
Combination Treatment Targeting mTOR and MAPK Pathways Has Synergistic Activity in Multiple Myeloma

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Cultures
2.2. Chemicals
2.3. Cell Viability Assay
2.4. Western Blotting
2.5. Apoptosis Assay
2.6. Cell Cycle Analysis
2.7. CFSE Proliferation Staining
2.8. Calculation of Combination Effect
2.9. Statistical Analysis
3. Results
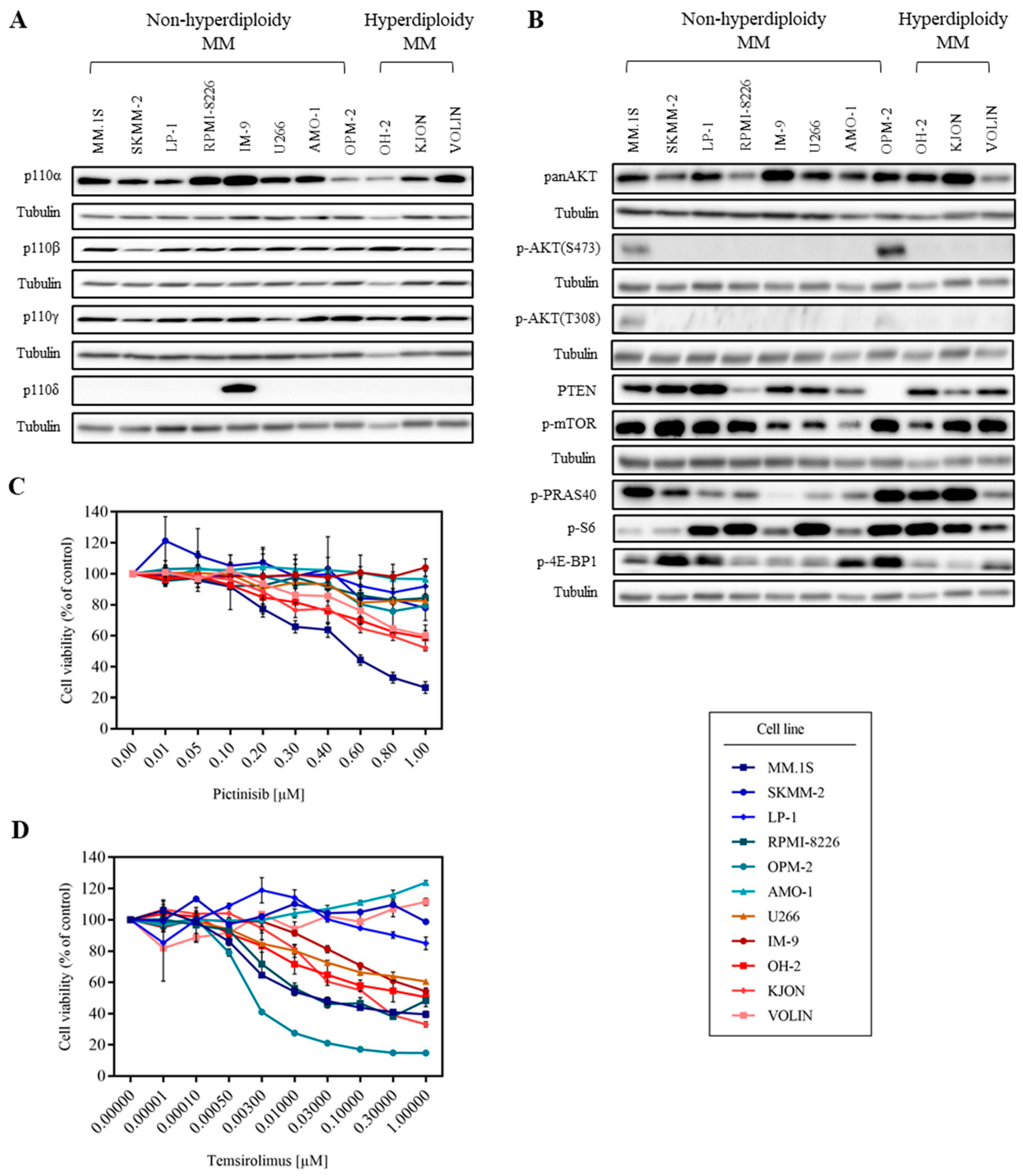
3.1. MM Cell Lines Show Various Sensitivities to PI3K and mTOR Inhibitors
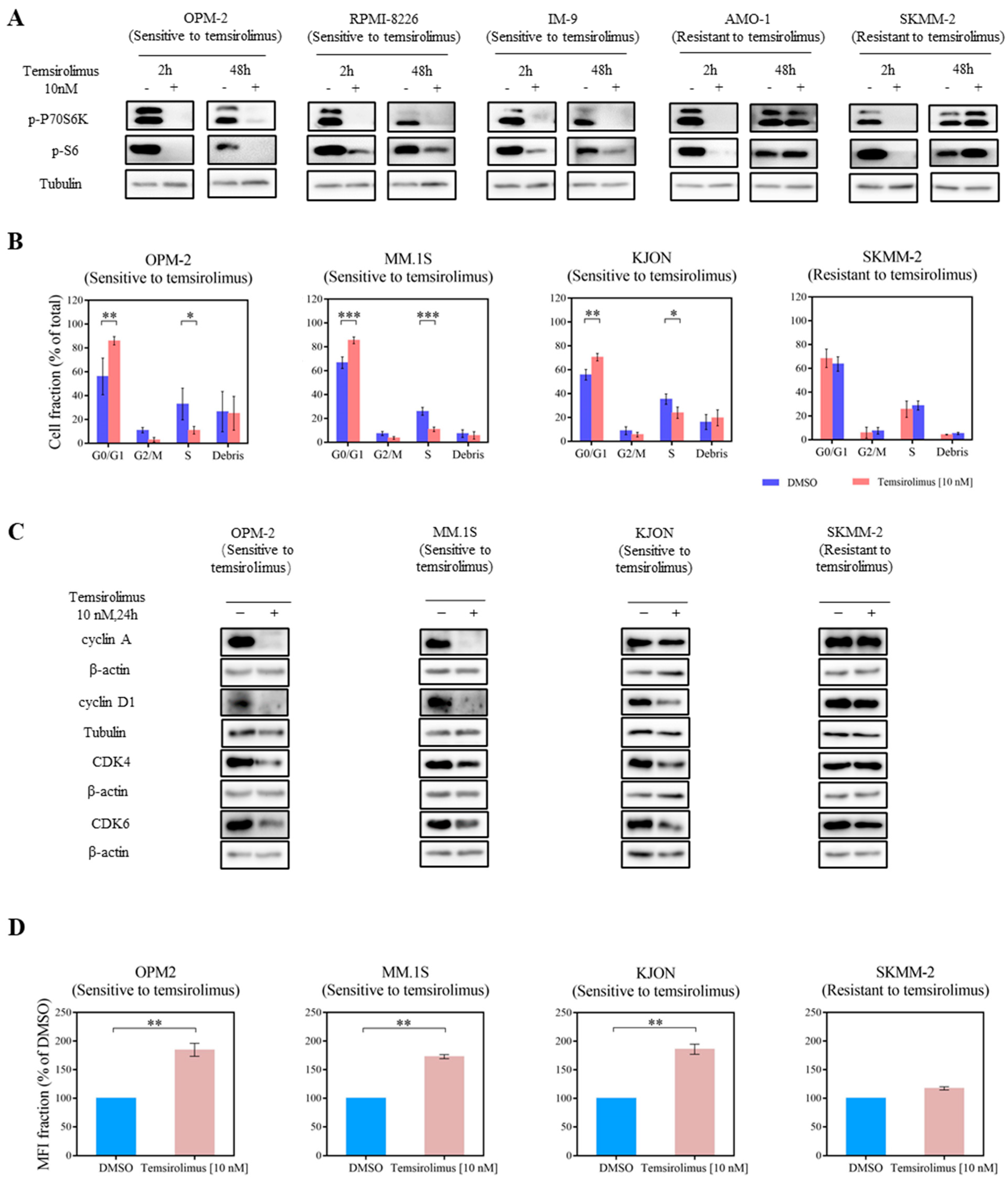
3.2. mTOR Inhibition Downregulates PI3K Pathway Key Molecules
3.3. Temsirolimus Induces Cell Cycle Arrest and Reduces Cell Proliferation
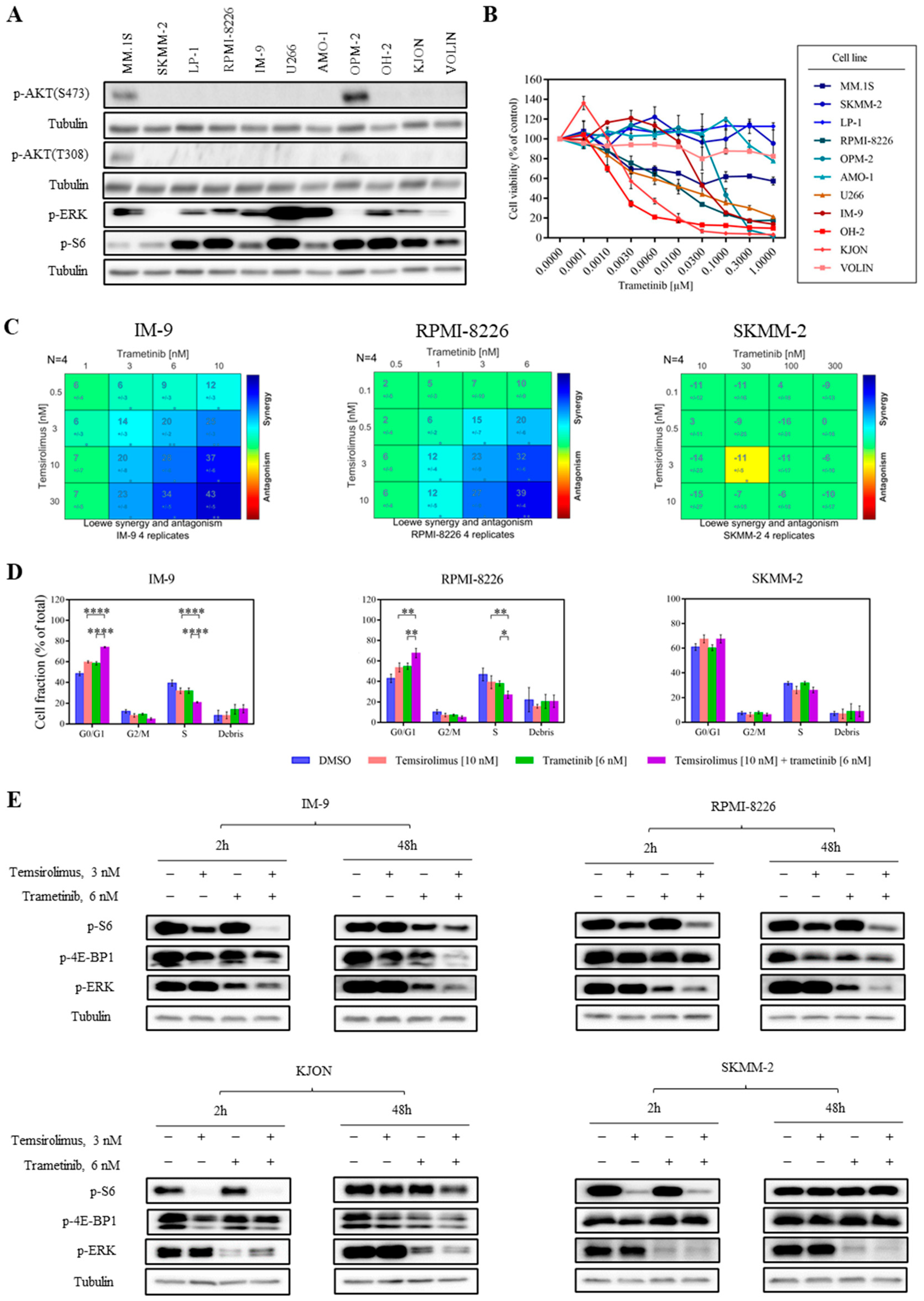
3.4. Combined mTOR and MEK Inhibition Is Synergistic in MM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kristinsson, S.Y.; Landgren, O.; Dickman, P.W.; Derolf, Å.R.; Björkholm, M. Patterns of survival in multiple myeloma: A population-based study of patients diagnosed in Sweden from 1973 to 2003. J. Clin. Oncol. 2007, 25, 1993–1999. [Google Scholar] [CrossRef]
- al Hamed, R.; Bazarbachi, A.H.; Malard, F.; Harousseau, J.L.; Mohty, M. Current status of autologous stem cell transplantation for multiple myeloma. Blood Cancer J. 2019, 9, 44. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Edavettal, S.; Mendonça, M.; Li, Y.; Tornetta, M.; Babich, A.; Majewski, N.; Husovsky, M.; Reeves, D.; Walsh, E.; et al. A T-cell–redirecting bispecific G-protein–coupled receptor class 5 member D × CD3 antibody to treat multiple myeloma. Blood 2020, 135, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Mailankody, S.; Giralt, S.A.; Landgren, C.O.; Smith, E.L.; Brentjens, R.J. CAR T Cell Therapy for Multiple Myeloma: Where are We Now and Where are We Headed? Leukemia and Lymphoma; Taylor and Francis Ltd.: Abingdon, UK, 2018; Volume 59, pp. 2056–2067. [Google Scholar]
- Bianchi, G.; Anderson, K.C. Understanding biology to tackle the disease: Multiple myeloma from bench to bedside, and back. CA Cancer J. Clin. 2014, 64, 422–444. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Kumar, S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Kaegi, C.; Wuest, B.; Schreiner, J.; Steiner, U.C.; Vultaggio, A.; Matucci, A.; Crowley, C.; Boyman, O. Systematic review of safety and efficacy of rituximab in treating immune-mediated disorders. Front. Immunol. 2019, 10, 1990. [Google Scholar] [CrossRef]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the first-line treatment of follicular lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef]
- Qiang, Y.W.; Yao, L.; Tosato, G.; Rudikoff, S. Insulin-like growth factor I induces migration and invasion of human multiple myeloma cells. Blood 2004, 103, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- Erdmann, T.; Klener, P.; Lynch, J.T.; Grau, M.; Vočková, P.; Molinsky, J.; Tuskova, D.; Hudson, K.; Polanska, U.M.; Grondine, M.; et al. Sensitivity to PI3K and AKT inhibitors is mediated by divergent molecular mechanisms in subtypes of DLBCL. Blood J. Am. Soc. Hematol. 2017, 130, 310–322. [Google Scholar] [CrossRef]
- Dreyling, M.; Santoro, A.; Mollica, L.; Leppä, S.; Follows, G.A.; Lenz, G.; Kim, W.S.; Nagler, A.; Panayiotidis, P.; Demeter, J.; et al. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J. Clin. Oncol. 2017, 35, 3898–3905. [Google Scholar] [CrossRef]
- Oki, Y.; Fanale, M.; Romaguera, J.; Fayad, L.; Fowler, N.; Copeland, A.; Samaniego, F.; Kwak, L.W.; Neelapu, S.; Wang, M.; et al. Phase II study of an AKT inhibitor MK2206 in patients with relapsed or refractory lymphoma. Br. J. Haematol. 2015, 171, 463–470. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.v.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Adachi, M.; Hoshino, Y.; Izumi, Y.; Sakai, H.; Takagi, S. Effects of inhibitors of vascular endothelial growth factor receptor 2 and downstream pathways of receptor tyrosine kinases involving phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin or mitogen-activated protein kinase in canine hemangiosarcoma cell lines. Can. J. Vet. Res. 2016, 80, 209–216. [Google Scholar]
- Günther, A.; Baumann, P.; Burger, R.; Kellner, C.; Klapper, W.; Schmidmaier, R.; Gramatzki, M. Activity of everolimus (RAD001) in relapsed and/or refractory multiple myeloma: A phase I study. Haematologica 2015, 100, 541. [Google Scholar] [CrossRef]
- Chen, H.; Huang, S.; Niu, P.; Zhu, Y.; Zhou, J.; Jiang, L.; Li, D.; Shi, D. Cardamonin suppresses pro-tumor function of macrophages by decreasing M2 polarization on ovarian cancer cells via mTOR inhibition. Mol. Ther. Oncolytics 2022, 26, 175–188. [Google Scholar] [CrossRef]
- Bjornsti, M.A.; Houghton, P.J. The TOR pathway: A target for cancer therapy. Nat. Rev. Cancer 2004, 4, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.H.; Park, C.; Ghosh, S.; Park, S.; Zivkovic, M.; Rascati, K.L. A network meta-analysis of efficacy and safety of first-line and second-line therapies for the management of metastatic renal cell carcinoma. J. Clin. Pharm. Ther. 2021, 46, 35–49. [Google Scholar] [CrossRef]
- Farag, S.S.; Zhang, S.; Jansak, B.S.; Wang, X.; Kraut, E.; Chan, K.; Dancey, J.; Grever, M. Phase II trial of temsirolimus in patients with relapsed or refractory multiple myeloma. Leuk. Res. 2009, 33, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Kumar, S.; Hideshima, T.; Ishitsuka, K.; Chauhan, D.; Mitsiades, C.; Podar, K.; Le Gouill, S.; Richardson, P.; Munshi, N.C.; et al. Combination of the mTOR inhibitor rapamycin and CC-5013 has synergistic activity in multiple myeloma. Blood 2004, 104, 4188–4193. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood J. Am. Soc. Hematol. 2018, 132, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef]
- Jokinen, E.; Koivunen, J.P. MEK and PI3K inhibition in solid tumors: Rationale and evidence to date. Ther. Adv. Med. Oncol. 2015, 7, 170–180. [Google Scholar] [CrossRef]
- Hoffner, B.; Benchich, K. Trametinib: A targeted therapy in metastatic melanoma. J. Adv. Pract. Oncol. 2018, 9, 741. [Google Scholar]
- Malyutina, A.; Majumder, M.M.; Wang, W.; Pessia, A.; Heckman, C.A.; Tang, J. Drug combination sensitivity scoring facilitates the discovery of synergistic and efficacious drug combinations in cancer. PLoS Comput. Biol. 2019, 15, e1006752. [Google Scholar] [CrossRef]
- Loewe, S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 1953, 3, 285–290. [Google Scholar] [PubMed]
- Di Veroli, G.Y.; Fornari, C.; Wang, D.; Mollard, S.; Bramhall, J.L.; Richards, F.M.; Jodrell, D.I. Combenefit: An interactive platform for the analysis and visualization of drug combinations. Bioinformatics 2016, 32, 2866–2868. [Google Scholar] [CrossRef] [PubMed]
- Pellat-Deceunynck, C.; Amiot, M.; Bataille, R.; Van Riet, I.; Van Camp, B.; Omede, P.; Boccadoro, M. Human myeloma cell lines as a tool for studying the biology of multiple myeloma: A reappraisal 18 years after. Blood 1995, 86, 4001–4002. [Google Scholar] [CrossRef]
- Moreaux, J.; Klein, B.; Bataille, R.; Descamps, G.; Maïga, S.; Hose, D.; Goldschmidt, H.; Jauch, A.; Rème, T.; Jourdan, M.; et al. A high-risk signature for patients with multiple myeloma established from the molecular classification of human myeloma cell lines. Haematologica 2011, 96, 574. [Google Scholar] [CrossRef]
- Våtsveen, T.K.; Børset, M.; Dikic, A.; Tian, E.; Micci, F.; Lid, A.H.B.; Meza-Zepeda, L.A.; Coward, E.; Waage, A.; Sundan, A.; et al. VOLIN and KJON—Two novel hyperdiploid myeloma cell lines. Genes Chromosomes Cancer 2016, 55, 890–901. [Google Scholar] [CrossRef]
- Våtsveen, T.K.; Tian, E.; Kresse, S.H.; Meza-Zepeda, L.A.; Gabrea, A.; Glebov, O.; Dai, H.Y.; Sundan, A.; Kuehl, W.M.; Børset, M. OH-2, a hyperdiploid myeloma cell line without an IGH translocation, has a complex translocation juxtaposing MYC near MAFB and the IGK locus. Leuk. Res. 2009, 33, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Vikova, V.; Jourdan, M.; Robert, N.; Requirand, G.; Boireau, S.; Bruyer, A.; Vincent, L.; Cartron, G.; Klein, B.; Elemento, O.; et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics 2019, 9, 540. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: Pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef]
- Munugalavadla, V.; Mariathasan, S.; Slaga, D.; Du, C.; Berry, L.; del Rosario, G.; Yan, Y.; Boe, M.; Sun, L.; Friedman, L.S.; et al. The PI3K inhibitor GDC-0941 combines with existing clinical regimens for superior activity in multiple myeloma. Oncogene 2014, 33, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Azab, F.; Vali, S.; Abraham, J.; Potter, N.; Muz, B.; de la Puente, P.; Fiala, M.; Paasch, J.; Sultana, Z.; Tyagi, A.; et al. PI3KCA plays a major role in multiple myeloma and its inhibition with BYL719 decreases proliferation, synergizes with other therapies and overcomes stroma-induced resistance. Br. J. Haematol. 2014, 165, 89–101. [Google Scholar] [CrossRef]
- Kim, J.; Hong, S.; Hong, S. Discovery of new aminopyrimidine-based phosphoinositide 3-kinase beta (PI3Kβ) inhibitors with selectivity over PI3Kα. Bioorg. Med. Chem. Lett. 2011, 21, 6977–6981. [Google Scholar] [CrossRef]
- Evans, C.A.; Liu, T.; Lescarbeau, A.; Nair, S.J.; Grenier, L.; Pradeilles, J.A.; Glenadel, Q.; Tibbitts, T.; Rowley, A.M.; DiNitto, J.P.; et al. Discovery of a selective phosphoinositide-3-kinase (PI3K)-γ inhibitor (IPI-549) as an immuno-oncology clinical candidate. ACS Med. Chem. Lett. 2016, 7, 862–867. [Google Scholar] [CrossRef]
- Somoza, J.R.; Koditek, D.; Villaseñor, A.G.; Novikov, N.; Wong, M.H.; Liclican, A.; Xing, W.; Lagpacan, L.; Wang, R.; Schultz, B.E.; et al. Structural, biochemical, and biophysical characterization of idelalisib binding to phosphoinositide 3-kinase δ. J. Biol. Chem. 2015, 290, 8439–8446. [Google Scholar] [CrossRef] [PubMed]
- Barlaam, B.; Cosulich, S.; Degorce, S.; Fitzek, M.; Green, S.; Hancox, U.; Lambert-van der Brempt, C.; Lohmann, J.J.; Maudet, M.; Morgentin, R.; et al. Discovery of (R)-8-(1-(3, 5-Difluorophenylamino) ethyl)-N, N-dimethyl-2-morpholino-4-oxo-4 H-chromene-6-carboxamide (AZD8186): A Potent and Selective Inhibitor of PI3Kβ and PI3Kδ for the Treatment of PTEN-Deficient Cancers. J. Med. Chem. 2015, 58, 943–962. [Google Scholar] [CrossRef] [PubMed]
- Mody, R.; Naranjo, A.; van Ryn, C.; Alice, L.Y.; London, W.B.; Shulkin, B.L.; Parisi, M.T.; Servaes, S.E.; Diccianni, M.B.; Sondel, P.M.; et al. Irinotecan–temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): An open-label, randomised, phase 2 trial. Lancet Oncol. 2017, 18, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, L.; Chi, Y.Y.; Hingorani, P.; Anderson, J.R.; Lyden, E.R.; Rodeberg, D.A.; Indelicato, D.J.; Kao, S.C.; Dasgupta, R.; Spunt, S.L.; et al. Randomized phase II trial of bevacizumab or temsirolimus in combination with chemotherapy for first relapse rhabdomyosarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 2866. [Google Scholar] [CrossRef]
- Abe, H.; Kikuchi, S.; Hayakawa, K.; Iida, T.; Nagahashi, N.; Maeda, K.; Sakamoto, J.; Matsumoto, N.; Miura, T.; Matsumura, K.; et al. Discovery of a highly potent and selective MEK inhibitor: GSK1120212 (JTP-74057 DMSO solvate). ACS Med. Chem. Lett. 2011, 2, 320–324. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Autophagosome Phagosome 2008, 445, 77–88. [Google Scholar]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox. Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Robiou-Du-Pont, S.; Cleynen, A.; Fontan, C.; Attal, M.; Munshi, N.; Corre, J.; Avet-Loiseau, H. Genomics of multiple myeloma. J. Clin. Oncol. 2017, 35, 963–967. [Google Scholar] [CrossRef]
- Zheng, Y.; Yang, J.; Qian, J.; Zhang, L.; Lu, Y.; Li, H.; Lin, H.; Lan, Y.; Liu, Z.; He, J.; et al. Novel phosphatidylinositol 3-kinase inhibitor NVP-BKM120 induces apoptosis in myeloma cells and shows synergistic anti-myeloma activity with dexamethasone. J. Mol. Med. 2012, 90, 695–706. [Google Scholar] [CrossRef]
- Strömberg, T.; Dimberg, A.; Hammarberg, A.; Carlson, K.; Osterborg, A.; Nilsson, K.; Jernberg-Wiklund, H. Rapamycin sensitizes multiple myeloma cells to apoptosis induced by dexamethasone. Blood 2004, 103, 3138–3147. [Google Scholar] [CrossRef]
- Okabe, S.; Tanaka, Y.; Tauchi, T.; Ohyashiki, K. Copanlisib, a novel phosphoinositide 3-kinase inhibitor, combined with carfilzomib inhibits multiple myeloma cell proliferation. Ann. Hematol. 2019, 98, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gera, J.; Hu, L.; Hsu, J.; hsin Bookstein, R.; Li, W.; Lichtenstein, A. Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res. 2002, 62, 5027–5034. [Google Scholar]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef]
- Cuyàs, E.; Corominas-Faja, B.; Joven, J.; Menendez, J.A. Cell Cycle Regulation by the Nutrient-Sensing Mammalian target of rapamycin (mTOR) pathway. Methods Mol. Biol. 2014, 1170, 113–144. [Google Scholar] [PubMed]
- Medema, R.H.; Kops, G.J.P.L.; Bos, J.L.; Burgering, B.M.T. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Nazim, U.M.; Bishayee, K.; Kang, J.; Yoo, D.; Huh, S.O.; Sadra, A. mTORC1-Inhibition Potentiating Metabolic Block by Tyrosine Kinase Inhibitor Ponatinib in Multiple Myeloma. Cancers 2022, 14, 2766. [Google Scholar] [CrossRef]
- Yee, A.J.; Hari, P.; Marcheselli, R.; Mahindra, A.K.; Cirstea, D.D.; Scullen, T.A.; Burke, J.N.; Rodig, S.J.; Hideshima, T.; Laubach, J.P.; et al. Outcomes in patients with relapsed or refractory multiple myeloma in a phase I study of everolimus in combination with lenalidomide. Br. J. Haematol. 2014, 166, 401–409. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, K.; Jin, L.; Karolová, J.; Vorwerk, J.; Hailfinger, S.; Opalka, B.; Zapukhlyak, M.; Lenz, G.; Khandanpour, C. Combination Treatment Targeting mTOR and MAPK Pathways Has Synergistic Activity in Multiple Myeloma. Cancers 2023, 15, 2373. https://doi.org/10.3390/cancers15082373
Sun K, Jin L, Karolová J, Vorwerk J, Hailfinger S, Opalka B, Zapukhlyak M, Lenz G, Khandanpour C. Combination Treatment Targeting mTOR and MAPK Pathways Has Synergistic Activity in Multiple Myeloma. Cancers. 2023; 15(8):2373. https://doi.org/10.3390/cancers15082373
Chicago/Turabian StyleSun, Kaiyan, Ling Jin, Jana Karolová, Jan Vorwerk, Stephan Hailfinger, Bertram Opalka, Myroslav Zapukhlyak, Georg Lenz, and Cyrus Khandanpour. 2023. "Combination Treatment Targeting mTOR and MAPK Pathways Has Synergistic Activity in Multiple Myeloma" Cancers 15, no. 8: 2373. https://doi.org/10.3390/cancers15082373
APA StyleSun, K., Jin, L., Karolová, J., Vorwerk, J., Hailfinger, S., Opalka, B., Zapukhlyak, M., Lenz, G., & Khandanpour, C. (2023). Combination Treatment Targeting mTOR and MAPK Pathways Has Synergistic Activity in Multiple Myeloma. Cancers, 15(8), 2373. https://doi.org/10.3390/cancers15082373