
The Therapeutic Landscape for KRAS-Mutated Colorectal Cancers


Abstract
Simple Summary
Abstract

1. Introduction
2. RAS Signaling
3. Prognostic Implications of KRAS Mutation
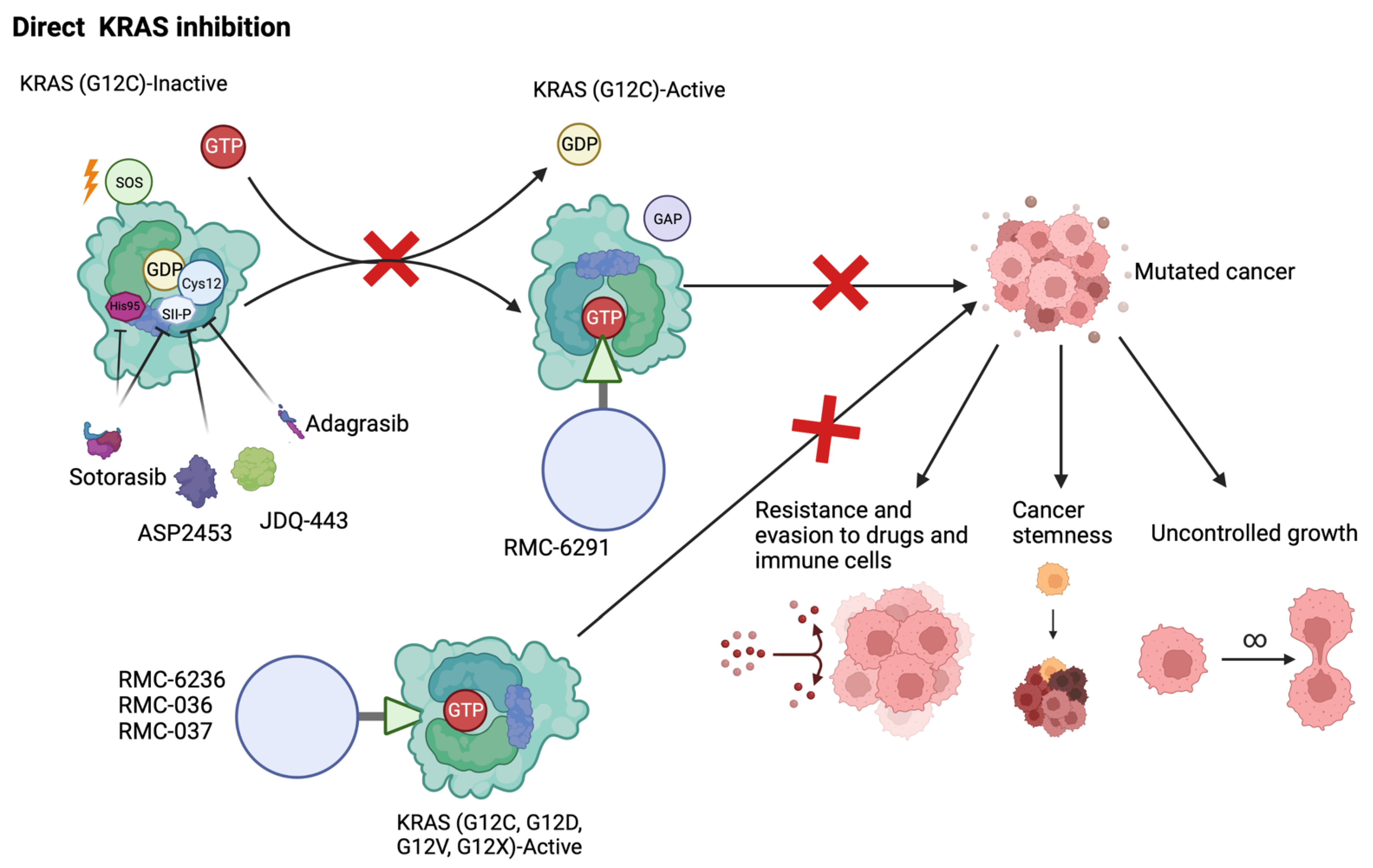
4. Direct Inhibition of Mutant KRAS
4.1. KRAS G12C Inhibition
4.1.1. Sotorasib: First Clinically Available KRAS Inhibitor
4.1.2. Adagrasib
4.1.3. ASP2453
4.1.4. JDQ443
4.2. Alternate KRAS-Mutant Inhibition
5. KRAS G12C Inhibitor Resistance
6. Indirectly Targeting KRAS-Mutant Cancers through Pan-Pathway Inhibition
7. Exploiting the Altered Metabolic Pathways to Target KRAS-Mutant Cancer
7.1. Ferroptosis
7.2. Glucose Metabolism
7.3. Glutamine Metabolism
8. Immunotherapy Options for KRAS-Mutant Cancers
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Fennell, L.; Dumenil, T.; Wockner, L.; Hartel, G.; Nones, K.; Bond, C.; Borowsky, J.; Liu, C.; McKeone, D.; Bowdler, L.; et al. Integrative Genome-Scale DNA Methylation Analysis of a Large and Unselected Cohort Reveals 5 Distinct Subtypes of Colorectal Adenocarcinomas. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 269–290. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.G.; Olson, P.; Briere, T.; Wiel, C.; Bergo, M.O. Targeting Kras(g12c)-mutant cancer with a mutation-specific inhibitor. J. Intern. Med. 2020, 288, 183–191. [Google Scholar] [CrossRef]
- Milburn, M.V.; Tong, L.; Devos, A.M.; Brünger, A.; Yamaizumi, Z.; Nishimura, S.; Kim, S.-H. Molecular Switch for Signal Transduction: Structural Differences between Active and Inactive Forms of Protooncogenic ras Proteins. Science 1990, 247, 939–945. [Google Scholar] [CrossRef]
- Cefalì, M.; Epistolio, S.; Palmarocchi, M.C.; Frattini, M.; De Dosso, S. Research progress on KRAS mutations in colorectal cancer. J. Cancer Metastasis Treat. 2021, 7, 26. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef]
- Castagnola, P.; Giaretti, W. Mutant KRAS, chromosomal instability and prognosis in colorectal cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2005, 1756, 115–125. [Google Scholar] [CrossRef]
- Modest, D.P.; Ricard, I.; Heinemann, V.; Hegewisch-Becker, S.; Schmiegel, W.; Porschen, R.; Stintzing, S.; Graeven, U.; Arnold, D.; von Weikersthal, L.F.; et al. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants: Pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann. Oncol. 2016, 27, 1746–1753. [Google Scholar] [CrossRef]
- Ozer, M.; Goksu, S.Y.; Sanford, N.N.; Ahn, C.; Beg, M.S.; Kazmi, S.M.A. Age-dependent prognostic value of KRAS mutation in metastatic colorectal cancer. Futur. Oncol. 2021, 17, 4883–4893. [Google Scholar] [CrossRef] [PubMed]
- Tsilimigras, D.I.; Ntanasis-Stathopoulos, I.; Bagante, F.; Moris, D.; Cloyd, J.; Spartalis, E.; Pawlik, T.M. Clinical significance and prognostic relevance of KRAS, BRAF, PI3K and TP53 genetic mutation analysis for resectable and unresectable colorectal liver metastases: A systematic review of the current evidence. Surg. Oncol. 2018, 27, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Hayama, T.; Hashiguchi, Y.; Okamoto, K.; Okada, Y.; Ono, K.; Shimada, R.; Ozawa, T.; Toyoda, T.; Tsuchiya, T.; Iinuma, H.; et al. G12V and G12C mutations in the gene KRAS are associated with a poorer prognosis in primary colorectal cancer. Int. J. Color. Dis. 2019, 34, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.P.; Sutton, P.A.; Evans, J.P.; Clifford, R.; McAvoy, A.; Lewis, J.; Rousseau, A.; Mountford, R.; McWhirter, D.; Malik, H.Z. Specific mutations in KRAS codon 12 are associated with worse overall survival in patients with advanced and recurrent colorectal cancer. Br. J. Cancer 2017, 116, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Morikawa, T.; Liao, X.; Lochhead, P.; Kuchiba, A.; Yamauchi, M.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A.; Haigis, K.M.; et al. Specific mutations in KRAS codons 12 and 13, and patient prognosis in 1075 BRAF wild-type colorectal cancers. Clin. Cancer Res. 2012, 18, 4753–4763. [Google Scholar] [CrossRef]
- Goldberg, R.M.; Montagut, C.; Wainberg, Z.A.; Ronga, P.; Audhuy, F.; Taieb, J.; Stintzing, S.; Siena, S.; Santini, D. Optimising the use of cetuximab in the continuum of care for patients with metastatic colorectal cancer. ESMO Open 2018, 3, e000353. [Google Scholar] [CrossRef]
- Lech, G.; Slotwinski, R.; Słodkowski, M.; Krasnodębski, I.W. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J. Gastroenterol. 2016, 22, 1745–1755. [Google Scholar] [CrossRef]
- Zocche, D.M.; Ramirez, C.; Fontao, F.M.; Costa, L.D.; Redal, M.A. Global impact of KRAS mutation patterns in FOLFOX treated metastatic colorectal cancer. Front. Genet. 2015, 6, 116. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- A Study of LY3499446 in Participants with Advanced Solid Tumors with KRAS G12C Mutation. 2019. Available online: clinicaltrials.gov (accessed on 8 May 2022).
- FIH Study of JAB-21822 in Adult Patients with Advanced Solid Tumors Harboring KRAS G12C Mutation in China. 2021. Available online: clinicaltrials.gov (accessed on 8 May 2022).
- JAB-21822 Activity in Adult Patients with Advanced Solid Tumors Harboring KRAS G12C Mutation. 2021. Available online: clinicaltrials.gov (accessed on 8 May 2022).
- First-in-Human Study of JNJ-74699157 in Participants with Tumors Harboring the KRAS G12C Mutation. 2019. Available online: clinicaltrials.gov (accessed on 8 May 2022).
- A Study of GFH925 in Patients with Advanced Solid Tumors with KRAS G12C Mutations. 2021. Available online: clinicaltrials.gov (accessed on 8 May 2022).
- A Phase 1, Study of YL-15293 in Subjects with Advanced Solid Tumors with a KRAS G12C Mutation. 2021. Available online: clinicaltrials.gov (accessed on 8 May 2022).
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Patricelli, M.P.; Janes, M.R.; Li, L.-S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Lito, P.; Solomon, M.; Li, L.-S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Faculty Opinions recommendation of Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2021, 28, 1482–1486. [Google Scholar] [CrossRef]
- A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of Sotorasib (AMG 510) in Subjects with Solid Tumors with a Specific KRAS Mutation (CodeBreaK 100). Available online: clinicaltrials.gov (accessed on 16 June 2022).
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Grants Accelerated Approval to Adagrasib for KRAS G12C-Mutated NSCLC. 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-adagrasib-kras-g12c-mutated-nsclc (accessed on 8 February 2023).
- Nakayama, A.; Nagashima, T.; Nishizono, Y.; Kuramoto, K.; Mori, K.; Homboh, K.; Yuri, M.; Shimazaki, M. Characterisation of a novel KRAS G12C inhibitor ASP2453 that shows potent anti-tumour activity in KRAS G12C-mutated preclinical models. Br. J. Cancer 2022, 126, 744–753. [Google Scholar] [CrossRef]
- Weiss, A.; Lorthiois, E.; Barys, L.; Beyer, K.S.; Bomio-Confaglia, C.; Burks, H.; Chen, X.; Cui, X.; de Kanter, R.; Dharmarajan, L.; et al. Discovery, Preclinical Characterization, and Early Clinical Activity of JDQ443, a Structurally Novel, Potent, and Selective Covalent Oral Inhibitor of KRASG12C. Cancer Discov. 2022, 12, 1500–1517. [Google Scholar] [CrossRef] [PubMed]
- Study of JDQ443 in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation (KontRASt-01). 2021. Available online: clinicaltrials.gov (accessed on 2 April 2023).
- Study of Efficacy and Safety of JDQ443 Single-Agent as First-Line Treatment for Patients with Locally Advanced or Metastatic KRAS G12C-Mutated Non-small Cell Lung Cancer with a PD-L1 Expression < 1% or a PD-L1 Expression ≥ 1% and an STK11 Co-Mutation. 2022. Available online: clinicaltrials.gov (accessed on 2 April 2023).
- Study of JDQ443 in Comparison with Docetaxel in Participants with Locally Advanced or Metastatic KRAS G12C Mutant Non-Small Cell Lung Cancer (KontRASt-02). 2021. Available online: clinicaltrials.gov (accessed on 2 April 2023).
- Platform Study of JDQ443 in Combinations in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation (KontRASt-03). 2022. Available online: clinicaltrials.gov (accessed on 2 April 2023).
- Kelsey, S. Discovery and Development of RAS(ON) Inhibitors Beyond KRAS-G12C. 2021. Available online: https://www.revmed.com/media/discovery-and-development-rason-inhibitors-beyond-kras-g12c (accessed on 8 September 2022).
- Smith, J. Combination Strategies to Defeat RAS-Addicted Cancers. 2021. Available online: https://www.revmed.com/media/combination-strategies-defeat-ras-addicted-cancers (accessed on 8 September 2022).
- Nichols, B. Targeting KRAS-G12C(ON) and Potential Application to Overcoming Drug Resistance in RAS-Addicted Tumors. 2021. Available online: https://www.revmed.com/media/targeting-kras-g12con-and-potential-application-overcoming-drug-resistance-ras-addicted (accessed on 8 September 2022).
- Vasta, J.D.; Peacock, D.M.; Zheng, Q.; Walker, J.A.; Zhang, Z.; Zimprich, C.A.; Thomas, M.R.; Beck, M.T.; Binkowski, B.F.; Corona, C.R.; et al. KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat. Chem. Biol. 2022, 18, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Blaquier, J.B.; Cardona, A.F.; Recondo, G. Resistance to KRAS(G12C) Inhibitors in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 787585. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Paniagua, G.; Jacob, H.K.; Brehey, O.; García-Alonso, S.; Lechuga, C.G.; Pons, T.; Musteanu, M.; Guerra, C.; Drosten, M.; Barbacid, M. KSR induces RAS-independent MAPK pathway activation and modulates the efficacy of KRAS inhibitors. Mol. Oncol. 2022, 16, 3066–3081. [Google Scholar] [CrossRef]
- Nguyen, A.; Burack, W.R.; Stock, J.L.; Kortum, R.; Chaika, O.V.; Afkarian, M.; Muller, W.J.; Murphy, K.M.; Morrison, D.K.; Lewis, R.E.; et al. Kinase Suppressor of Ras (KSR) Is a Scaffold Which Facilitates Mitogen-Activated Protein Kinase Activation in vivo. Mol. Cell. Biol. 2002, 22, 3035–3045. [Google Scholar] [CrossRef]
- Ou SH, I.; Jänne, P.A.; Leal, T.A.; Rybkin, I.I.; Sabari, J.K.; Barve, M.A.; Bazhenova, L.; Johnson, M.L.; Velastegui, K.L.; Cilliers, C.; et al. First-in-Human Phase I/IB Dose-Finding Study of Adagrasib (MRTX849) in Patients with Advanced KRAS(G12C) Solid Tumors (KRYSTAL-1). J. Clin. Oncol. 2022, 40, 2530–2538. [Google Scholar]
- Adagrasib in Combination with BI 1701963 in Patients with Cancer (KRYSTAL 14). 2021. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- Adagrasib in Combination with Palbociclib in Patients with Advanced Solid Tumors (KRYSTAL-16). 2022. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- Adagrasib in Combination with TNO155 in Patients with Cancer (KRYSTAL 2). 2020. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- Phase 3 Study of MRTX849 (Adagrasib) vs. Docetaxel in Patients with Advanced Non-Small Cell Lung Cancer with KRAS G12C Mutation (KRYSTAL-12). 2021. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- Testing the Use of Targeted Treatment (AMG 510) for KRAS G12C Mutated Advanced Non-Squamous Non-Small Cell Lung Cancer (A Lung-MAP Treatment Trial). 2020. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- A Phase I/II Study of AMG 510 in Combination with MVASI in Patients with Advanced, Unresectable or Metastatic KRAS G12C Mutant NSCLC with Asymptomatic Brain Metastasis. 2022. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- Study to Compare AMG 510 “Proposed INN Sotorasib” with Docetaxel in Non Small Cell Lung Cancer (NSCLC) (CodeBreak 200). 2020. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- A Study of Sotorasib (AMG 510) in Participants with Stage IV NSCLC Whose Tumors Harbor a KRAS p.G12C Mutation in Need of First-Line Treatment (CodeBreaK201). 2021. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- Phase ½ Study of VS-6766 + Sotorasib in G12C NSCLC Patients (RAMP203). 2021. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- Combination Study of RMC-4630 and Sotorasib for NSCLC Subjects with KRASG12C Mutation after Failure of Prior Standard Therapies. 2021. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- RW Efficacy of Sotorasib in KRAS G12C-Mutated Metastatic NSCLC (LungKG12Ci). 2022. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- A Phase II Study of Neoadjuvant Sotorasib in Combination with Cisplatin or Carboplatin and Pemetrexed for Surgically Resectable Stage IIA-IIIB Non-Squamous Non-Small Cell Lung Cancer with a KRAS p.G12C Mutation. 2021. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- Sotorasib and Panitumumab Versus Investigator’s Choice for Participants with Kirsten Rat Sarcoma (KRAS) p.G12C Mutation (CodeBreak 300). 2022. Available online: clinicaltrials.gov (accessed on 16 October 2022).
- Han, J.; Liu, Y.; Yang, S.; Wu, X.; Li, H.; Wang, Q. MEK inhibitors for the treatment of non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Van Schaeybroeck, S.; Kalimutho, M.; Dunne, P.; Carson, R.; Allen, W.; Jithesh, P.V.; Redmond, K.L.; Sasazuki, T.; Shirasawa, S.; Blayney, J.; et al. ADAM17-Dependent c-MET-STAT3 Signaling Mediates Resistance to MEK Inhibitors in KRAS Mutant Colorectal Cancer. Cell Rep. 2014, 7, 1940–1955. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Luo, D.; Yu, J.; Zhang, M.; Zheng, X.; Xu, G.; Wang, J.; Wang, H.; Xu, Y.; Jiang, K.; et al. Genome-wide CRISPR-cas9 knockout screening identifies GRB7 as a driver for MEK inhibitor resistance in KRAS mutant colon cancer. Oncogene 2021, 41, 191–203. [Google Scholar] [CrossRef]
- Pek, M.; Yatim, S.M.J.M.; Chen, Y.; Li, J.; Gong, M.; Jiang, X.; Zhang, F.; Zheng, J.; Wu, X.; Yu, Q. Oncogenic KRAS-associated gene signature defines co-targeting of CDK4/6 and MEK as a viable therapeutic strategy in colorectal cancer. Oncogene 2017, 36, 4975–4986. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Häussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 2197. [Google Scholar] [CrossRef] [PubMed]
- Balmanno, K.; Chell, S.D.; Gillings, A.S.; Hayat, S.; Cook, S.J. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int. J. Cancer. 2009, 125, 2332–2341. [Google Scholar] [CrossRef]
- Wee, S.; Jagani, Z.; Xiang, K.X.; Loo, A.; Dorsch, M.; Yao, Y.-M.; Sellers, W.R.; Lengauer, C.; Stegmeier, F. PI3K Pathway Activation Mediates Resistance to MEK Inhibitors in KRAS Mutant Cancers. Cancer Res. 2009, 69, 4286–4293. [Google Scholar] [CrossRef]
- Migliardi, G.; Sassi, F.; Torti, D.; Galimi, F.; Zanella, E.R.; Buscarino, M.; Ribero, D.; Muratore, A.; Massucco, P.; Pisacane, A.; et al. Inhibition of MEK and PI3K/mTOR Suppresses Tumor Growth but Does Not Cause Tumor Regression in Patient-Derived Xenografts of RAS-Mutant Colorectal Carcinomas. Clin. Cancer Res. 2012, 18, 2515–2525. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Alsina, M.; Soares, H.P.; Braña, I.; Britten, C.D.; Del Conte, G.; Ezeh, P.; Houk, B.; Kern, K.A.; Leong, S.; et al. A Multi-Arm Phase I Study of the PI3K/mTOR Inhibitors PF-04691502 and Gedatolisib (PF-05212384) plus Irinotecan or the MEK Inhibitor PD-0325901 in Advanced Cancer. Target. Oncol. 2017, 12, 775–785. [Google Scholar] [CrossRef]
- Do, K.; Speranza, G.; Bishop, R.; Khin, S.; Rubinstein, L.; Kinders, R.J.; Datiles, M.; Eugeni, M.; Lam, M.H.; Austin Doyle, L.; et al. Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Investig. New Drugs 2015, 33, 720–728. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Von Hoff, D.D.; Eskens, F.; Blumenschein, G.; Richards, D.; Genvresse, I.; Reschke, S.; Granvil, C.; Skubala, A.; Peña, C.; et al. Phase Ib Trial of the PI3K Inhibitor Copanlisib Combined with the Allosteric MEK Inhibitor Refametinib in Patients with Advanced Cancer. Target. Oncol. 2020, 15, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Gounder, M.; Rodon, J.; Janku, F.; Lolkema, M.P.; Stephenson, J.J.; Bedard, P.L.; Schuler, M.; Sessa, C.; LoRusso, P.; et al. Phase Ib Study of Combination Therapy with MEK Inhibitor Binimetinib and Phosphatidylinositol 3-Kinase Inhibitor Buparlisib in Patients with Advanced Solid Tumors with RAS/RAF Alterations. Oncologist 2020, 25, e160–e169. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; LoRusso, P.; Cho, D.C.; Musib, L.; Yan, Y.; Wongchenko, M.; Chang, I.; Patel, P.; Chan, I.T.; Sanabria-Bohorquez, S.; et al. A phase Ib open-label dose escalation study of the safety, pharmacokinetics, and pharmacodynamics of cobimetinib (GDC-0973) and ipatasertib (GDC-0068) in patients with locally advanced or metastatic solid tumors. Investig. New Drugs 2020, 39, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Arend, R.C.; Davis, A.M.; Chimiczewski, P.; O’Malley, D.M.; Provencher, D.; Vergote, I.; Ghamande, S.; Birrer, M.J. EMR 20006-012: A phase II randomized double-blind placebo controlled trial comparing the combination of pimasertib (MEK inhibitor) with SAR245409 (PI3K inhibitor) to pimasertib alone in patients with previously treated unresectable borderline or low grade ovarian cancer. Gynecol. Oncol. 2019, 156, 301–307. [Google Scholar] [CrossRef]
- Shin, D.H.; Kim, S.H.; Choi, M.; Bae, Y.-K.; Han, C.; Choi, B.K.; Kim, S.S.; Han, J.-Y. Oncogenic KRAS promotes growth of lung cancer cells expressing SLC3A2-NRG1 fusion via ADAM17-mediated shedding of NRG1. Oncogene 2022, 41, 280–292. [Google Scholar] [CrossRef]
- Meng, X.; Hu, B.; Hossain, M.M.; Chen, G.; Sun, Y.; Zhang, X. ADAM17-siRNA inhibits MCF-7 breast cancer through EGFR-PI3K-AKT activation. Int. J. Oncol. 2016, 49, 682–690. [Google Scholar] [CrossRef]
- MEK and MET Inhibition in Colorectal Cancer (MErCuRIC1). Available online: clinicaltrials.gov (accessed on 16 October 2022).
- A Phase Ia/b Study of MEK1/2 Inhibitor PD-0325901 or MEK-162 with cMET Inhibitor PF-02341066 in RASMT and RASWT (with aberrant c-MET) Colorectal Cancer Patients. 2020. Available online: https://cordis.europa.eu/project/id/602901 (accessed on 9 October 2022).
- Phase Ib/II Study of Efficacy and Safety of MEK162 and Panitumumab, in Adult mCRC Patients with Mutant or Wild-Type RAS Tumors. 2021. Available online: clinicaltrials.gov (accessed on 12 November 2022).
- Lee, M.S.; Helms, T.L.; Feng, N.; Gay, J.; Chang, Q.E.; Tian, F.; Wu, J.Y.; Toniatti, C.; Heffernan, T.P.; Powis, G.; et al. Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 2016, 7, 39595–39608. [Google Scholar] [CrossRef]
- Binimetinib and Palbociclib or TAS-102 in Treating Patients with KRAS and NRAS Mutant Metastatic or Unresectable Colorectal Cancer. 2019. Available online: clinicaltrials.gov (accessed on 12 November 2022).
- PALBOCICLIB + PD-0325901 for NSCLC & Solid Tumors. 2013. Available online: clinicaltrials.gov (accessed on 12 November 2022).
- Study of the CDK4/6 Inhibitor Palbociclib (PD-0332991) in Combination with the MEK Inhibitor Binimetinib (MEK162) for Patients with Advanced KRAS Mutant Non-Small Cell Lung Cancer. 2017. Available online: clinicaltrials.gov (accessed on 12 November 2022).
- A Trail of CDK4/6 Inhibitor and MEK Inhibitor in the Treatment of Metastatic Digestive System Tumors. 2020. Available online: clinicaltrials.gov (accessed on 12 November 2022).
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1–KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef]
- Chen, P.; Li, X.; Zhang, R.; Liu, S.; Xiang, Y.; Zhang, M.; Chen, X.; Pan, T.; Yan, L.; Feng, J.; et al. Combinative treatment of β-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 2020, 10, 5107–5119. [Google Scholar] [CrossRef]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021, 12, 1079. [Google Scholar] [CrossRef]
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965. [Google Scholar] [CrossRef] [PubMed]
- Kerk, S.A.; Papagiannakopoulos, T.; Shah, Y.M.; Lyssiotis, C.A. Metabolic networks in mutant KRAS-driven tumours: Tissue specificities and the microenvironment. Nat. Rev. Cancer 2021, 21, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Heiden, M.G.V.; McCormick, F. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; He, M.M.; Xiao, J.; Zhang, Y.Q.; Yuan, X.L.; Fang, W.J.; Zhang, Y.; Wang, W.; Hu, X.H.; Ma, Z.G.; et al. A Randomized, Open-Label, Multicenter, Phase 3 Study of High-Dose Vitamin C Plus FOLFOX ± Bevacizumab versus FOLFOX ± Bevacizumab in Unresectable Untreated Metastatic Colorectal Cancer (VITALITY Study). Clin. Cancer Res. 2022, 28, 4232–4239. [Google Scholar] [CrossRef]
- Kandasamy, P.; Zlobec, I.; Nydegger, D.T.; Pujol-Giménez, J.; Bhardwaj, R.; Shirasawa, S.; Tsunoda, T.; Hediger, M.A. Oncogenic KRAS mutations enhance amino acid uptake by colorectal cancer cells via the hippo signaling effector YAP1. Mol. Oncol. 2021, 15, 2782–2800. [Google Scholar] [CrossRef]
- Najumudeen, A.K.; CRUK Rosetta Grand Challenge Consortium; Ceteci, F.; Fey, S.K.; Hamm, G.; Steven, R.T.; Hall, H.; Nikula, C.J.; Dexter, A.; Murta, T.; et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat. Genet. 2021, 53, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Xu, J.; Bian, X.; Wu, J.-L.; Kang, W.; Qian, Y.; Li, W.; Chen, H.; Gou, H.; Liu, D.; et al. In Colorectal Cancer Cells with Mutant KRAS, SLC25A22-Mediated Glutaminolysis Reduces DNA Demethylation to Increase WNT Signaling, Stemness, and Drug Resistance. Gastroenterology 2020, 159, 2163–2180. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Tannir, N.M.; Iliopoulos, O.; Lee, R.J.; Telli, M.L.; Fan, A.C.; DeMichele, A.; Haas, N.B.; Patel, M.R.; Harding, J.J.; et al. Telaglenastat Plus Cabozantinib or Everolimus for Advanced or Metastatic Renal Cell Carcinoma: An Open-Label Phase I Trial. Clin. Cancer Res. 2022, 28, 1540–1548. [Google Scholar] [CrossRef]
- Tannir, N.M.; Agarwal, N.; Porta, C.; Lawrence, N.J.; Motzer, R.; McGregor, B.; Lee, R.J.; Jain, R.K.; Davis, N.; Appleman, L.J.; et al. Efficacy and Safety of Telaglenastat Plus Cabozantinib vs Placebo Plus Cabozantinib in Patients with Advanced Renal Cell Carcinoma: The CANTATA Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1411–1418. [Google Scholar] [CrossRef]
- Study of CB-839 (Telaglenastat) in Combination with Talazoparib in Patients with Solid Tumors. Available online: clinicaltrials.gov (accessed on 9 April 2023).
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Kamphues, C.; Kadowaki, S.; Amini, N.; van den Berg, I.; Wang, J.; Andreatos, N.; Sakamoto, Y.; Ogura, T.; Kakuta, M.; Pikouli, A.; et al. The interplay of KRAS mutational status with tumor laterality in non-metastatic colorectal cancer: An international, multi-institutional study in patients with known KRAS, BRAF, and MSI status. J. Surg. Oncol. 2021, 123, 1005–1014. [Google Scholar] [CrossRef]
- Andre, T.; Amonkar, M.; Norquist, J.M.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.; Garcia-Carbonero, R.; et al. Health-related quality of life in patients with microsatellite instability-high or mismatch repair deficient metastatic colorectal cancer treated with first-line pembrolizumab versus chemotherapy (KEYNOTE-177): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A.; Shiu, K.-K.; Kim, T.-W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): Final analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2022, 23, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Casak, S.J.; Marcus, L.; Fashoyin-Aje, L.; Mushti, S.L.; Cheng, J.; Shen, Y.L.; Pierce, W.F.; Her, L.; Goldberg, K.B.; Theoret, M.R.; et al. FDA Approval Summary: Pembrolizumab for the First-line Treatment of Patients with MSI-H/dMMR Advanced Unresectable or Metastatic Colorectal Carcinoma. Clin. Cancer Res. 2021, 27, 4680–4684. [Google Scholar] [CrossRef]
- Liu, C.; Liu, R.; Wang, B.; Lian, J.; Yao, Y.; Sun, H.; Zhang, C.; Fang, L.; Guan, X.; Shi, J.; et al. Blocking IL-17A enhances tumor response to anti-PD-1 immunotherapy in microsatellite stable colorectal cancer. J. Immunother. Cancer 2021, 9, e001895. [Google Scholar] [CrossRef]
- Liu, C.; Liu, R.; Wang, B.; Lian, J.; Yao, Y.; Sun, H.; Zhang, C.; Fang, L.; Guan, X.; Shi, J.; et al. TRIB3 reduces CD8+ T cell infiltration and induces immune evasion by repressing the STAT1-CXCL10 axis in colorectal cancer. Sci. Transl. Med. 2022, 14, eabf0992. [Google Scholar]
- Chen, J.; Hou, S.; Liang, Q.; He, W.; Li, R.; Wang, H.; Zhu, Y.; Zhang, B.; Chen, L.; Dai, X.; et al. Localized Degradation of Neutrophil Extracellular Traps by Photoregulated Enzyme Delivery for Cancer Immunotherapy and Metastasis Suppression. ACS Nano 2022, 16, 2585–2597. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.-J.; Tsao, L.-C.; Acharya, C.R.; Trotter, T.; Agarwal, P.; Wei, J.; Wang, T.; Yang, X.-Y.; Lei, G.; Osada, T.; et al. Sensitizing immune unresponsive colorectal cancers to immune checkpoint inhibitors through MAVS overexpression. J. Immunother. Cancer 2022, 10, e003721. [Google Scholar] [CrossRef] [PubMed]
- El-Sayes, N.; Vito, A.; Salem, O.; Workenhe, S.T.; Wan, Y.; Mossman, K. A Combination of Chemotherapy and Oncolytic Virotherapy Sensitizes Colorectal Adenocarcinoma to Immune Checkpoint Inhibitors in a cDC1-Dependent Manner. Int. J. Mol. Sci. 2022, 23, 1754. [Google Scholar] [CrossRef] [PubMed]
- Dosset, M.; Vargas, T.R.; Lagrange, A.; Boidot, R.; Végran, F.; Roussey, A.; Chalmin, F.; Dondaine, L.; Paul, C.; Marie-Joseph, E.L.; et al. PD-1/PD-L1 pathway: An adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology 2018, 7, e1433981. [Google Scholar] [CrossRef]
- Limagne, E.; Thibaudin, M.; Nuttin, L.; Spill, A.; Derangère, V.; Fumet, J.-D.; Amellal, N.; Peranzoni, E.; Cattan, V.; Ghiringhelli, F. Trifluridine/Tipiracil plus Oxaliplatin Improves PD-1 Blockade in Colorectal Cancer by Inducing Immunogenic Cell Death and Depleting Macrophages. Cancer Immunol. Res. 2019, 7, 1958–1969. [Google Scholar] [CrossRef]
- Mathur, D.; Root, A.R.; Bugaj-Gaweda, B.; Bisulco, S.; Tan, X.; Fang, W.; Kearney, J.C.; Lucas, J.; Guffroy, M.; Golas, J.; et al. A Novel GUCY2C-CD3 T-Cell Engaging Bispecific Construct (PF-07062119) for the Treatment of Gastrointestinal Cancers. Clin. Cancer Res. 2020, 26, 2188–2202. [Google Scholar] [CrossRef]
- Immunotherapy Using Tumor Infiltrating Lymphocytes for Patients with Metastatic Cancer. 2010. Available online: clinicaltrials.gov (accessed on 10 November 2022).
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, J.-A.; Zhang, H.-X.; Jiang, Y.-N.; Luo, W.-H. Cancer vaccines: Targeting KRAS-driven cancers. Expert Rev. Vaccines 2020, 19, 163–173. [Google Scholar] [CrossRef]
- Toubaji, A.; Achtar, M.; Provenzano, M.; Herrin, V.E.; Behrens, R.; Hamilton, M.; Bernstein, S.; Venzon, D.; Gause, B.; Marincola, F.; et al. Pilot study of mutant ras peptide-based vaccine as an adjuvant treatment in pancreatic and colorectal cancers. Cancer Immunol. Immunother. 2008, 57, 1413–1420. [Google Scholar] [CrossRef]
- A Study of a Personalized Cancer Vaccine Targeting Shared Neoantigens. 2019. Available online: clinicaltrials.gov (accessed on 10 April 2023).
- Pooled Mutant KRAS-Targeted Long Peptide Vaccine Combined with Nivolumab and Ipilimumab for Patients with Resected MMR-p Colorectal and Pancreatic Cancer. 2016. Available online: clinicaltrials.gov (accessed on 10 April 2023).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Alternate Names | Manufacturer | Target | Development Stage |
|---|---|---|---|---|
| ARS-853 | Wellspring Biosciences | G12C | In vivo | |
| ARS-1620 | Wellspring Biosciences | G12C | In vivo | |
| AMG510 | Sotorasib | Amgen | G12C | FDA-approved |
| MRTX849 | Adagrasib | Mirati therapeutics | G12C | FDA-approved |
| MRTX-EX185 | Mirati therapeutics | G12D | In vivo | |
| MRTX-1133 | Mirati therapeutics | G12D | Phase ½ Clinical trials | |
| ASP2453 | Astellas Pharma Inc | G12C | In vivo | |
| RMC-6291 | Revolution Medicines | G12C | Phase 1 Clinical trial | |
| RMC-6236 | Revolution Medicines | G12C, G12D, G12V, G12X | In vivo (not published) | |
| RMC-036 | Revolution Medicines | G12D | In vivo (not published) | |
| RMC-037 | Revolution Medicines | G12D | In vivo (not published) | |
| BBO-8520 | Bridgebio Pharma | G12C | In vivo (not published) | |
| ERAS-3490 | Erasca | G12C | In vivo (not published) | |
| JDQ443 | Novartis | G12C | Phase 1–3 Clinical trials |
| Drug | Clinical Trial Identifier | Phase | Treatment | Cancer Type | Status and Estimated Primary Completion | Enrollment Number |
|---|---|---|---|---|---|---|
| Direct Inhibitors | ||||||
| Adagrasib | NCT04793958 | III | In combination with Cetuximab against FOLFOX or FOLFIRI | KRAS-G12C-mutant advanced/metastatic colorectal cancer | Recruiting-April 2024 | 420 |
| NCT03785249 | I/II | Single agent or with Cetuximab/Pembrolizumab/Afatinib | Advanced KRAS G12C mutated solid cancer | Recruiting-December 2023 | 822 | |
| NCT05375994 | I/II | In combination with Avutometinib | G12C mutated NSCLC | Recruiting-July 2024 | 85 | |
| NCT05578092 | I/II | In combination with MRTX0902 (SOSi) | KRAS G12C mutated advanced solid cancer | Recruiting-July 2026 | 225 | |
| NCT05178888 | I | In combination with Palbociclib | KRAS G12C mutated advanced solid cancer | Active-December 2023 | 11 | |
| NCT05472623 | III | Single agent or with Nivolumab | KRAS G12C NSCLC | Not yet recruiting-November 2025 | 42 | |
| NCT04613596 | II | Single agent or in combination with Pembrolizumab | KRAS-G12C-mutant advanced/metastatic NSCLC | Recruiting-March 2028 | 950 | |
| III | Combination with Pembrolizumab versus Pembrolizumab plus chemotherapy | |||||
| NCT04330664 | I/II | In combination with TNO155 | KRAS G12C mutated advanced solid cancer | Active-September 2022 | 86 | |
| NCT05722327 | I | In combination with Cetuximab and Irinotecan | KRAS-G12C-mutant colorectal cancer | Not yet recruiting-September 2025 | 24 | |
| NCT05609578 | II | In combination with Pembrolizumab | KRAS-G12Cmutant advanced/metastatic NSCLC | Recruiting-June 2025 | 90 | |
| NCT04685135 | III | Single agent versus Docetaxel | KRAS-G12C-mutant advanced/metastatic NSCLC | Recruiting-August 2023 | 340 | |
| NCT05634525 | I | Single agent | KRAS-G12C-mutated pancreatic cancer | Not yet recruiting-November 2025 | 14 | |
| Sotorasib | NCT05451056 | II | Single agent | KRAS-G12C-mutant NSCLC | Not yet recruiting-January 2026 | 37 |
| NCT05118854 | II | In combination with Cisplatin or Carboplatin and Pemetrexed | KRAS-G12C-mutant NSCLC | Recruiting-October 2023 | 27 | |
| NCT05374538 | I | In combination with VIC-1911 (Aurora Kinase A inhibitor) | KRAS-G12C-mutant advanced/metastatic NSCLC | Recruiting-March 2026 | 140 | |
| NCT04933695 | II | Single agent | KRAS-G12C-mutant advanced/metastatic NSCLC | Active-May 2024 | 42 | |
| NCT05054725 | II | In combination with RMC-4630 | KRAS-G12C-mutant NSCLC | Active-September 2023 | 47 | |
| NCT05638295 | II | Single agent or with Panitumumab | KRAS-G12C-mutant advanced/metastatic solid cancers | Not yet recruiting-December 2025 | 105 | |
| NCT05198934 | III | In combination with Panitumumab versus chemotherapy (Tas-102 or Regorafenib) | KRAS-G12C-mutated metastatic colorectal cancer | Active-May 2023 | 160 | |
| NCT04185883 | I/II | Single agent or in combination with several anti-cancer therapies | KRAS-G12C-mutant advanced solid cancers | Recruiting-July 2026 | 1143 | |
| NCT03600883 | I/II | Single agent or with Anti-PD1/L1 or Midazolam | KRAS-G12C-mutant advanced solid cancers | Active-May 2026 | 713 | |
| NCT05480865 | I | In combination with BBP-398 (SHP2 Inhibitor) | KRAS-G12C-mutant metastatic solid cancers | Recruiting-June 2024 | 85 | |
| NCT05074810 | I/II | In combination with Avutometinib | KRAS G12C. Mutant NSCLC | Recruiting-December 2023 | 53 | |
| NCT05313009 | I/II | In combination with Tarlozotinib (HER kinase inhibitor) | KRAS-G12C-mutant NSCLC | Recruiting-December 2023 | 30 | |
| NCT04303780 | III | Single agent versus Docetaxel | KRAS-G12C-mutant advanced/metastatic NSCLC | Active-August 2022 | 345 | |
| MRTX-1133 | NCT05737706 | I/II | Single agent | KRAS-G12C-mutant advanced solid cancers | Recruiting-August 2026 | 304 |
| RMC-6291 | NCT05462717 | I | Single agent | KRAS-G12C-mutant advanced solid cancers | Recruiting-November 2024 | 117 |
| RMC_6236 | NCT05379985 | I | Single agent | KRAS-G12C-mutant advanced solid cancers | Recruiting-June 2024 | 141 |
| JDQ443 | NCT05132075 | III | Single agent versus Docetaxel | KRAS-G12C-mutant advanced NSCLC | Recruiting-April 2025 | 360 |
| NCT05445843 | II | Single agent | KRAS-G12C-mutant advanced/metastatic NSCLC | Recruiting-November 2026 | 120 | |
| NCT04699188 | I/II | Single agent and in combination with TNO155 and/or Tislelizumab | KRAS-G12C-mutant advanced solid cancers | Recruiting-May 2025 | 375 | |
| NCT05714891 | II | Single agent | Surgically resectable NSCLC | Not yet recruiting-August 2026 | 27 | |
| NCT05358249 | I/II | In combination with Trametinib, Ribociclib, or Cetuximab | KRAS-G12C-mutant advanced solid cancers | Recruiting-June 2025 | 346 | |
| IBI351 | NCT05497336 | I | Single agent or in combination with Cetuximab | KRAS-G12C-mutant advanced/metastatic colorectal cancer | Recruiting-August 2023 | 80 |
| JAB-21822 | NCT05194995 | I/II | In combination with Cetuximab | KRAS-G12C-mutant colorectal, small intestine and appendiceal cancer | Recruiting-December 2023 | 62 |
| NCT05288205 | I/II | In combination with JAB-3122 (SHP2i) | KRAS-G12C-mutant advanced solid cancers | Recruiting-March 2026 | 124 | |
| GDC-6036 | NCT04449874 | I | Single agent or in combination with several anti-cancer therapies | KRAS-G12C-mutant advanced/metastatic solid cancers | Recruiting-November 2024 | 498 |
| HBI-2438 | NCT05485974 | I | Single agent | KRAS-G12C-mutant advanced solid cancers | Recruiting-August 2025 | 44 |
| ELI-002 | NCT05726864 | I/II | Single agent | KRAS/NRAS-mutated solid cancers | Recruiting-November 2026 | 156 |
| NCT04853017 | I | Single agent | KRAS/NRAS-mutated solid cancers | Recruiting-January 2023 | 18 | |
| BPI-421286 | NCT05315180 | I | Single agent | KRAS-G12C-mutant and not advanced solid cancers | Recruiting-July 2023 | 80 |
| DCC-3116 | NCT04892017 | I/II | Single agent or in combination with Trametinib, Binimetinib, or Sotorasib | RAS/MAPK-mutant advanced/metastatic solid cancers | Recruiting-April 2024 | 323 |
| BDTX-4933 | NCT05786924 | I | Single agent | BRAF-mutant, select KRAS and NRAS-mutant advanced/metastatic solid cancers | Recruiting-June 2026 | 140 |
| Indirect targeting | ||||||
| RMC-4630 | NCT04916236 | I | In combination with LY3214996 | KRAS-mutant metastatic solid cancers | Recruiting-January 2024 | 55 |
| Vitamin C | NCT03146962 | II | Single agent | RAS/BRAF-mutant and wildtype advanced/metastatic solid cancers | Recruiting-June 2023 | 78 |
| Telaglenastat | NCT03965845 | I | Single agent or in combination with Palbociclib | KRAS-mutant advanced solid cancers | Completed-September 2021 | 53 |
| Immunotherapy | ||||||
| Sintilimab | NCT04745130 | II | In combination with Regorafenib and Cetuximab | Metastatic colorectal cancer (KRAS wt and mutant) | Recruiting-February 2023 | 90 |
| KRAS peptide vaccine | NCT04117087 | I | In combination with Nivolumab and Ipilumab | KRAS-mutant PDAC or metastatic colorectal cancer | Recruiting-December 2024 | 30 |
| NCT03953235 | I/II | In combination with Nivolumab and Ipilumab | RAS/BRAF/TP53-mutant MSS-CRC, NCLC, PDAC | Active-December 2023 | 30 | |
| Molecule/Therapy | Mechanism to Aid Immunotherapy |
|---|---|
| Blocking interleukin 17A (IL-17A) | Decreases PD-L1 expression and increases cytotoxic T-cells |
| Degrading Tribbles homolog 3 (TRIB3) | Increases tumor lymphocyte infiltration |
| Degrading neutrophil extracellular traps (NETS) | Removes a physical barrier between immunotherapy and cancer cells |
| Introducing mitochondrial antiviral signaling gene (MAVS) | Increases CD8 T-cells cytotoxicity, and the expression of PD-L1 |
| 5-FU + oxaliplatin combination | Induces immunogenic cell death that stimulates the immune system |
| Tas-102 + oxaliplatin combination | Induces immunogenic cell death that stimulates the immune system |
| PF-07062119 treatment | Bispecific antibody that acts as a scaffold to bring T-cells in close proximity to cancer cells also increases PD-1 and PD-L1 expression |
| Adoptive T-cell transfer | Expands cancer cell specific T-cells ex vivo for treatment |
| Sotorasib | Increases T-cell priming and infiltration, antigen presentation, and activation |
| Vaccine | Primes immune system to KRAS-mutant proteins for adaptive killing |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tria, S.M.; Burge, M.E.; Whitehall, V.L.J. The Therapeutic Landscape for KRAS-Mutated Colorectal Cancers. Cancers 2023, 15, 2375. https://doi.org/10.3390/cancers15082375
Tria SM, Burge ME, Whitehall VLJ. The Therapeutic Landscape for KRAS-Mutated Colorectal Cancers. Cancers. 2023; 15(8):2375. https://doi.org/10.3390/cancers15082375
Chicago/Turabian StyleTria, Simon Manuel, Matthew E. Burge, and Vicki L. J. Whitehall. 2023. "The Therapeutic Landscape for KRAS-Mutated Colorectal Cancers" Cancers 15, no. 8: 2375. https://doi.org/10.3390/cancers15082375
APA StyleTria, S. M., Burge, M. E., & Whitehall, V. L. J. (2023). The Therapeutic Landscape for KRAS-Mutated Colorectal Cancers. Cancers, 15(8), 2375. https://doi.org/10.3390/cancers15082375