
Co-Occurrence of Beckwith–Wiedemann Syndrome and Early-Onset Colorectal Cancer
 , , ,
, , ,  , , , ,
, , , ,  , , and
, , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Ethics
2.2. Tumor Diagnostics
2.3. Genetic and Epigenetic Analysis of Blood, Neoplastic and Perineoplastic Tissues
2.4. DNA Methylation Analysis
2.5. Copy Number Analysis
2.6. DNA Sequencing
2.7. Expression Analysis
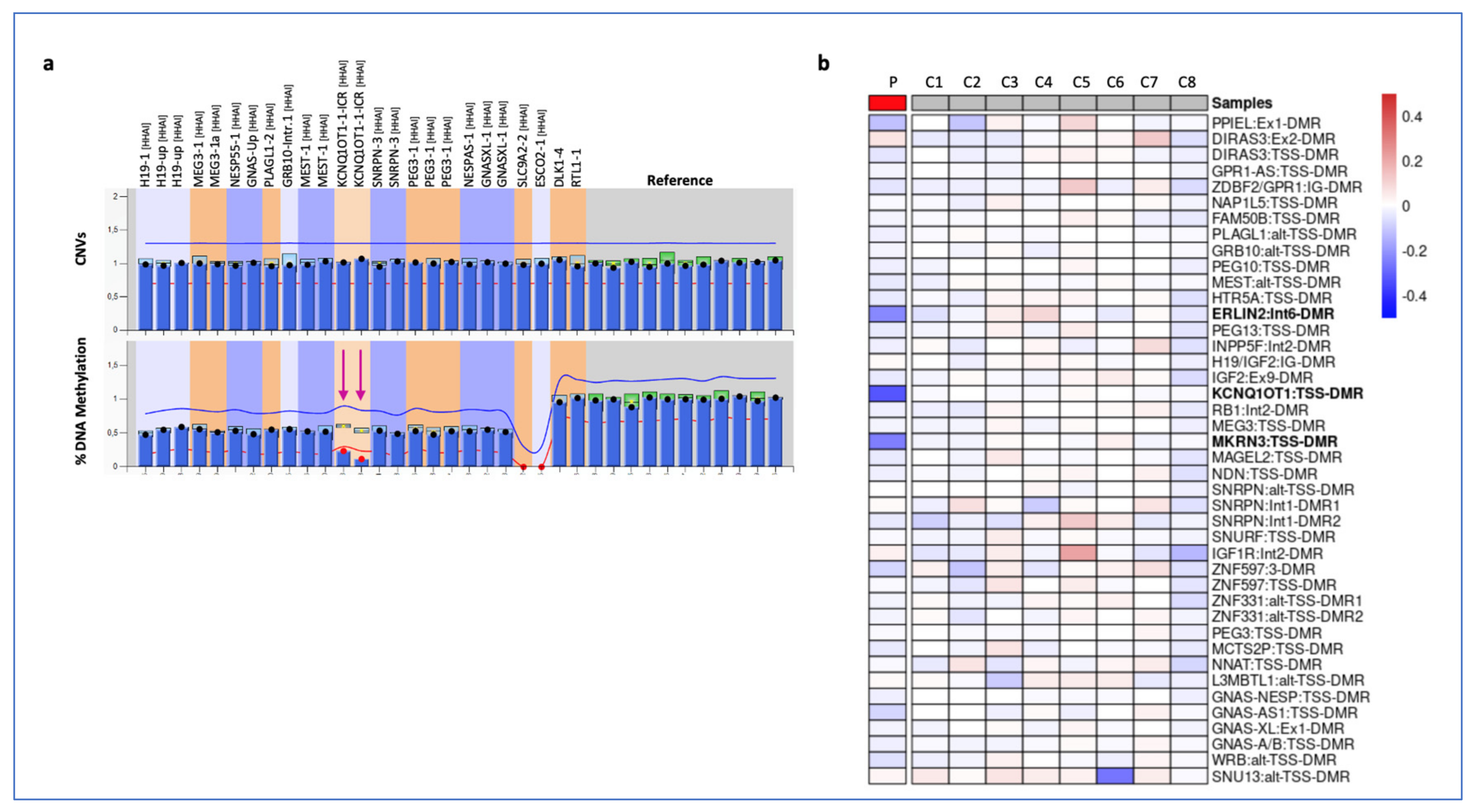
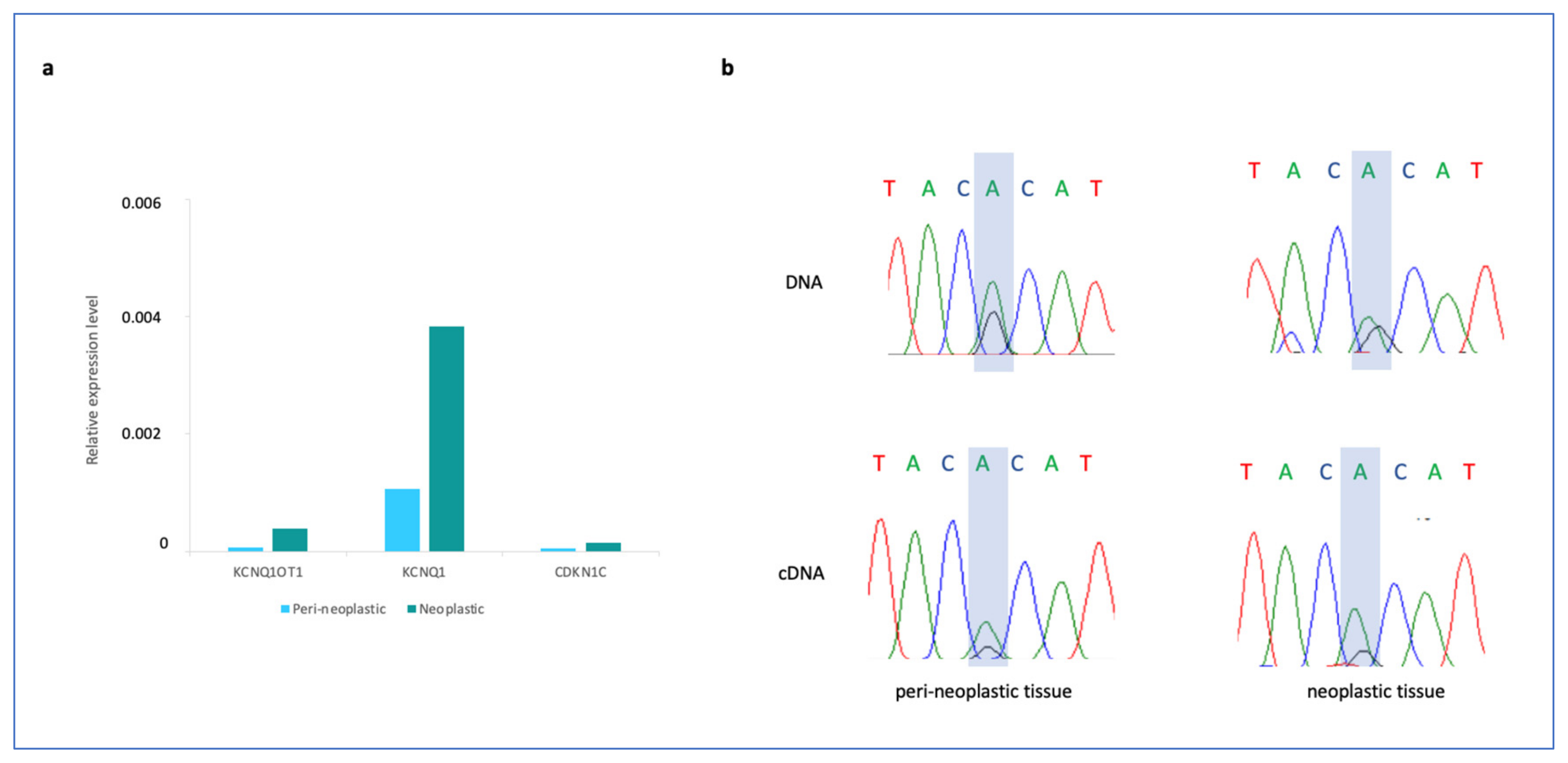
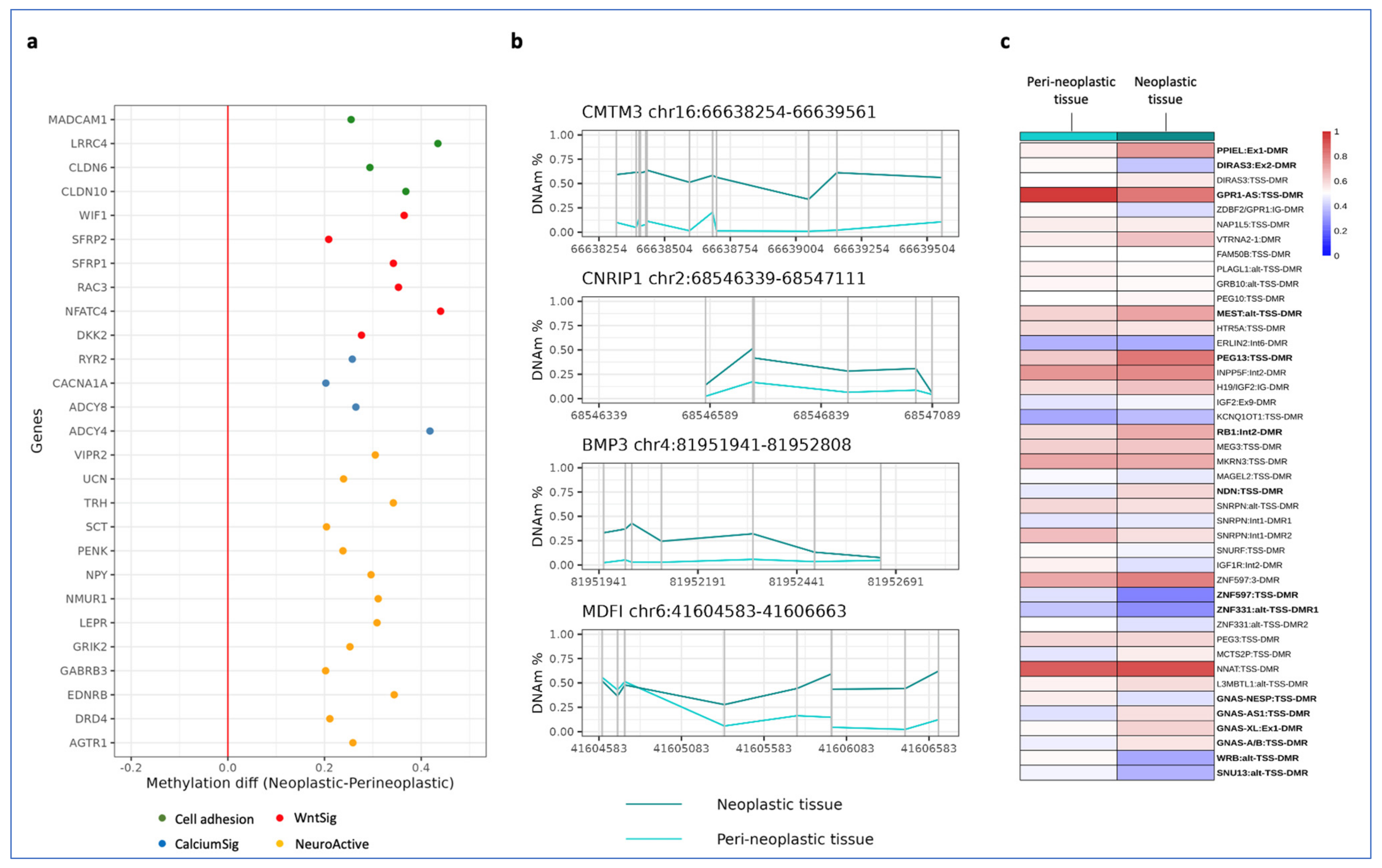
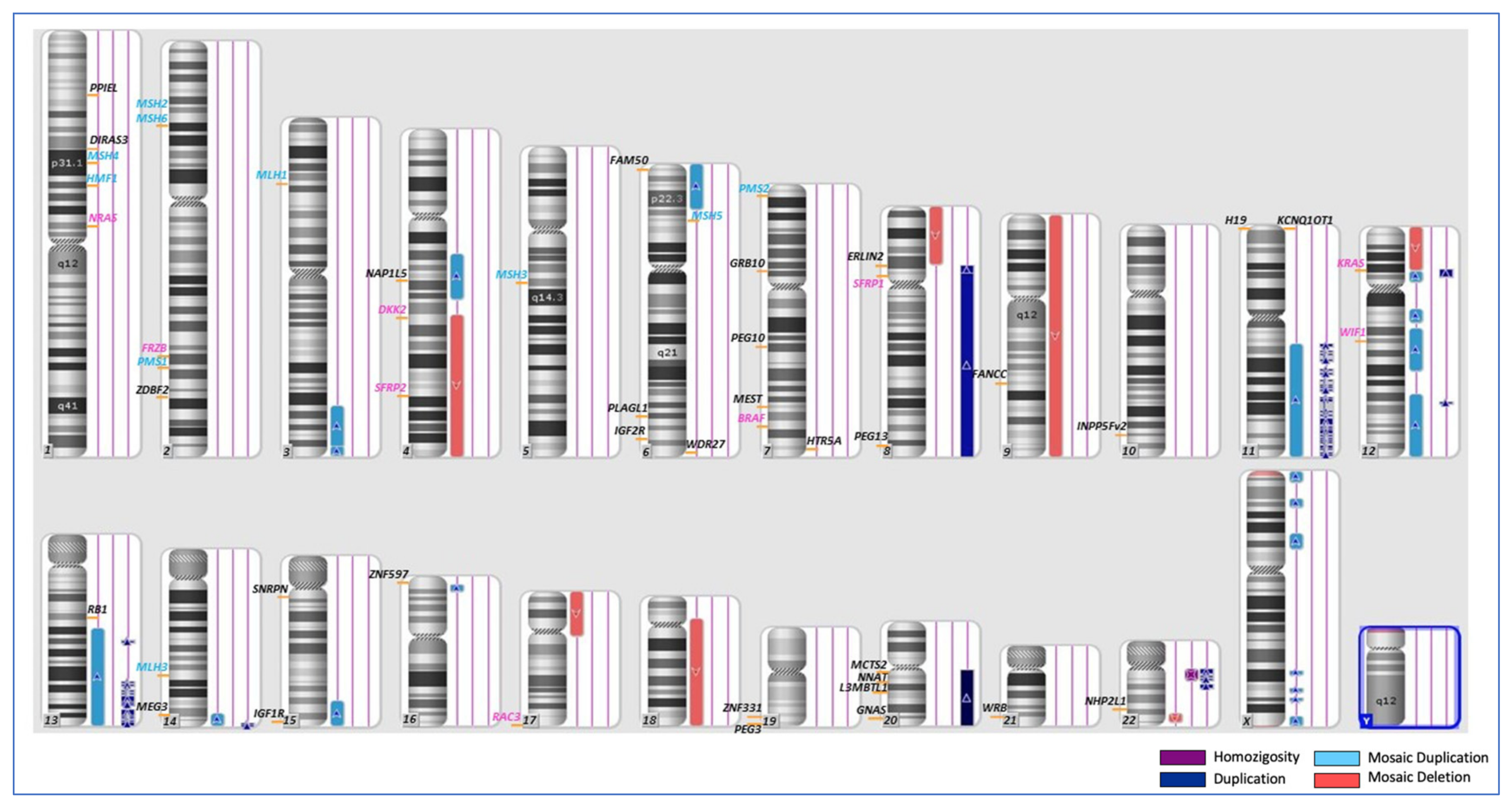
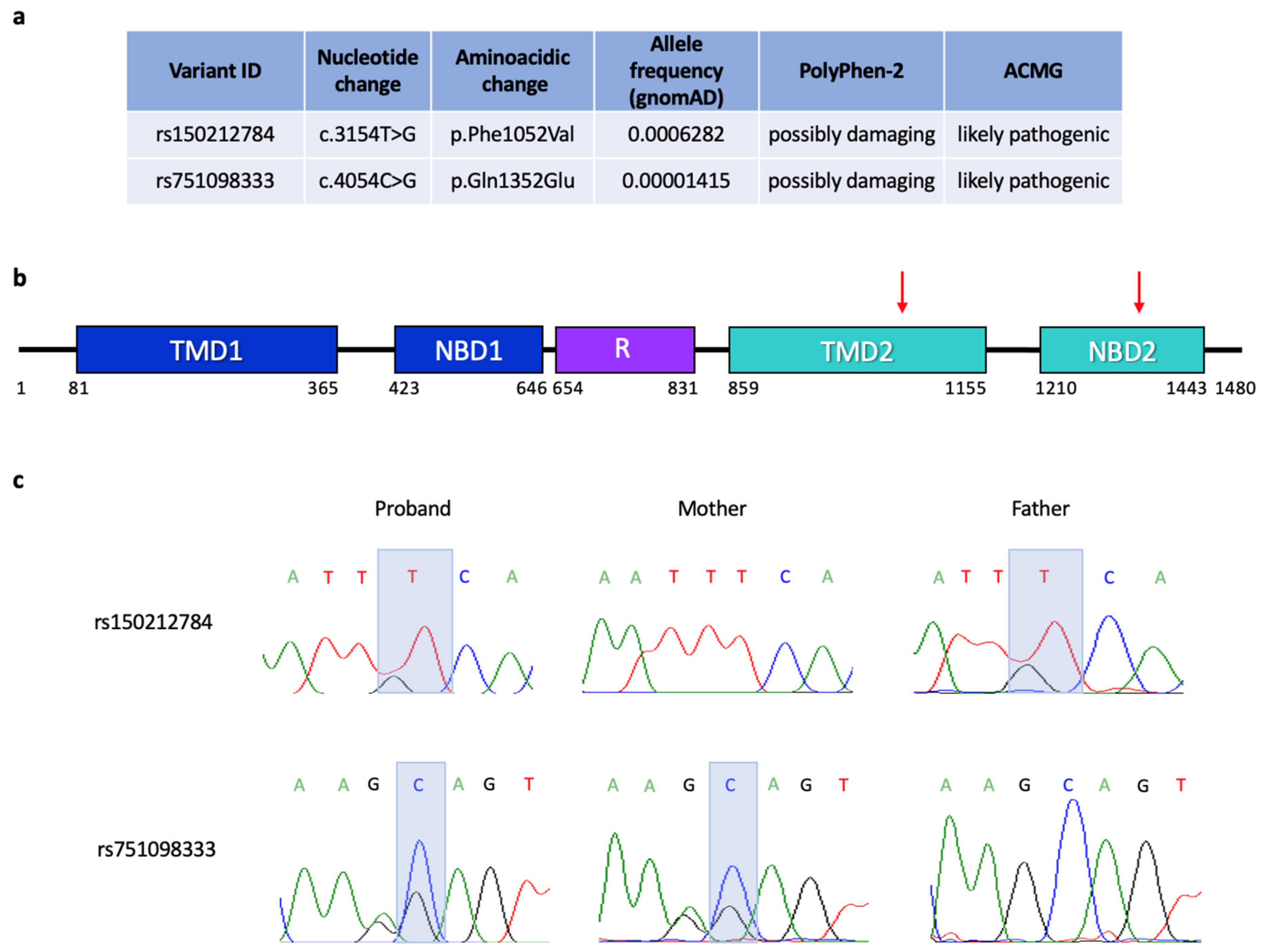
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef]
- Monk, D.; Mackay, D.J.G.; Eggermann, T.; Maher, E.R.; Riccio, A. Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019, 20, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Choufani, S.; Shuman, C.; Weksberg, R. Molecular findings in Beckwith-Wiedemann syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Duffy, K.A.; Getz, K.D.; Hathaway, E.R.; Byrne, M.E.; MacFarland, S.P.; Kalish, J.M. Characteristics Associated with Tumor Development in Individuals Diagnosed with Beckwith-Wiedemann Spectrum: Novel Tumor-(epi)Genotype-Phenotype Associations in the BWSp Population. Genes 2021, 12, 1839. [Google Scholar] [CrossRef]
- Eggermann, T.; Maher, E.R.; Kratz, C.P.; Prawitt, D. Molecular Basis of Beckwith-Wiedemann Syndrome Spectrum with Associated Tumors and Consequences for Clinical Practice. Cancers 2022, 14, 3083. [Google Scholar] [CrossRef] [PubMed]
- Brioude, F.; Nicolas, C.; Marey, I.; Gaillard, S.; Bernier, M.; Das Neves, C.; Le Bouc, Y.; Touraine, P.; Netchine, I. Hypercortisolism due to a Pituitary Adenoma Associated with Beckwith-Wiedemann Syndrome. Horm. Res. Paediatr. 2016, 86, 206–211. [Google Scholar] [CrossRef]
- Romanelli, V.; Nevado, J.; Fraga, M.; Trujillo, A.M.; Mori, M.; Fernández, L.; Pérez de Nanclares, G.; Martínez-Glez, V.; Pita, G.; Meneses, H.; et al. Constitutional mosaic genome-wide uniparental disomy due to diploidisation: An unusual cancer-predisposing mechanism. J. Med. Genet. 2011, 48, 212–216. [Google Scholar] [CrossRef]
- Bertoin, F.; Letouzé, E.; Grignani, P.; Patey, M.; Rossignol, S.; Libé, R.; Pasqual, C.; Lardière-Deguelte, S.; Hoeffel-Fornes, C.; Gaillard, D.; et al. Genome-wide paternal uniparental disomy as a cause of Beckwith-Wiedemann syndrome associated with recurrent virilizing adrenocortical tumors. Horm. Metab. Res. 2015, 47, 497–503. [Google Scholar] [CrossRef]
- Clouston, W.M.; Cannell, G.C.; Fryar, B.G.; Searle, J.W.; Martin, N.I.; Mortimer, R.H. Virilizing adrenal adenoma in an adult with the Beckwith-Wiedemann syndrome: Paradoxical response to dexamethasone. Clin. Endocrinol. 1989, 31, 467–473. [Google Scholar] [CrossRef]
- Bémurat, L.; Gosse, P.; Ballanger, P.; Tauzin-Fin, P.; Barat, P.; Lacombe, D.; Lemétayer, P.; Clémenty, J. Successful laparoscopic operation of bilateral pheochromocytoma in a patient with Beckwith-Wiedemann syndrome. J. Hum. Hypertens. 2002, 16, 281–284. [Google Scholar] [CrossRef]
- Gazzin, A.; Carli, D.; Sirchia, F.; Molinatto, C.; Cardaropoli, S.; Palumbo, G.; Zampino, G.; Ferrero, G.B.; Mussa, A. Phenotype evolution and health issues of adults with Beckwith-Wiedemann syndrome. Am. J. Med. Genet. A 2019, 179, 1691–1702. [Google Scholar] [CrossRef]
- Cui, H.; Cruz-Correa, M.; Giardiello, F.M.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; He, X.; Powe, N.R.; Feinberg, A.P. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science 2003, 299, 1753–1755. [Google Scholar] [CrossRef]
- Goovaerts, T.; Steyaert, S.; Vandenbussche, C.A.; Galle, J.; Thas, O.; Van Criekinge, W.; De Meyer, T. A comprehensive overview of genomic imprinting in breast and its deregulation in cancer. Nat. Commun. 2018, 9, 4120. [Google Scholar] [CrossRef]
- Ito, Y.; Koessler, T.; Ibrahim, A.E.; Rai, S.; Vowler, S.L.; Abu-Amero, S.; Silva, A.L.; Maia, A.T.; Huddleston, J.E.; Uribe-Lewis, S.; et al. Somatically acquired hypomethylation of IGF2 in breast and colorectal cancer. Hum. Mol. Genet. 2008, 17, 2633–2643. [Google Scholar] [CrossRef]
- Martin-Trujillo, A.; Vidal, E.; Monteagudo-Sánchez, A.; Sanchez-Delgado, M.; Moran, S.; Hernandez Mora, J.R.; Heyn, H.; Guitart, M.; Esteller, M.; Monk, D. Copy number rather than epigenetic alterations are the major dictator of imprinted methylation in tumors. Nat. Commun. 2017, 8, 467. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Meltzer, S.J.; James, S.P. Colon polyps in Beckwith-Wiedmann syndrome: Role of imprinted genes. Gastroenterology 2000, 118, 637. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Mauri, G.; Sartore-Bianchi, A.; Russo, A.G.; Marsoni, S.; Bardelli, A.; Siena, S. Early-onset colorectal cancer in young individuals. Mol. Oncol. 2019, 13, 109–131. [Google Scholar] [CrossRef]
- Sparago, A.; Verma, A.; Patricelli, M.G.; Pignata, L.; Russo, S.; Calzari, L.; De Francesco, N.; Del Prete, R.; Palumbo, O.; Carella, M.; et al. The phenotypic variations of multi-locus imprinting disturbances associated with maternal-effect variants of NLRP5 range from overt imprinting disorder to apparently healthy phenotype. Clin. Epigenet. 2019, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Pignata, L.; Cecere, F.; Verma, A.; Hay Mele, B.; Monticelli, M.; Acurzio, B.; Giaccari, C.; Sparago, A.; Hernandez Mora, J.R.; Monteagudo-Sánchez, A.; et al. Novel genetic variants of KHDC3L and other members of the subcortical maternal complex associated with Beckwith-Wiedemann syndrome or Pseudohypoparathyroidism 1B and multi-locus imprinting disturbances. Clin. Epigenet. 2022, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Pignata, L.; Palumbo, O.; Cerrato, F.; Acurzio, B.; de Álava, E.; Roma, J.; Gallego, S.; Mora, J.; Carella, M.; Riccio, A.; et al. Both Epimutations and Chromosome Aberrations Affect Multiple Imprinted Loci in Aggressive Wilms Tumors. Cancers 2020, 12, 3411. [Google Scholar] [CrossRef] [PubMed]
- Cubellis, M.V.; Pignata, L.; Verma, A.; Sparago, A.; Del Prete, R.; Monticelli, M.; Calzari, L.; Antona, V.; Melis, D.; Tenconi, R.; et al. Loss-of-function maternal-effect mutations of PADI6 are associated with familial and sporadic Beckwith-Wiedemann syndrome with multi-locus imprinting disturbance. Clin. Epigenet. 2020, 12, 139. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Mackay, D.; Bliek, J.; Kagami, M.; Tenorio-Castano, J.; Pereda, A.; Brioude, F.; Netchine, I.; Papingi, D.; de Franco, E.; Lever, M.; et al. First step towards a consensus strategy for multi-locus diagnostic testing of imprinting disorders. Clin. Epigenet. 2022, 14, 143. [Google Scholar] [CrossRef]
- Müller, D.; Győrffy, B. DNA methylation-based diagnostic, prognostic, and predictive biomarkers in colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188722. [Google Scholar] [CrossRef]
- Tapial, S.; Olmedillas-López, S.; Rueda, D.; Arriba, M.; García, J.L.; Vivas, A.; Pérez, J.; Pena-Couso, L.; Olivera, R.; Rodríguez, Y.; et al. Cimp-Positive Status is More Representative in Multiple Colorectal Cancers than in Unique Primary Colorectal Cancers. Sci. Rep. 2019, 9, 10516. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Chromosome 20q11.21 Amplifications in Colorectal Cancer. Cancer Genom. Proteom. 2021, 18, 487–496. [Google Scholar] [CrossRef]
- Wang, G.; Wang, F.; Meng, Z.; Wang, N.; Zhou, C.; Zhang, J.; Zhao, L.; Shan, B. Uncovering potential genes in colorectal cancer based on integrated and DNA methylation analysis in the gene expression omnibus database. BMC Cancer 2022, 22, 138. [Google Scholar] [CrossRef]
- Ye, Z.; Li, Y.; Xie, J.; Feng, Z.; Yang, X.; Wu, Y.; Pu, Y.; Gao, J.; Xu, X.; Zhu, Z.; et al. Integrated bioinformatics identifies the dysregulation induced by aberrant gene methylation in colorectal carcinoma. Genes Dis. 2021, 8, 521–530. [Google Scholar] [CrossRef]
- Wang, W.; Yu, S.; Huang, S.; Deng, R.; Ding, Y.; Wu, Y.; Li, X.; Wang, A.; Wang, S.; Chen, W.; et al. A Complex Role for Calcium Signaling in Colorectal Cancer Development and Progression. Mol. Cancer Res. 2019, 17, 2145–2153. [Google Scholar] [CrossRef]
- Brocardo, M.; Henderson, B.R. APC shuttling to the membrane, nucleus and beyond. Trends Cell Biol. 2008, 18, 587–596. [Google Scholar] [CrossRef]
- Nomura, R.; Saito, T.; Mitomi, H.; Hidaka, Y.; Lee, S.Y.; Watanabe, S.; Yao, T. GNAS mutation as an alternative mechanism of activation of the Wnt/β-catenin signaling pathway in gastric adenocarcinoma of the fundic gland type. Hum. Pathol. 2014, 45, 2488–2496. [Google Scholar] [CrossRef]
- Miller, A.C.; Comellas, A.P.; Hornick, D.B.; Stoltz, D.A.; Cavanaugh, J.E.; Gerke, A.K.; Welsh, M.J.; Zabner, J.; Polgreen, P.M. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc. Natl. Acad. Sci. USA 2020, 117, 1621–1627. [Google Scholar] [CrossRef]
- Scott, P.; Anderson, K.; Singhania, M.; Cormier, R. Cystic Fibrosis, CFTR, and Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 2891. [Google Scholar] [CrossRef]
- Niccum, D.E.; Billings, J.L.; Dunitz, J.M.; Khoruts, A. Colonoscopic screening shows increased early incidence and progression of adenomas in cystic fibrosis. J. Cyst. Fibros. 2016, 15, 548–553. [Google Scholar] [CrossRef]
- Nakano, S.; Murakami, K.; Meguro, M.; Soejima, H.; Higashimoto, K.; Urano, T.; Kugoh, H.; Mukai, T.; Ikeguchi, M.; Oshimura, M. Expression profile of LIT1/KCNQ1OT1 and epigenetic status at the KvDMR1 in colorectal cancers. Cancer Sci. 2006, 97, 1147–1154. [Google Scholar] [CrossRef]
- Lin, Z.B.; Long, P.; Zhao, Z.; Zhang, Y.R.; Chu, X.D.; Zhao, X.X.; Ding, H.; Huan, S.W.; Pan, Y.L.; Pan, J.H. Long Noncoding RNA KCNQ1OT1 is a Prognostic Biomarker and mediates CD8. Int. J. Biol. Sci. 2021, 17, 1757–1768. [Google Scholar] [CrossRef]
- Zhu, S.; Chen, C.Y.; Hao, Y. LncRNA KCNQ1OT1 acts as miR-216b-5p sponge to promote colorectal cancer progression via up-regulating ZNF146. J. Mol. Histol. 2021, 52, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Sunamura, N.; Ohira, T.; Kataoka, M.; Inaoka, D.; Tanabe, H.; Nakayama, Y.; Oshimura, M.; Kugoh, H. Regulation of functional KCNQ1OT1 lncRNA by β-catenin. Sci. Rep. 2016, 6, 20690. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772. [Google Scholar] [CrossRef] [PubMed]
- Rapetti-Mauss, R.; Bustos, V.; Thomas, W.; McBryan, J.; Harvey, H.; Lajczak, N.; Madden, S.F.; Pellissier, B.; Borgese, F.; Soriani, O.; et al. Bidirectional KCNQ1:β-catenin interaction drives colorectal cancer cell differentiation. Proc. Natl. Acad. Sci. USA 2017, 114, 4159–4164. [Google Scholar] [CrossRef]
- Li, J.Q.; Wu, F.; Usuki, H.; Kubo, A.; Masaki, T.; Fujita, J.; Bandoh, S.; Saoo, K.; Takeuchi, H.; Kuriyama, S.; et al. Loss of p57KIP2 is associated with colorectal carcinogenesis. Int. J. Oncol. 2003, 23, 1537–1543. [Google Scholar] [CrossRef]
- Sun, K.; Wang, W.; Zeng, J.J.; Wu, C.T.; Lei, S.T.; Li, G.X. MicroRNA-221 inhibits CDKN1C/p57 expression in human colorectal carcinoma. Acta Pharmacol. Sin. 2011, 32, 375–384. [Google Scholar] [CrossRef]
- Feinberg, A.P. The Epigenetics of cancer etiology. Semin. Cancer Biol. 2004, 14, 427–432. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term_Name | Term_Size | Intersection_Size | p_Value |
|---|---|---|---|
| Neuroactive ligand–receptor interaction | 227 | 44 | 2.137936−9 |
| Calcium signaling pathway | 198 | 30 | 8.469467−4 |
| WNT signaling pathway | 155 | 23 | 1.356191−2 |
| Cell adhesion molecules | 104 | 17 | 3.260210−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cecere, F.; Pignata, L.; Hay Mele, B.; Saadat, A.; D’Angelo, E.; Palumbo, O.; Palumbo, P.; Carella, M.; Scarano, G.; Rossi, G.B.; et al. Co-Occurrence of Beckwith–Wiedemann Syndrome and Early-Onset Colorectal Cancer. Cancers 2023, 15, 1944. https://doi.org/10.3390/cancers15071944
Cecere F, Pignata L, Hay Mele B, Saadat A, D’Angelo E, Palumbo O, Palumbo P, Carella M, Scarano G, Rossi GB, et al. Co-Occurrence of Beckwith–Wiedemann Syndrome and Early-Onset Colorectal Cancer. Cancers. 2023; 15(7):1944. https://doi.org/10.3390/cancers15071944
Chicago/Turabian StyleCecere, Francesco, Laura Pignata, Bruno Hay Mele, Abu Saadat, Emilia D’Angelo, Orazio Palumbo, Pietro Palumbo, Massimo Carella, Gioacchino Scarano, Giovanni Battista Rossi, and et al. 2023. "Co-Occurrence of Beckwith–Wiedemann Syndrome and Early-Onset Colorectal Cancer" Cancers 15, no. 7: 1944. https://doi.org/10.3390/cancers15071944
APA StyleCecere, F., Pignata, L., Hay Mele, B., Saadat, A., D’Angelo, E., Palumbo, O., Palumbo, P., Carella, M., Scarano, G., Rossi, G. B., Angelini, C., Sparago, A., Cerrato, F., & Riccio, A. (2023). Co-Occurrence of Beckwith–Wiedemann Syndrome and Early-Onset Colorectal Cancer. Cancers, 15(7), 1944. https://doi.org/10.3390/cancers15071944