liu, R.; Lu, M.; Li, T.; Fu, Y.; Guo, M.; Huang, H.; Ou, Y.; et al. A20 Targets PFKL and Glycolysis to Inhibit the Progression of Hepatocellular Carcinoma. Cell Death Dis. 2020, 11, 89. [Google Scholar] [CrossRef]
- Kreuzaler, P.; Panina, Y.; Segal, J.; Yuneva, M. Adapt and Conquer: Metabolic Flexibility in Cancer Growth, Invasion and Evasion. Mol. Metab. 2020, 33, 83–101. [Google Scholar] [CrossRef]
- Kohnhorst, C.L.; Schmitt, D.L.; Sundaram, A.; An, S. Subcellular Functions of Proteins under Fluorescence Single-Cell Microscopy. Biochim. Biophys. Acta 2016, 1864, 77–84. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).